La gluconeogenesi è la via metabolica che permette, anche agli organismi non fotosintetizzanti, di produrre glucosio a partire dal piruvato, la base coniugata dell’acido piruvico, e da altri precursori non glucidici.[1][6]
E’ presente in tutti gli animali, piante, funghi e microorganismi, con essenzialmente le stesse reazioni, che portano, da due molecole di piruvato, alla sintesi di una molecola di glucosio. Dunque è essenzialmente l’inverso della glicolisi, che invece procede dal glucosio fino al piruvato, e con essa condivide molti enzimi.[5]
La glicogenolisi è distinta dalla gluconeogenesi, non corrispondendo a una sintesi de novo del monosaccaride, come si può facilmente evidenziare osservando la sua reazione complessiva:
Glicogeno o (glucosio)n → n molecole di glucosio
La discussione successiva verterà sulla gluconeogenesi che avviene negli animali superiori, e in particolare nel fegato dei mammiferi.
Indice
- Perché è importante?
- Dove avviene?
- Tappe irreversibili
- Conversione del piruvato in fosfoenolpiruvato
- Conversione del fruttosio-1,6-bisfosfato in fruttosio-6-fosfato
- Conversione del glucosio-6-fosfato a glucosio
- La gluconeogenesi è energeticamente costosa
- Regolazione coordinata della glicolisi e gluconeogenesi
- Regolazione della gluconeogenesi
- Precursori
- Bibliografia
Perché è importante?
La gluconeogenesi è una via metabolica di grande importanza per almeno due motivi.
- Assicura il mantenimento di una adeguata concentrazione ematica di glucosio quando le riserve epatiche di glicogeno sono prossime all’esaurimento e lo zucchero non viene assunto con l’alimentazione.[6]
Il mantenimento della glicemia entro il range di normalità, 3,3-5,5 mmol/L (60 e i 99 mg/dL), è essenziale in quanto molte cellule e tessuti dipendono largamente o totalmente dal glucosio per soddisfare le loro richieste di ATP; tra questi i globuli rossi, i neuroni, il muscolo scheletrico quando lavora in anaerobiosi, la midollare del rene, i testicoli, la lente e la cornea dell’occhio, e i tessuti embrionali. Considerando ad esempio il cervello, il suo fabbisogno giornaliero di glucosio è di circa 120 g, una quantità pari a:
oltre il 50% delle riserve corporee totali del monosaccaride, circa 210 g, di cui 190 g immagazzinato come glicogeno muscolare ed epatico, e 20 g in forma libera nei fluidi corporei;
circa il 75% del fabbisogno giornaliero di glucosio dell’intero organismo, quindi sui 160 g.[1]
Nel digiuno breve, come nell’intervallo tra i pasti o durante la notte, la glicemia è mantenuta entro il range di normalità grazie alla glicogenolisi epatica e al rilascio di acidi grassi dal tessuto adiposo e corpi chetonici dal fegato. Acidi grassi e corpi chetonici, utilizzati di preferenza dal muscolo scheletrico, consentono un risparmio di glucosio che sarà disponibile per le cellule e i tessuti che da esso dipendono. Tuttavia, dopo circa 18 ore di digiuno, le riserve di glicogeno sono prossime all’esaurimento, riserve che possono divenire insufficienti anche durante l’attività fisica intensa e prolungata. Ed è a questo punto che, se non sono assunti carboidrati con l’alimentazione, la gluconeogenesi diviene essenziale.
E, a sottolineare ulteriormente l’importanza della sintesi de novo del glucosio il fatto che se i valori della glicemia scendono al di sotto di 2 mmol/L si verifica la perdita di coscienza.
- L’eventuale escrezione del piruvato comporterebbe la perdita della possibilità di produrre ATP attraverso la sua ossidazione aerobica, ossia più di 10 molecole di ATP per ogni piruvato ossidato.
Dove avviene?
Negli animali superiori la gluconeogenesi avviene nel fegato, nella corticale del rene e nelle cellule epiteliali dell’intestino tenue, gli enterociti.[5]
Quantitativamente il fegato rappresenta la sede principale, producendo circa il 90% di tutto il glucosio sintetizzato, seguito dalla corticale del rene, con circa il 10%.[1] La maggiore capacità di sintesi del fegato rispetto alla corticale del rene è dovuta solo alle sue maggiori dimensioni; se infatti si considera il rapporto peso/sintesi, la corticale del rene produce più glucosio del fegato.
A livello della corticale del rene, le cellule che portano a termine la gluconeogenesi sono quelle del tubulo prossimale, la porzione del nefrone successiva al glomerulo. Molto del glucosio prodotto nel rene viene utilizzato dalla midollare del rene stesso, mentre l’azione dell’organo sul mantenimento della omeostasi glicemica diviene più importante durante il digiuno prolungato e nell’insufficienza epatica. Va tuttavia sottolineato che il rene, essendo privo di depositi di glicogeno, può contribuire alla regolazione della glicemia solo attraverso la gluconeogenesi e non anche attraverso la glicogenolisi, come invece può fare il fegato.
Parte della via gluconeogenetica può verificarsi anche nel muscolo scheletrico e cardiaco e nel cervello, anche se a velocità estremamente ridotta. Nell’adulto il muscolo ha circa 18 volte la massa del fegato, per cui la sua sintesi di glucosio potrebbe avere una qualche importanza quantitativa.
Tuttavia in questi tessuti la sintesi de novo del glucosio non porta alla sua liberazione in circolo, essendo assente la glucosio-6-fosfatasi (EC 3.1.3.9), l’enzima che catalizza l’ultima tappa della via.[6] Quindi, l’eventuale produzione di glucosio-6-fosfato, compreso quello derivante dalla glicogenolisi, non contribuirà al mantenimento della glicemia ma aiuterà solo a ripristinare le scorte del glicogeno, per la verità nel cervello piccole e limitate per lo più agli astrociti. L’unico contributo diretto al mantenimento della glicemia operato da questi tessuti, e in particolare dal muscolo scheletrico, vista la sua grande massa, deriva dalla piccola quota di glucosio rilasciata dall’enzima deramificante (EC 3.2.1.33) della glicogenolisi.
Per quello che riguarda la localizzazione cellulare, la maggior parte delle reazioni della gluconeogenesi avvengono nel citosol, alcune nel mitocondrio, e la tappa finale all’interno delle cisterne del reticolo endoplasmatico.
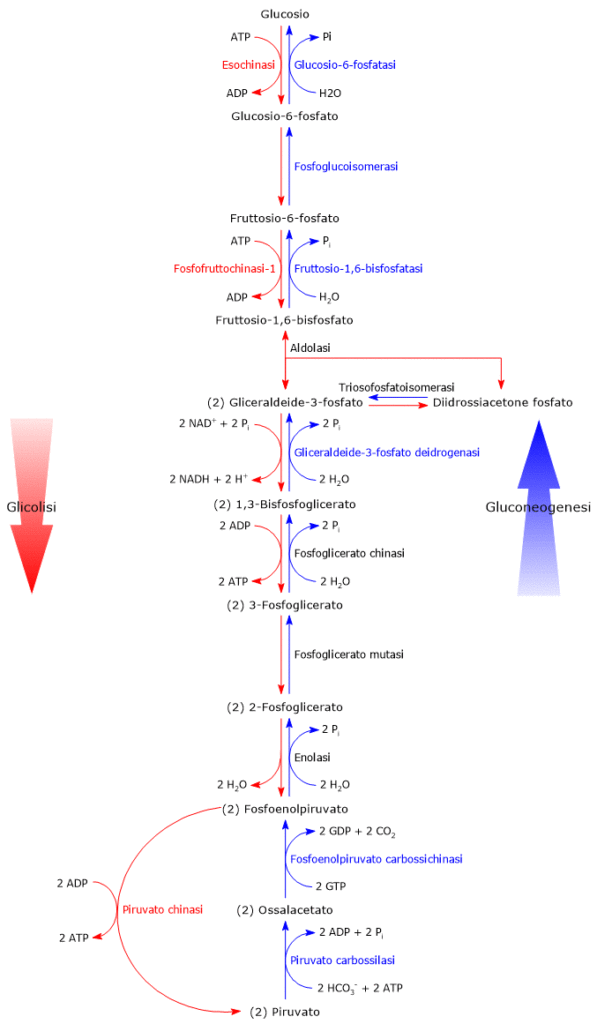
Tappe irreversibili
Come detto, glicolisi e gluconeogenesi sono essenzialmente una l’inverso dell’altra. E, delle dieci reazioni che costituiscono la gluconeogenesi, ben 7 sono in comune con la glicolisi. Si tratta di reazioni caratterizzate da un ΔG prossimo allo zero, per cui facilmente reversibili. Ma nelle normali condizioni cellulari, il ΔG complessivo della glicolisi è pari a circa -63 kJ/mole (-15 kcal/mole) e quello della gluconeogenesi a circa -16 kJ/mole (-3,83 kcal/mole), ossia si tratta di processi irreversibili.[5]
Nel caso della glicolisi l’irreversibilità è conseguenza di tre reazioni fortemente esoergoniche, che non potranno essere utilizzate nella gluconeogenesi, e di seguito elencate.
- La fosforilazione del glucosio a glucosio-6 fosfato, catalizzata dalla esochinasi (EC 2.7.1.1) o dalla glucochinasi (EC 2.7.1.2).
ΔG = -33,4 kJ/mole (-8 kcal/mole)
ΔG°’ = -16,7 kJ/mole (-4 kcal/mole) - La fosforilazione del fruttosio-6-fosfato a fruttosio-1,6-bisfosfato, catalizzata dalla fosfofruttochinasi-1 o PFK-1 (EC 2.7.1.11).
ΔG = -22,2 kJ/mole (-5,3 kcal/mole)
ΔG°’ = -14,2 kJ/mole (-3,4 kcal/mole) - La conversione del fosfoenolpiruvato o PEP, acronimo dell’inglese phosphoenolpyruvate, in piruvato, catalizzata dalla piruvato chinasi (EC 2.7.1.40).
ΔG = -16,7 kJ/mole (-4,0 kcal/mole)
ΔG°’ = -31,4 kJ/mole (-7,5 kcal/mole)
Nella gluconeogenesi queste tre tappe unidirezionali sono superate grazie a enzimi specifici che catalizzano passaggi irreversibili nella direzione della sintesi del glucosio.[6] In questo modo è assicurata l’irreversibilità dell’intera via metabolica.
Di seguito sono analizzate tali reazioni.
Conversione del piruvato in fosfoenolpiruvato
Il primo passaggio della gluconeogenesi che by-passa una tappa irreversibile della glicolisi, nello specifico quella catalizzata dalla piruvato chinasi, è la conversione del piruvato in fosfoenolpiruvato.[8]
La sintesi del fosfoenolpiruvato è ottenuta attraverso una sequenza di due reazioni catalizzate nell’ordine dagli enzimi:
- piruvato carbossilasi (EC 6.4.1.1);
- fosfoenolpiruvato carbossichinasi o PEP carbossichinasi (EC 4.1.1.32).
Piruvato → Ossalacetato → Fosfoenolpiruvato
La piruvato carbossilasi catalizza la carbossilazione del piruvato in ossalacetato, con consumo di una molecola di ATP. L’enzima richiede la presenza di ioni manganese o magnesio.
Piruvato + HCO3– + ATP → Ossalacetato + ADP + Pi
L’enzima, scoperto nel 1960 da Merton Utter, è una proteina mitocondriale formata da quattro subunità identiche, ognuna dotata di attività catalitica. Le subunità hanno come coenzima la biotina, legata attraverso legame ammidico al gruppo amminico ε di un residuo di lisina, e la cui funzione è quella di trasportatore di CO2 attivata nel corso della reazione enzimatica. In ogni subunità è presente anche un sito di legame per l’acetil-CoA.
Va notato che la reazione catalizzata dalla piruvato carbossilasi, portando alla produzione di ossalacetato, fornisce intermedi anche al ciclo dell’acido citrico o di Krebs.
La fosfoenolpiruvato carbossichinasi è presente, all’incirca nelle stesse quantità, sia nel mitocondrio che nel citosol dell’epatocita. Le due forme isoenzimatiche sono codificate da distinti geni nucleari.
L’enzima catalizza la decarbossilazione e fosforilazione dell’ossalacetato a dare fosfoenolpiruvato, in una reazione in cui il GTP funge da donatore di un gruppo fosfato, e richiede la presenza sia di ioni manganese che magnesio. La reazione nelle normali condizioni cellulari è reversibile.
Ossalacetato + GTP ⇄ PEP + CO2 + GDP
Nella reazione la CO2 aggiunta nella tappa catalizzata dalla piruvato carbossilasi viene rimossa. La sequenza di carbossilazione e decarbossilazione è un modo per “attivare” il piruvato, poiché la decarbossilazione dell’ossalacetato facilita, rende termodinamicamente possibile la formazione del fosfoenolpiruvato.
Più in generale le sequenze carbossilazione-decarbossilazione sono utilizzate per favorire reazioni che altrimenti sarebbero fortemente endoergoniche, e sono utilizzate anche nel ciclo dell’acido citrico, nella via del pentoso fosfato, detta via dell’esoso monofosfato, e nella sintesi degli acidi grassi.
Prima della nascita i livelli di PEP carbossichinasi sono molto bassi, mentre, poche ore dopo il parto la sua attività aumenta di diverse volte. Questo è il motivo per cui la gluconeogenesi è attiva solo dopo la nascita.
La somma delle reazioni catalizzate dalla piruvato carbossilasi e dalla fosfoenolpiruvato carbossichinasi è:
Piruvato + ATP + GTP + HCO3– → PEP + ADP + GDP + Pi + CO2
Il ΔG°’ della reazione è pari a 0,9 kJ/mole (0,2 kcal/mole), mentre la variazione di energia libera standard associata alla formazione di piruvato dal fosfoenolpiruvato per semplice inversione della reazione catalizzata dalla piruvato chinasi è di + 31,4 kJ/mole (7,5 kcal/mole).
Sebbene il ΔG’° dei due passaggi che portano alla formazione di fosfoenolpiruvato dal piruvato sia leggermente positivo, il ΔG calcolato dalle concentrazioni intracellulari degli intermedi è molto negativo, -25 kJ/mole (-6 kcal/mole), grazie al rapido consumo del fosfoenolpiruvato in altre reazioni, il che mantiene la sua concentrazione molto bassa. Quindi, nelle condizioni esistenti nella cellula la suddetta sintesi del PEP dal piruvato è un processo irreversibile.[5]
Particolarità della sintesi del fosfoenolpiruvato dal piruvato è che la via seguita dipende dal precursore prevalente: piruvato o alanina, oppure il lattato.
Substrato prevalente piruvato o alanina
I passaggi di seguito descritti prevalgono quando i substrati gluconeogenetici prevalenti sono piruvato o alanina.
La piruvato carbossilasi è un enzima mitocondriale, per cui il piruvato dovrà essere trasportato dal citosol nell’organello. Ciò avviene grazie a due trasportatori presenti sulla membrana mitocondriale interna, indicati come MPC1 and MPC2, che, associandosi, formano un eteropolimero che facilita il passaggio della molecola.[4]
Il piruvato può anche essere prodotto direttamente all’interno del mitocondrio dall’alanina per transaminazione, nella reazione catalizzata dalla alanina aminotransferasi o ALT (EC 2.6.1.2), mentre il gruppo amminico sarà infine convertito in urea attraverso il ciclo dell’urea.
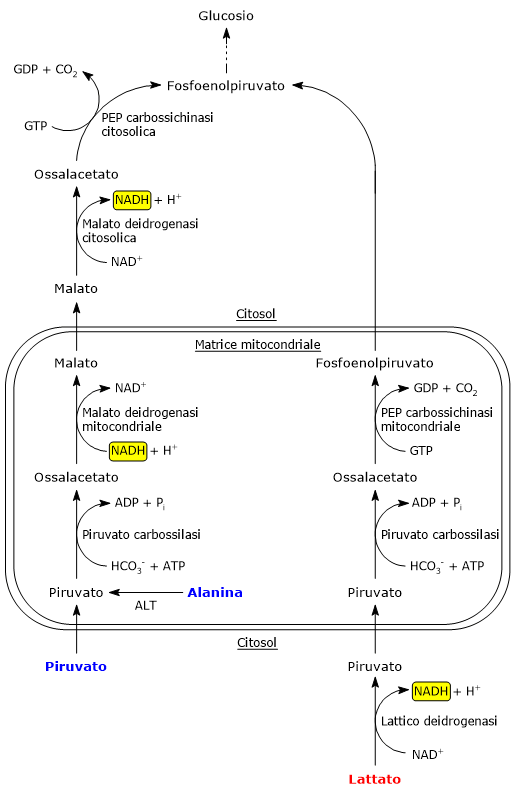
Poiché gli enzimi che intervengono nelle tappe successive della gluconeogenesi, fino alla formazione del glucosio-6-fosfato, sono citosolici, l’ossalacetato prodotto nei mitocondri dovrà essere trasportato nel citosol. La membrana mitocondriale interna è però priva di trasportatori per l’ossalacetato. Il suo passaggio nel citosol avviene a seguito della sua riduzione a malato, che al contrario può attraversare la membrana mitocondriale interna. La reazione è catalizzata dalla malato deidrogenasi mitocondriale (EC 1.1.1.37), un enzima che interviene anche nel ciclo dell’acido citrico dove però il flusso dei metaboliti procede in direzione opposta. Nella reazione il NADH viene ossidato a NAD+.
Ossalacetato + NADH + H+ ⇄ Malato + NAD+
Sebbene ΔG°’ della reazione sia piuttosto elevato, il ΔG calcolato sulla base della concentrazione intracellulare dell’ossalacetato, molto bassa, è prossimo allo zero per cui la reazione è facilmente reversibile.
Il malato prodotto attraversa la membrana mitocondriale interna grazie a un componente dello shuttle del malato-aspartato, il trasportatore malato-α-chetoglutarato. Una volta nel citosol il malato è riossidato a ossalacetato nella reazione catalizzata dalla malato deidrogenasi citosolica, con produzione di NADH.
Malato + NAD+ → Ossalacetato + NADH + H+
Nota: lo shuttle del malato-aspartato è il più attivo nel trasporto degli equivalenti riducenti del NADH dal citosol all’interno del mitocondrio, ed è presente nei mitocondri del fegato, rene, e cuore.
Grazie a questa reazione equivalenti riducenti mitocondriali, in forma di NADH, sono trasferiti nel citosol.[8] Tale trasferimento è necessario per il proseguimento della gluconeogenesi, in quanto nel citosol il NADH, utilizzato nella reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12), è presente in bassa concentrazione, con un rapporto [NADH]/[NAD+] pari a 8 x 10-4, circa 100000 volte più basso di quello osservato nei mitocondri.[5]
Infine l’ossalacetato viene convertito in fosfoenolpiruvato nella reazione catalizzata dalla PEP carbossichinasi.
Substrato prevalente lattato
Il lattato è un importante precursore gluconeogenico. Esempi di cellule e tessuti che lo producono in grande quantità sono i globuli rossi, che, completamente dipendenti dalla glicolisi anaerobica, lo producono continuamente, e il muscolo scheletrico in forte attività, quando la velocità della glicolisi supera la velocità del ciclo dell’acido citrico e della fosforilazione ossidativa.[5]
Quando il lattato è il precursore gluconeogenico prevalente la sintesi del PEP segue una via differente rispetto a quanto visto in precedenza.
Nel citosol dell’epatocita, dove come detto la concentrazione del NAD+ è elevata, il lattato viene ridotto a piruvato nella reazione catalizzata dall’isoenzima epatico della lattato deidrogenasi (EC 1.1.1.27). Nella reazione il NAD+ viene ridotto a NADH.
Lattato + NAD+ → Piruvato + NADH + H+
La produzione citosolica di NADH rende non necessaria l’esportazione di equivalenti riducenti dal mitocondrio.
Il piruvato passa nella matrice mitocondriale per essere convertito in ossalacetato nella reazione catalizzata dalla piruvato carbossilasi. Nel mitocondrio l’ossalacetato è convertito in fosfoenolpiruvato nella reazione catalizzata dall’isoenzima mitocondriale della piruvato carbossilasi, che, a mezzo di un trasportatore anionico della membrana mitocondriale interna, esce dal mitocondrio per continuare nella via gluconeogenetica.
Nota: la sintesi del glucosio dal lattato può essere considerata anche come una parte del “ramo epatico” del ciclo di Cori.
Conversione del fruttosio-1,6-bisfosfato in fruttosio-6-fosfato
Il secondo passaggio della gluconeogenesi che supera una reazione irreversibile della via glicolitica, quella catalizzata dalla PFK-1, è la defosforilazione del fruttosio-1,6-bisfosfato a fruttosio-6-fosfato.
La reazione, catalizzata dalla fruttosio-1,6-bisfosfatasi o FBPasi-1 (EC 3.1.3.11), enzima citosolico e Mg2+ dipendente, comporta l’idrolisi del fosfato sul C-1, senza alcuna produzione di ATP.
Fruttosio-1,6-bisfosfato + H2O → Fruttosio-6-fosfato + Pi
Il ΔG°’ della reazione è pari a -16,3 kJ/mol (-3,9 kcal/mol), dunque una reazione irreversibile.
Conversione del glucosio-6-fosfato a glucosio
Il terzo passaggio esclusivo della gluconeogenesi permette di superare la reazione della glicolisi catalizzata dalla esochinasi o dalla glucochinasi. Nello specifico si tratta della defosforilazione del glucosio-6-fosfato a glucosio catalizzata dall’unità catalitica della glucosio-6-fosfatasi, un complesso proteico presente nella membrana del reticolo endoplasmatico degli epatociti, enterociti e cellule del tubulo prossimale del rene.[8] La glucosio-6-fosfatasi è composta da una subunità catalitica dotata di attività fosfatasica e un trasportatore bidirezionale specifico per il glucosio-6-fosfato detto glucosio-6-fosfato traslocasi o T1.
La subunità catalitica della glucosio-6-fosfatasi presenta il sito attivo rivolto verso la superficie luminale dell’organello: quindi l’enzima catalizza una idrolisi intraluminale del substrato.
Il glucosio-6-fosfato, sia quello derivante dalla gluconeogenesi, rilasciato dalla reazione catalizzata dalla glucosio-6-fosfato isomerasi o fosfoglucosio isomerasi (EC 5.3.1.9), che dalla glicogenolisi, rilasciato dalla reazione catalizzata dalla fosfoglucomutasi (EC 5.4.2.2), è prodotto nel citosol, e dovrà entrare nel lume del reticolo endoplasmatico per essere defosforilato. Il suo passaggio è operato dalla glucosio-6-fosfato traslocasi.
La subunità catalitica della glucosio-6-fosfatasi, un enzima Mg2+-dipendente, catalizza la reazione corrisponde all’ultima tappa sia della gluconeogenesi che della glicogenolisi. E, al pari della reazione che porta alla sintesi del fruttosio-6-fosfato, anche questa è una semplice idrolisi di un estere fosforico.
Glucosio-6-fosfato + H2O → Glucosio + Pi
Va inoltre sottolineato che, grazie all’orientamento del sito attivo, la cellula separa questa reazione dal citosol, e dunque dalla glicolisi, che verrebbe bloccata dall’azione dell’enzima sul glucosio-6-fosfato.
Il ΔG°’ della reazione è pari a -13,8 kJ/mol (-3,3 kcal/mol), dunque una reazione irreversibile. Se invece la reazione fosse l’inverso di quella catalizzata dalla esochinasi/glucochinasi, comporterebbe il trasferimento di un gruppo fosforico all’ADP dal glucosio-6-fosfato, una reazione con ΔG pari a +33,4 kJ/mole (+8 kcal/mol), quindi fortemente endoergonica. Analoghe considerazioni possono essere estese alla reazione catalizzata dalla FBPasi-1.
Sembra che il glucosio e il gruppo Pi prodotti siano trasportati nel citosol da due distinti trasportatori, indicati rispettivamente come T2 e T3, quest’ultimo un trasportatore anionico.
Infine, grazie al trasportatore di membrana GLUT2, il glucosio potrà lasciare l’epatocita ed entrare in circolo per essere trasportato ai tessuti che lo richiedano. Come accennato in precedenza invece, il glucosio prodotto nel rene, in condizioni di normale efficienza epatica, viene utilizzato per la maggior parte dalla midollare del rene stesso.
La gluconeogenesi è energeticamente costosa
Al pari di quanto accade nella glicolisi, sono le tappe irreversibili della gluconeogenesi le responsabili del consumo della maggior parte dell’energia necessaria al processo.[5]
Sono consumati sei legami fosforici ad alta energia, due forniti dal GTP e quattro dall’ATP, cui vanno aggiunte due molecole di NADH per la riduzione di altrettante molecole di 1,3-bisfosfoglicerato nella reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi. Il consumo dei due NADH comporta la mancata produzione di 5 molecole di ATP che sarebbero potute essere sintetizzate nel caso in cui gli elettroni del coenzima ridotto fossero stati utilizzati per la sintesi di ATP nel mitocondrio attraverso la fosforilazione ossidativa.
Anche da queste considerazioni prettamente energetiche emerge che la gluconeogenesi non è semplicemente l’inverso della glicolisi, nel qual caso necessiterebbe del consumo di sole due molecole di ATP, come si evince dalla equazione glicolitica complessiva.
Glucosio + 2 ADP + 2 Pi + 2 NAD+ → 2 Piruvato + 2 ATP + 2 NADH + 2 H+ + 2 H2O
Per la gluconeogenesi invece:
2 Piruvato + 4 ATP + 2 GTP + 2 NADH + 2 H+ + 4 H2O → Glucosio + 4 ADP + 2 GDP + 6 Pi + 2 NAD+
Almeno nel fegato, l’ATP necessario a sostenere la gluconeogenesi deriva di solito dall’ossidazione degli acidi grassi, o dall’ossidazione degli scheletri carboniosi degli amminoacidi, a seconda del “carburante” disponibile.
Regolazione coordinata della glicolisi e gluconeogenesi
Se la glicolisi e la gluconeogenesi procedessero simultaneamente ad alta velocità nella stessa cellula, ne risulterebbe solo il consumo di ATP e la produzione di calore, in particolare in corrispondenza delle tappe irreversibili dei due processi, e nulla di più.
Ad esempio, considerando PFK-1 e FBPasi-1:
ATP + Fruttosio-6-fosfato → ADP + Fruttosio-1,6-bisfosfato
Fruttosio 1,6-bisfosfato + H2O → Fruttosio 6-fosfato + Pi
E, dalla somma delle due reazioni:
ATP + H2O → ADP + Pi + Calore
Quando reazioni contrapposte di questo tipo procedono simultaneamente si parla di ciclo futile o ciclo del substrato.
La contemporanea attività ad alta velocità di enzimi che catalizzano reazioni contrapposte è evitata grazie al controllo dell’attività degli enzimi stessi, che può avvenire mediante:
- meccanismi allosterici;
- modificazioni covalenti, in particolare fosforilazioni e defosforilazioni;
- modificazioni nella concentrazione degli enzimi coinvolti, conseguenti a variazioni del rapporto tra la loro sintesi e degradazione.
I meccanismi allosterici sono molto rapidi e istantaneamente reversibili, avvenendo in un arco temporale di millisecondi. Gli altri, innescati da segnali che provengono dall’esterno della cellula e trasportati da ormoni quali insulina, glucagone, o adrenalina, richiedono tempi più lunghi, dai secondi ai minuti per le modificazioni covalenti, e fino a ore per le modificazioni della concentrazione degli enzimi.
Grazie a questi meccanismi regolatori è possibile ottenere una regolazione coordinata delle due vie, tale da assicurare che quando il flusso di piruvato procede attraverso la gluconeogenesi, il flusso del glucosio attraverso la glicolisi rallenta, e viceversa.
Regolazione della gluconeogenesi
La regolazione della gluconeogenesi e della glicolisi avviene attraverso controlli esercitati sugli enzimi specifici delle singole vie, e non su quelli comuni.[1]
Mentre i principali punti di regolazione della glicolisi sono le reazioni catalizzate dagli enzimi PFK-1 e piruvato chinasi, i principali punti di regolazione della via gluconeogenetica sono le reazioni catalizzate dagli enzimi fruttosio-1,6-bisfosfatasi e piruvato carbossilasi. Anche gli altri due enzimi esclusivi della via gluconeogenetica, glucosio-6-fosfatasi e PEP carbossichinasi, sono soggetti a regolazione ma solo a livello trascrizionale.
Piruvato carbossilasi
Nel mitocondrio il piruvato può essere convertito in:
- acetil-CoA, nelle reazioni catalizzate dal complesso della piruvato deidrogenasi, che lega la glicolisi al ciclo di Krebs;
- ossalacetato, nella reazione catalizzata dalla privato carbossilasi, l’enzima che catalizza la prima tappa della gluconeogenesi.
Il destino metabolico del piruvato dipende dai livelli dell’acetil-CoA e dunque dalla disponibilità di acidi grassi nel mitocondrio.
Quando gli acidi grassi sono disponibili, la loro beta-ossidazione porta alla liberazione di molecole di acetil-CoA, che entrano nel ciclo di Krebs per dar luogo alla formazione di GTP e NADH. Una volta che i fabbisogni energetici della cellula sono soddisfatti, la fosforilazione ossidativa rallenta, il rapporto NADH/NAD+ cresce, il NADH inibisce il ciclo dell’acido citrico e si verifica un accumulo di acetil-CoA nella matrice mitocondriale.[1][5]
L’acetil-CoA è un effettore allosterico positivo della piruvato carbossilasi e un effettore allosterico negativo della piruvato chinasi. Inoltre inibisce il complesso della piruvato deidrogenasi sia attraverso una inibizione da prodotto finale che mediante la sua fosforilazione, a seguito dell’attivazione di una specifica chinasi.
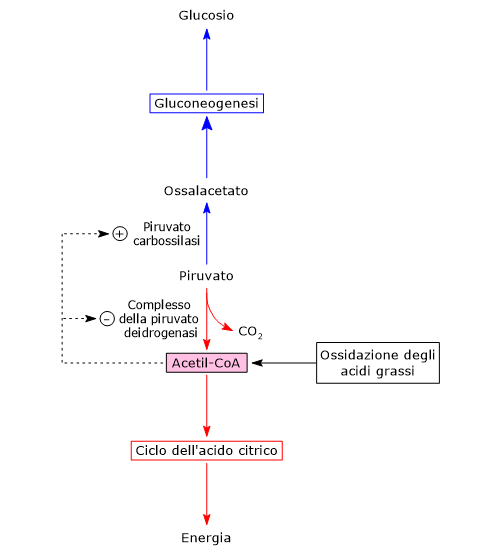
Dunque, quando la carica energetica della cellula è alta, quello che accade è che la formazione di acetil-CoA dal piruvato è rallentata, mentre viene stimolata la conversione del piruvato in glucosio. L’acetil-CoA è quindi un segnale metabolico che indica che un’ulteriore ossidazione del glucosio a scopo energetico non è necessaria e che i metaboliti carboniosi possono essere utilizzati per la sintesi e il deposito di glucosio.
Viceversa, quando i livelli di acetil-CoA si riducono, l’attività della piruvato chinasi e del complesso della piruvato deidrogenasi aumentano, e quindi anche il flusso di metaboliti attraverso il ciclo di Krebs, il tutto per rifornire di energia la cellula.
Si potrebbe riassumere dicendo che la piruvato carbossilasi è attiva quando la carica energetica della cellula è alta e che il primo punto di controllo della gluconeogenesi determina quello che sarà il destino del piruvato nel mitocondrio.
Fruttosio-1,6-bisfosfatasi
Il secondo punto principale di controllo della gluconeogenesi è rappresentato dalla reazione catalizzata dalla fruttosio-1,6-bisfosfatasi. L’enzima è inibito allostericamente dall’AMP. Quindi quando la concentrazione dell’AMP è alta, e di conseguenza quella dell’ATP è bassa, la gluconeogenesi rallenta. Ossia, come visto in precedenza, l’enzima è attivo quando la carica energetica della cellula è adeguata a sostenere la sintesi de novo del glucosio.
Di contro la PFK-1, l’enzima glicolitico corrispondente, è stimolata allostericamente dall’AMP e dall’ADP e inibita dall’ATP e dal citrato, quest’ultimo derivante dalla condensazione tra l’acetil-CoA e l’ossalacetato. Quindi riassumendo:
- quando la concentrazione dell’AMP è alta la gluconeogenesi rallenta, mentre la glicolisi accelera;
- quando la concentrazione dell’ATP è elevata, e quindi è bassa quella di ADP e AMP, o quando l’acetil-CoA o il citrato sono presenti in concentrazioni adeguate, viene promossa la gluconeogenesi mentre rallenta la glicolisi.
L’aumento della concentrazione del citrato segnala che l’attività del ciclo dell’acido citrico può rallentare; in questo modo il piruvato potrà essere utilizzato per la sintesi del glucosio.
Fruttosio-1,6-bisfosfatasi, PFK-1 e fruttosio-2,6-bisfosfato
Il fegato ha un ruolo centrale nel mantenimento della glicemia a valori costanti: questo richiede la presenza di meccanismi regolatori che coordinino il consumo e la produzione del glucosio. Due sono gli ormoni principalmente coinvolti: il glucagone e l’insulina.
Il glucagone, rilasciato in circolo quando la glicemia scende, segnala al fegato di ridurre il suo consumo di glucosio e di aumentarne la produzione de novo e il rilascio dal glicogeno.[6]
L’azione degli ormoni suddetti è mediata da un effettore allosterico della PFK-1 e della fruttosio-1,6-bisfosfatasi: il fruttosio-2,6-bisfosfato, una molecola strutturalmente correlata al fruttosio-1,6-bisfosfato ma che non è ne un intermedio della glicolisi nel della gluconeogenesi.[1]
La molecola fu scoperta nel 1980 da Emile Van Schaftingen and Henri-Gery Hers come un potente stimolatore della PFK-1; l’anno successive gli stessi ricercatori dimostrarono che è anche un potente inibitore dalla fruttosio-1,6-bisfosfatasi.[9][10]
A seguito del legame allo specifico sito allosterico sulla PFK-1, il fruttosio-2,6-bisfosfato svolge un duplice effetto: riduce l’affinità della proteina per i suoi inibitori allosterici ATP e citrato e ne aumenta l’affinità per il fruttosio-6-fosfato, il suo substrato. La PFK-1, in assenza di fruttosio-2,6-bisfosfato, e in presenza di concentrazioni fisiologiche di ATP, fruttosio-6-fosfato, e dei suoi effettori allosterici AMP, ATP e citrato, è praticamente inattiva. La presenza di fruttosio-2,6-bisfosfato ha invece l’effetto di attivare l’enzima quindi stimolare la glicolisi nell’epatocita. Nel contempo la molecola rallenta la gluconeogenesi, inibendo la fruttosio-1,6-bisfosfatasi, anche in assenza di AMP. Tuttavia gli effetti di AMP e fruttosio-2,6-bisfosfato sull’inibizione di FBPasi-1 sono sinergici.
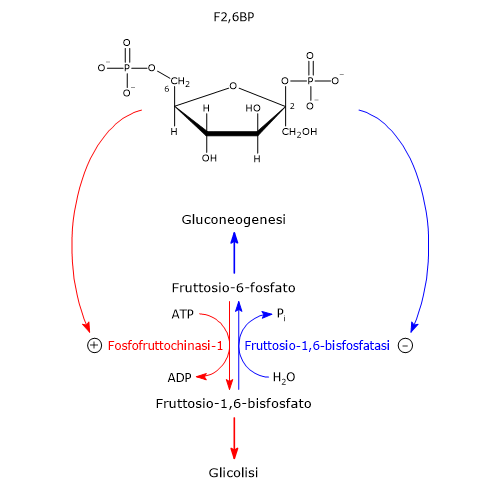
La concentrazione di fruttosio-2,6-bisfosfato è regolata dalle velocità relative della sua sintesi e degradazione. Viene sintetizzato a partire dal fruttosio-6-fosfato nella reazione catalizzata dalla fosfofruttochinasi 2 o PFK-2 (EC 2.7.1.105), e idrolizzato a fruttosio-6-fosfato nella reazione catalizzata dalla fruttosio-2,6-bisfosfatasi o FBPasi-2 (EC 3.1.3.46). Le due attività enzimatiche sono presenti su una stessa proteina, che dunque è un enzima bifunzionale, anche detto enzima tandem, e nel fegato sono regolate dall’insulina e dal glucagone, nel modo di seguito descritto.
- Il glucagone, a seguito del legame allo specifico recettore di membrana, stimola la adenilato ciclasi (EC 4.6.1.1) presente sulla membrana plasmatica a produrre 3’-5’ AMP ciclico o cAMP, che, a seguito del legame alla protein chinasi cAMP-dipendente o protein chinasi A o PKA (EC 2.7.11.11), la attiva. La chinasi attivata catalizza la fosforilazione, a spese di una molecola di ATP, di uno specifico residuo di serina (Ser32) di PFK-2/FBPasi-2. La fosforilazione comporta un aumento dell’attività fosfatasica a spese di quella chinasica, che si riduce in conseguenza di un aumento della Km per il fruttosio-6-fosfato. Tutto ciò porta a una riduzione dei livelli di fruttosio-2,6-bisfosfato, con conseguente stimolazione della gluconeogenesi e inibizione della glicolisi. Quindi, in risposta al segnale trasportato dal glucagone, aumenta la produzione epatica di glucosio attraverso la gluconeogenesi, il che rende l’organo capace di contrastare la riduzione della glicemia segnalata dall’ormone.
Nota: il glucagone, al pari dell’adrenalina, stimola la gluconeogenesi in parte anche aumentando la disponibilità di substrati quali il glicerolo e gli amminoacidi.
- L’insulina, a seguito del legame agli specifici recettori di membrana dell’epatocita, va ad attivare una protein fosfatasi, la fosfoprotein fosfatasi 2A che catalizza la rimozione del gruppo fosforico dalla PFK-2/FBPasi-2, attivando così la PFK-2 e riducendo l’attività della FBPasi-2. (Nel contempo stimola anche una cAMP fosfodiesterasi che idrolizza il cAMP ad AMP). Il risultato è l’aumento dei livelli intracellulari di fruttosio-2,6-bisfosfato e la conseguente inibizione della gluconeogenesi e attivazione della glicolisi.
Inoltre il fruttosio-6-fosfato inibisce allostericamente la FBPasi-2 mentre attiva la PFK-2. Riguardo l’attività dell’enzima bifunzionale PFK-2/FBPasi-2 va sottolineato che entrambe le attività sono inibite dai rispettivi prodotti di reazione, e tuttavia i fattori predominanti sono la concentrazione del fruttosio-6-fosfato e lo stato di fosforilazione dell’enzima stesso.
Glucosio-6-fosfatasi
A differenza della piruvato carbossilasi e della fruttosio-1,6-bisfosfatasi, la subunità catalitica della glucosio-6-fosfatasi non è soggetta a regolazione allosterica o covalente, mentre viene regolata a livello trascrizionale.[7] La bassa glicemia e il glucagone, dunque fattori che determinano una maggiore produzione di glucosio, e i glucocorticoidi ne stimolano la sintesi, che al contrario è inibita dall’insulina.
Inoltre la sua Km per il glucosio-6-fosfato è decisamente più alta rispetto al normale intervallo di concentrazione della molecola stessa. Ne risulta che l’attività dell’enzima mostra una dipendenza quasi lineare rispetto alla concentrazione del substrato. Per questo si dice che l’enzima è sotto controllo da parte della concentrazione del substrato.
PEP carbossichinasi
La regolazione dell’enzima avviene principalmente a livello della sua sintesi e demolizione. Ad esempio elevati livelli di glucagone o il digiuno ne aumentano la produzione, a seguito della stabilizzazione del suo mRNA e dell’aumento della sua velocità di trascrizione. Glicemie elevate o l’insulina hanno effetto opposto.
Xilulosio-5-fosfato
Anche un altro meccanismo regolatorio scoperto di recente, attraverso l’azione dello xilulosio-5-fosfato, un intermedio della via del pentoso fosfato, stimola la glicolisi e inibisce la gluconeogenesi, intervenendo nel controllo della concentrazione del fruttosio-2,6-bisfosfato nell’epatocita.
Quando la concentrazione ematica del glucosio aumenta, come dopo un pasto ricco di carboidrati, nel fegato si verifica l’attivazione della glicolisi e della via dell’esoso monofosfato. In quest’ultima via metabolica viene prodotto anche xilulosio-5-fosfato che è in grado di attivare la protein fosfatasi 2A. Ciò porta alla defosforilazione della PFK-2/FBPasi-2, inibendo così la FBPasi-2 e stimolando la PFK-2. Ne risulta un aumento della concentrazione del fruttosio-2,6-bisfosfato, e quindi l’inibizione della gluconeogenesi e la stimolazione della glicolisi, con conseguente aumento della produzione di acetil-CoA, il principale substrato per la sintesi dei lipidi.[2]
Il concomitante aumento del flusso attraverso la via dell’esoso monofosfato produce NADPH, fonte di elettroni per la sintesi dei lipidi. Infine, la protein fosfatasi 2A defosforila anche ChREBP, un fattore di trascrizione che attiva l’espressione dei geni epatici per la sintesi dei lipidi. Quindi, in risposta a un aumento della glicemia, a livello epatico sarà stimolata la sintesi dei lipidi.
Risulta dunque evidente che lo xilulosio-5-fosfato è un regolatore chiave del metabolismo sia dei carboidrati che dei grassi.
Precursori
Oltre al piruvato, i principali precursori gluconeogenici sono il lattato, di cui si è parlato in precedenza, il glicerolo, la maggior parte degli amminoacidi, e comunque qualunque composto che possa essere convertito in piruvato od ossalacetato.[1]
Glicerolo
Il glicerolo deriva dalla lipolisi nel tessuto adiposo. Con l’esclusione del propionil-CoA, è l’unica parte delle molecole dei lipidi che negli animali possa essere utilizzata per la sintesi del glucosio.
Il suo punto di ingresso nella gluconeogenesi, o nella glicolisi, a seconda delle condizioni energetiche in cui si trova la cellula, è rappresentato dal diidrossiacetone fosfato, la cui sintesi avviene in due passaggi.
Nel primo il glicerolo è fosforilato a glicerolo-3-fosfato, nella reazione catalizzata dalla glicerolo chinasi (EC 2.7.1.30). La reazione consuma una molecola di ATP.
Glicerolo + ATP → Glicerolo-3-fosfato + ADP + Pi
L’enzima assente negli adipociti, ma presente nel fegato. Ciò significa che il glicerolo dovrà raggiungere il fegato prima di essere ulteriormente metabolizzato.
Il glicerolo-3-fosfato viene quindi ossidato a diidrossiacetone fosfato, nella reazione catalizzata dalla glicerolo-3-fosfato deidrogenasi (EC 1.1.1.8). Nella reazione il NAD+ viene ridotto a NADH.
Glicerolo-3-fosfato + NAD+ ⇄ Diidrossiacetone fosfato + NADH + H+
Durante il digiuno prolungato il glicerolo è il principale precursore gluconeogenetico, essendo responsabile della produzione di circa il 20% del glucosio.[3]
Amminoacidi glucogenici
Piruvato e ossalacetato rappresentano i punti di ingresso per gli amminoacidi glucogenici, ossia quelli il cui scheletro carbonioso o parte di esso può essere utilizzato per la sintesi del glucosio.
Gli amminoacidi derivano dalla demolizione delle proteine, sia di origine alimentare che endogena, come quelle del muscolo scheletrico nel corso del digiuno o dell’attività fisica intensa e prolungata.
I processi catabolici a carico di ognuno dei venti amminoacidi che compongono le proteine convergono verso la sintesi di sette prodotti principali: acetil-CoA, acetoacetil-CoA, α-chetoglutarato, succinil-CoA, fumarato, ossalacetato e piruvato.
Con l’esclusione di acetil-CoA e acetoacetil-CoA, le altre cinque molecole possono essere utilizzate per la sintesi del glucosio; quindi gli amminoacidi glucogenici possono essere definiti anche come quelli il cui scheletro carbonioso, in parte o in toto, può essere convertito in una o più delle suddette molecole.
Solo leucina e lisina sono esclusivamente chetogenici, essendo il loro scheletro carbonioso catabolizzato ad acetil-CoA e/o acetoacetil-CoA.
Di seguito sono elencati gli amminoacidi glucogenici con i rispettivi punti di ingresso.
- Piruvato: alanina, cisteina, glicina, serina, treonina e triptofano.
- Ossalacetato: aspartato e asparagina.
- α-Chetoglutarato: glutammato, arginina, glutammina, istidina e prolina.
- Succinil-CoA: isoleucina, metionina, treonina e valina.
- Fumarato: fenilalanina e tirosina.
α-Chetoglutarato, succinil-CoA e fumarato, tutti intermedi del ciclo dell’acido citrico, entrano nella via gluconeogenetica previa conversione in ossalacetato.
Propionato
Il propionato, che fa parte del gruppo degli acidi grassi a catena corta, in forma di propionil-CoA è un precursore gluconeogenetico in quanto può essere convertito in succinil-CoA.
Di seguito sono analizzate le diverse fonti di propionato.
- Può derivare dalla beta-ossidazione di acidi grassi a catena dispari, come ad esempio l’acido margarico, acido grasso saturo con 17 atomi di carbonio. Tali acidi grassi sono molto rari rispetto a quelli a catena pari, e presenti in quantità significative nei lipidi di alcuni organismi marini, delle piante, e nel grasso dei ruminanti. Nell’ultimo passaggio del loro ciclo di ossidazione, il substrato è non a 4 ma a 5 atomi di carbonio, per cui una volta ossidato e scisso in due frammenti, darà origine a un acetil-CoA e un propionil-CoA.
- Altra fonte è la ossidazione degli acidi grassi a catena ramificata, con ramificazioni costituite da gruppi alchilici con un numero dispari di atomi di carbonio. Un esempio è l’acido fitanico, prodotto nei ruminanti dall’ossidazione del fitolo, un derivato della degradazione della clorofilla.
- Nei ruminanti, è prodotto anche a partire dal glucosio liberato dall’idrolisi della cellulosa grazie all’azione di batteri presenti nel rumine, una delle quattro camere che compongono lo stomaco di questi animali. Sempre nel rumine, gli stessi batteri convertono, attraverso fermentazione, il glucosio in propionato, che potrà, una volta assorbito, essere utilizzato per la gluconeogenesi, essere ossidato per la produzione di energia, o essere utilizzato per la sintesi degli acidi grassi.[5]
Nei ruminanti, dove la gluconeogenesi tende a essere un processo continuo, il propionato è il più importante precursore gluconeogenetico. - Il propionato può derivare anche catabolismo della valina, leucina e isoleucina.
- Nel colon, il propionato viene prodotto dai batteri del microbiota intestinale attraverso la fermentazione anaerobica di carboidrati non digeribili, come l’amido resistente e le fibre. Assorbito dagli enterociti, per la maggior parte non è ivi metabolizzato, passa nel circolo portale e raggiunge il fegato, dove può entrare nella gluconeogenesi.
L’ossidazione del propionil-CoA a succinil-CoA avviene attraverso tre reazioni che si verificano nel fegato e in altri tessuti.
Nella prima reazione il propionil-CoA è carbossilato a dare D-metilmalonil-CoA, nella reazione catalizzata dalla propionil-CoA carbossilasi (EC 6.4.1.3), enzima che ha come cofattore la biotina. La reazione consuma un ATP.
Propionil-CoA + HCO3– + ATP → D-Metilmalonil-CoA+ ADP + Pi
Nella reazione successiva il D-metilmalonil-CoA viene epimerizzato nello stereoisomero L. La reazione è catalizzata dalla metilmalonil-CoA epimerasi (EC 5.1.99.1).
D-Metilmalonil-CoA ⇄ L-Metilmalonil-CoA
Infine l’L-metilmalonil-CoA, nella reazione catalizzata dalla metilmalonil-CoA mutasi (EC 5.4.99.2), enzima che richiede come coenzima la 5-deossiadenosilcobalamina, un derivato della cobalamina o vitamina B12, subisce un riarrangiamento intramolecolare a dare succinil-CoA.
L-Metilmalonil-CoA ⇄ Succinil-CoA
Bibliografia
- ^ a b c d e f g Garrett R.H., Grisham C.M. Biochemistry. 4th Edition. Brooks/Cole, Cengage Learning, 2010
- ^ Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003;100:5107-5112. doi:10.1073/pnas.0730817100
- ^ Kuriyama H. et all. Coordinated regulation of fat-specific and liver-specific glycerol channels, aquaporin adipose and aquaporin 9. Diabetes 2002;51(10):2915-2921. 10.2337/diabetes.51.10.2915
- ^ McCommis K.S. and Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J 2015;466(3):443-454. doi:10.1042/BJ20141171
- ^ a b c d e f g h i Nelson D.L., M. M. Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
- ^ a b c d e Rosenthal M.D., Glew R.H. Medical biochemistry – Human metabolism in health and disease. John Wiley J. & Sons, Inc., Publication, 2009
- ^ Soty M., Chilloux J., Delalande F., Zitoun C., Bertile F., Mithieux G., and Gautier-Stein A. Post-Translational regulation of the glucose-6-phosphatase complex by cyclic adenosine monophosphate is a crucial determinant of endogenous glucose production and is controlled by the glucose-6-phosphate transporter. J Proteome Res 2016;15(4):1342-1349. doi:10.1021/acs.jproteome.6b00110
- ^ a b c Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
- ^ Van Schaftingen E., and Hers H-G. Inhibition of fructose-1,6-bisphosphatase by fructose-2,6-bisphosphate. Proc Natl Acad Sci USA 1981;78(5):2861-2863. doi:10.1073/pnas.78.5.2861
- ^ Van Schaftingen E., Jett M-F., Hue L., and Hers H-G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA 1981;78(6):3483-3486. doi:10.1073/pnas.78.6.3483