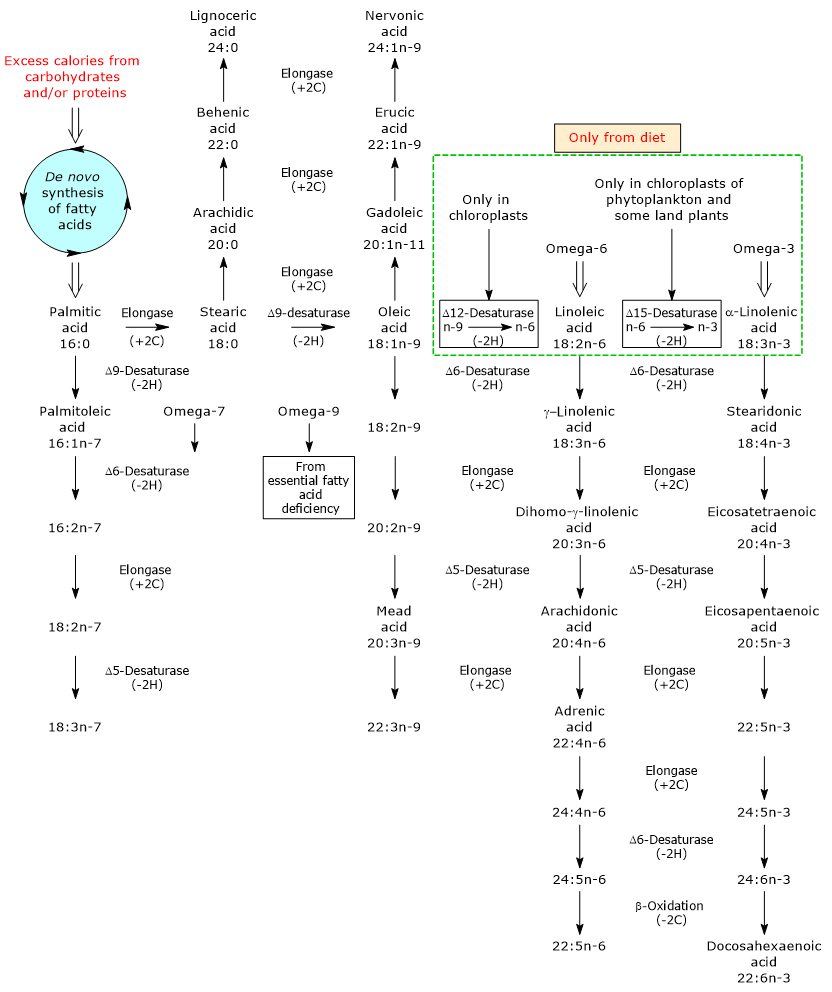
Biochemistry, or biological chemistry, studies the chemical processes that occur in animals, plants, and microorganisms.
It is one of the five main branches of chemistry, along with inorganic chemistry, organic chemistry, physical chemistry, and analytical chemistry. As the name suggests, it can also be considered a branch of biology.
Biochemistry became a distinct discipline in the early 1900s. Scientists began combining biology, physiology, and chemistry to study living organisms. This development built on knowledge from the previous century, thanks to researchers like Justus von Liebig, Eduard Buchner, Louis Pasteur, and Emil Fischer.
Since the mid-1900s, biochemists have gained deeper insight into cellular metabolic pathways. They achieved this by applying advances in chemistry, physics, and mathematics. These pathways allow organisms to obtain energy from food or sunlight. Examples include glycolysis, the citric acid cycle, lipolysis, oxidative phosphorylation, and photosynthesis.
Scientists have also explained how cells use this energy to produce what they need to live and reproduce. As a result, cellular metabolism has become clearer. It includes anabolic and catabolic pathways that synthesize and break down molecules and macromolecules such as carbohydrates, proteins, lipids, and nucleic acids.
The structure of these macromolecules has also been revealed. For example, the discovery of DNA’s structure was made possible by the work of Rosalind Elsie Franklin, James Dewey Watson, and Francis Harry Compton Crick.
Together with metabolism and enzymology, structural biology forms one of the three main branches of biochemistry.
Glucose 6-phosphate (G6P) is a key metabolite in carbohydrate metabolism and, more broadly, in cellular metabolism.[1]
In animal and bacterial cells, it mainly derives from the reaction catalyzed by the enzyme hexokinase, which catalyzes the first step of the glycolytic pathway. Alternatively, it can be produced through glycogenolysis, a catabolic process in which glycogen is degraded to provide glucose in response to specific metabolic needs.[2] A third, although quantitatively less significant, synthesis pathway involves the conversion of galactose via the Leloir pathway.[3]
Glucose 6-phosphate represents a crucial cellular metabolic hub. Depending on the cell’s needs, it can proceed through glycolysis, be diverted into the hexose monophosphate shunt, or be used for glycogen synthesis and macromolecular glycosylation.[4] Furthermore, in the liver, it can be converted into free glucose or participate in detoxification processes via glucuronidation.[5]
Summary: Key Points
Central metabolic hub: glucose 6-phosphate is a crucial intermediate that sits at the intersection of major metabolic pathways.
Cellular trapping: the phosphorylation of glucose to G6P adds a negative charge to the molecule, trapping it inside the cell.
Primary sources: it is synthesized through three main routes: the hexokinase/glucokinase reaction (from glucose), glycogenolysis (from glycogen stores), or the Leloir pathway (from galactose).
Metabolic fates: depending on energy needs, it can be oxidized for ATP, used to generate NADPH and ribose-5-phosphate, stored as glycogen, or used as a precursor for glucuronidation.
Glucose 6-phosphate has a molecular weight of 260.14 g/mol and a molecular formula of C6H13O9P.
According to IUPAC nomenclature, its systematic name is [(2R,3R,4S,5R)-2,3,4,5-tetrahydroxy-6-oxohexyl] dihydrogen phosphate.
Its pKa1 and pKa2, at 25 °C are 1.65 and 6.11, respectively. Therefore, at physiological pH, it exists almost exclusively in its ionized form and carries a charge of −2.[6]
How glucose 6-phosphate is formed
In animal and bacterial cells (excluding photosynthesis), glucose 6-phosphate is primarily produced through two pathways: the direct phosphorylation of glucose and, in animal cells, glycogenolysis. It can also be synthesized from galactose.[2]
Glucose 6-phosphate as a Central Metabolic Hub
Hexokinase
Once glucose enters the cell, its main metabolic fate is to be phosphorylated to glucose 6-phosphate.
This reaction is the first of the ten steps of glycolysis and also the first of the two phosphorylation steps that the monosaccharide undergoes during this metabolic pathway.
Hexokinase (EC 2.7.1.1), an enzyme found in the cells of all organisms and present in four isoforms in humans, catalyzes this irreversible reaction, phosphorylating glucose at the C-6 position using one ATP molecule.
Glucose + ATP → Glucose 6-phosphate + ADP
This glucose 6-phosphate synthesis pathway is particularly important when the monosaccharide is available from external sources, such as after a meal.[1]
Glycogenolysis
Glucose 6-phosphate can also be derived from glycogenolysis, the metabolic pathway by which glucose molecules stored in glycogen, a polysaccharide, are released when needed.
During fasting, for example, at night or between meals, but also during physical activity, glycogenolysis, through the intervention of three enzymatic activities, namely, glycogen phosphorylase (EC 2.4.1.1), alpha-(1,4)-glucan-6-glycosyltransferase (EC 2.4.1.25), and amylo-alpha-(1,6)-glucosidase, or debranching enzyme (EC 3.2.1.33), leads to the release of glucose: approximately 90% in the form of glucose 1-phosphate (G1P), and the remaining 10% directly as glucose.
Glucose 1-phosphate is then isomerized to glucose 6-phosphate in the reversible reaction catalyzed by phosphoglucomutase (EC 5.4.2.2).[2]
Galactose
Galactose is a monosaccharide present in milk and dairy products, where it is mainly found as a component, along with glucose, of lactose. Through the Leloir pathway, it can be converted into glucose 1-phosphate, which in turn can be isomerized to glucose 6-phosphate.[7]
The Leloir pathway allows the formation of metabolites that can enter various metabolic routes, both anabolic and catabolic, depending on the tissue and the metabolic conditions of the cell. Among the possible fates of galactose along this pathway is its conversion into glucose 1-phosphate, which can then be isomerized to glucose 6-phosphate.[3]
Why the phosphorylation of glucose to glucose 6-phosphate is important
The phosphorylation of glucose to glucose 6-phosphate is metabolically important for several reasons.
Glucose is a neutral molecule whose passage through the plasma membrane occurs via facilitated diffusion, mediated by specific protein transporters belonging to the GLUT or SLC2A family of membrane transport proteins.[8]
Phosphorylation at C-6 gives glucose a negative charge, preventing it from crossing the membrane, partly because of the absence of specific transporters for phosphorylated sugars. Therefore, phosphorylation traps the sugar inside the cell.[4]
Thanks to the phosphorylation at C-6 and the lack of transporters for phosphorylated sugars, it is no longer necessary to expend energy to retain glucose 6-phosphate inside the cell, despite the large difference between its intracellular and extracellular concentrations.[9]
The rapid conversion of glucose into glucose 6-phosphate also helps maintain a low intracellular concentration of free glucose, thereby facilitating its entry by facilitated diffusion.[4]
The addition of a phosphate group increases the energy content of glucose, that is, it begins to destabilize the molecule, facilitating its subsequent metabolism.[9]
Metabolic fate of glucose 6-phosphate
Glucose 6-phosphate is a central hub in carbohydrate metabolism. As a common intermediate in various metabolic pathways, its fate depends on the metabolic needs of the cell. It can participate in several metabolic routes, both anabolic and catabolic, such as glycolysis, gluconeogenesis, the pentose phosphate pathway, glycogen synthesis, and hexosamine pathway. Finally, it is also involved in detoxification processes through the glucuronidation of molecules destined for excretion.[10]
Glycolysis
Glycolysis is a fundamental metabolic pathway, not only for energy production but also as a source of intermediates for other metabolic pathways.[11]
Glucose 6-phosphate can continue along the pathway, becoming a substrate for the reaction catalyzed by phosphoglucose isomerase (PGI; EC 5.3.1.9). This enzyme catalyzes its reversible isomerization to fructose 6-phosphate, an example of functional group isomerism. However, fructose 6-phosphate is not an exclusive metabolite of glycolysis.
In the third step of the glycolytic pathway, fructose 6-phosphate undergoes a second phosphorylation to form fructose 1,6-bisphosphate. This reaction, catalyzed by phosphofructokinase-1 (PFK-1; EC 2.7.1.11), is irreversible and leads to the formation of a metabolite that is exclusive to glycolysis, and, in the reverse direction, to gluconeogenesis.[9]
Hexosamine pathway
Fructose 6-phosphate, produced by the isomerization of glucose 6-phosphate, is one of the key substrates of the hexosamine pathway. This metabolic pathway accounts for approximately 2–5% of glucose metabolism and is essential for the glycosylation of macromolecules.[10]
In the first step, fructose 6-phosphate is converted to glucosamine 6-phosphate in a reaction catalyzed by the enzyme glutamine:fructose-6-phosphate aminotransferase (GFAT; EC 2.6.1.16), which catalyzes the first committed step of the pathway. In this reaction, the amino acid glutamine serves as the donor of the amino group.[12]
The subsequent steps ultimately lead to the synthesis of UDP-N-acetylglucosamine (UDP-GlcNAc), which is used in both N-linked and O-linked glycosylation of proteins, lipids, and nucleic acids.[13]
Pentose phosphate pathway
Glucose 6-phosphate can enter the pentose phosphate pathway, also known as the hexose monophosphate shunt, an alternative to glycolysis for the oxidative metabolism of glucose.[14]
Entry into the pathway occurs at the level of the reaction catalyzed by glucose 6-phosphate dehydrogenase (G6PD; EC 1.1.1.49). In this reaction, G6P is oxidized to 6-phosphoglucono-δ-lactone, which then continues along the metabolic pathway.
The pentose phosphate pathway serves a dual function: the synthesis, in variable ratios depending on the metabolic conditions of the cell, of NADPH and ribose-5-phosphate.
NADPH is a reduced coenzyme essential for biosynthetic reactions such as the synthesis of fatty acids and cholesterol, as well as for antioxidant defense by reducing oxidized glutathione.[1]
Ribose 5-phosphate, a five-carbon sugar, is essential for the synthesis of nucleotides and nucleic acids, and is therefore particularly important during periods of growth.[4]
It is estimated that over 10% of metabolized glucose is channeled through this pathway, which, interestingly, while oxidizing the monosaccharide, neither produces nor directly consumes ATP.[15]
Glycogen synthesis
Glucose 6-phosphate can be used to replenish cellular glycogen reserves. The anabolic pathway leading to polysaccharide biosynthesis occurs when carbohydrates are available, glucose demand is low, and there is an energy surplus. Glycogen synthesis is particularly important in the liver and muscle.[16]
During glycogen synthesis, glucose 6-phosphate is isomerized to glucose 1-phosphate in a reaction catalyzed by phosphoglucomutase. This enzyme also functions in glycogenolysis, catalyzing the reverse reaction. The direction of the reaction is determined by the relative concentrations of the two sugar phosphates.
In the next step, the carbon skeleton is activated by the transfer of a UDP group in a reaction catalyzed by UDP-glucose pyrophosphorylase (EC 2.7.7.9).
The resulting UDP-glucose then serves as the substrate for the reaction catalyzed by glycogen synthase (EC 2.4.1.11) and is used to elongate glycogen chains. UDP-glucose is also used as a sugar donor in the synthesis of glycolipids.[4]
Detoxification processes
Detoxification processes enable the elimination of xenobiotics, drugs, and catabolic by-products such as bilirubin. These metabolic pathways occur primarily in the liver, where glucose 6-phosphate acts as a precursor in the synthesis of glucuronic acid.[17]
G6P, similar to its role in glycogen synthesis, is first activated to UDP-glucose. UDP-glucose is then oxidized to UDP-glucuronate in a reaction catalyzed by UDP-glucose 6-dehydrogenase (EC 1.1.1.22). UDP-glucuronate serves as a glucuronic acid donor in reactions catalyzed by UDP-glucuronosyltransferase (EC 2.4.1.17). This process, known as glucuronidation, increases the hydrophilicity of otherwise lipophilic compounds, thereby facilitating their elimination via bile or urine.[5]
References
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ abc Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
^ ab Conte F., van Buuringen N., Voermans N.C., Lefeber D.J. Galactose in human metabolism, glycosylation and congenital metabolic diseases: time for a closer look. Biochim Biophys Acta Gen Subj 2021;1865(8):129898. doi:10.1016/j.bbagen.2021.129898
^ ab Rowland A., Miners J.O., Mackenzie P.I. The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int J Biochem Cell Biol 2013;45(6):1121-32. doi:10.1016/j.biocel.2013.02.019
^ Leloir L.F., de Fekete M.A., Cardini C.E. Starch and oligosaccharide synthesis from uridine diphosphate glucose. J Biol Chem 1961;236:636-41. doi:10.1016/S0021-9258(18)64280-2
^ Thorens B., Mueckler M. Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab 2010;298(2):E141-5. doi:10.1152/ajpendo.00712.2009
^ abc Berg J.M., Tymoczko J.L., Gatto J.G., Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
^ ab Rajas F., Gautier-Stein A., Mithieux G. Glucose-6 phosphate, a central hub for liver carbohydrate metabolism. Metabolites 2019;9(12):282. doi:10.3390/metabo9120282
^ Kierans S.J., Taylor C.T. Glycolysis: a multifaceted metabolic pathway and signaling hub. J Biol Chem 2024;300(11):107906. doi:10.1016/j.jbc.2024.107906
^ Michal G., Schomburg D. Biochemical pathways. An atlas of biochemistry and molecular biology. 2nd Edition. John Wiley J. & Sons, Inc. 2012.
^ Adeva-Andany M.M., González-Lucán M., Donapetry-García C., Fernández-Fernández C., Ameneiros-Rodríguez E. Glycogen metabolism in humans. BBA Clin 2016;5:85-100. doi:10.1016/j.bbacli.2016.02.001
^ Yang G., Ge S., Singh R., Basu S., Shatzer K., Zen M., Liu J., Tu Y., Zhang C., Wei J., Shi J., Zhu L., Liu Z., Wang Y., Gao S., Hu M. Glucuronidation: driving factors and their impact on glucuronide disposition. Drug Metab Rev 2017;49(2):105-138. doi:10.1080/03602532.2017.1293682
Substrate-level phosphorylation is defined as the production of ATP or GTP following the transfer of a phosphate group from a substrate to ADP or GDP.[1]
The energy released from the cleavage of a high-energy bond of the substrate facilitates the formation of the phosphoanhydride bond.[2] Part of this energy drives the reaction toward nucleotide triphosphate formation, while the remainder is stored in the newly formed bond.[3]
This process involves soluble enzymes and chemical intermediates, differing from oxidative phosphorylation and photophosphorylation, which rely on membrane-bound enzymes and transmembrane proton gradients.[2]
Examples of substrate-level phosphorylation are found in the glycolytic pathway and the citric acid cycle.[3]
Although under aerobic conditions most ATP is produced by oxidative phosphorylation and by photophosphorylation in photosynthetic organisms, substrate-level phosphorylation remains essential in energy metabolism, particularly under anaerobic conditions.[4][5]
Summary: Key Points
Definition: the direct production of ATP or GTP by transferring a high-energy phosphate group from a metabolic substrate to ADP or GDP.
Mechanism: driven by the cleavage of high-energy chemical bonds; it uses soluble enzymes and occurs independently of proton gradients or membrane-bound complexes.
Metabolic pathways: occurs in glycolysis (reactions catalyzed by phosphoglycerate kinase and pyruvate kinase) and the citric acid cycle (reaction catalyzed by succinyl-CoA synthetase).
Biological significance: essential for energy metabolism, it is the primary mechanism for ATP synthesis under anaerobic conditions.
Two well-known examples of substrate-level phosphorylation occur in glycolysis.
During the energy recovery phase of glycolysis, two of the five reactions directly extract and store chemical energy from metabolic intermediates as ATP.[5]
These reactions are catalyzed by phosphoglycerate kinase (EC 2.7.2.3) and pyruvate kinase (EC 2.7.1.40).
In these reactions, a high-energy phosphate group is transferred to ADP from the following substrates:
1,3-bisphosphoglycerate, resulting in the production of ATP and 3-phosphoglycerate;
phosphoenolpyruvate, resulting in the production of ATP and pyruvate.[6]
These two glycolytic substrate-level phosphorylation reactions enable ATP synthesis even under anaerobic conditions.[3]
The citric acid cycle and GDP phosphorylation
Another example of substrate-level phosphorylation occurs in in the reaction of the citric acid cycle catalyzed by succinyl-CoA synthetase, also known as succinate-CoA ligase (EC 6.2.1.4). This enzyme catalyzes the cleavage of the high-energy thioester bond in succinyl-CoA, similar to that in acetyl-CoA. The energy released drives the formation of a phosphoanhydride bond, leading to the conversion of GDP to GTP.[5]
References
^ Ha C.E., Bhagavan N.V. Chapter 11 – Carbohydrate metabolism I: glycolysis and the tricarboxylic acid cycle. Editor(s): Chung Eun Ha, N.V. Bhagavan. Essentials of Medical Biochemistry. 3rd Edition. Academic Press, 2023, pages 203-227. doi:10.1016/B978-0-323-88541-6.00030-2
Ketogenic amino acids are amino acids whose carbon skeleton can be fully or partially catabolized into acetoacetyl-CoA or acetyl-CoA, molecules that serve as precursors for the synthesis of ketone bodies, hence the name, or of fatty acids.[1]
Amino acids are the fundamental constituents of proteins.
Since the body lacks specific amino acid reserves comparable to glycogen or triglycerides, excess amino acids are converted into citric acid cycle intermediates to produce energy once protein synthesis demands are met.[2]
When cellular energy requirements are satisfied, the carbon residues from amino acid catabolism are redirected towards the synthesis of glucose, fatty acids, or ketone bodies.[3]
Proteinogenic amino acids are classified as ketogenic amino acids or glucogenic based on the metabolic fate of their carbon skeleton.[4] Ketogenic amino acids yield acetoacetyl-CoA and/or acetyl-CoA, while glucogenic ones are catabolized into five precursor metabolites: pyruvate, oxaloacetate, α-ketoglutarate, succinyl-CoA and fumarate.[5]
However, this classification is not unique, as five amino acids are both ketogenic and glucogenic, originating at least one glucogenic and one ketogenic precursor.[6]
Summary: Key Points
Metabolic fate: ketogenic amino acids are catabolized into acetyl-CoA or acetoacetyl-CoA, serving as direct precursors for ketone bodies and fatty acids synthesis.
Exclusively ketogenic: leucine and lysine are the only two strictly ketogenic amino acids, meaning they cannot contribute to net glucose production in animals.
Biochemical barrier: acetyl-CoA cannot generate glucose due to the loss of two carbon atoms in the citric acid cycle and the irreversibility of the pyruvate dehydrogenase complex reaction.
Fasting dynamics: during prolonged fasting, their role in ketogenesis is minor compared to fatty acid beta-oxidation; the body prioritizes glucogenic amino acids to maintain glucose homeostasis.
Only two of the twenty amino acids that make up proteins are exclusively ketogenic: leucine and lysine. The catabolism of their carbon skeleton results in the production of acetoacetyl-CoA and acetyl-CoA.[6]
Classification of ketogenic and glucogenic amino acids based on their catabolites
Amino acid
Ketogenic
Glucogenic
Acetyl-CoA
Acetoacetyl-CoA
Phenylalanine
✔
Fumarate
Isoleucine
✔
Succinyl-CoA
Leucine *
✔
✔
Lysine *
✔
Tyrosine
✔
Fumarate
Threonine
✔
Succinyl-CoA
Tryptophan
✔
✔
Pyruvate
* Exclusively ketogenic amino acids
Another five amino acids, namely isoleucine, phenylalanine, threonine, tryptophan, and tyrosine, are both ketogenic and glucogenic as their carbon skeleton catabolism leads to the formation of acetyl-CoA and/or acetoacetyl-CoA and a glucogenic precursor.[5]
The utilization of amino acid carbon skeletons is preceded by the removal of the amino group. Alanine and glutamate, glucogenic amino acids, play a key role in the transport of amino groups from extrahepatic tissues to the liver. In particular, alanine is transported from muscle and other peripheral tissues to the liver via the glucose-alanine cycle.[7]
Biochemical basis
Acetyl-CoA and acetoacetyl-CoA are not glucogenic precursors. The explanation lies in the stoichiometry of the citric acid cycle and the inability of animals to convert acetyl-CoA into pyruvate.[8]
Metabolic Map of Ketogenic Amino Acid Entry Points into the Krebs Cycle.
Acetyl-CoA enters the citric acid cycle through the reaction catalyzed by citrate synthase (EC 2.3.3.1). The enzyme catalyzes the condensation of acetyl-CoA with oxaloacetate to form citrate. Thus, a four-carbon compound is converted into a six-carbon compound, with a net gain of two carbon atoms.
However, in the two subsequent oxidative decarboxylations of the cycle, catalyzed respectively by isocitrate dehydrogenase (EC 1.1.1.42) and the α-ketoglutarate dehydrogenase multienzyme complex, two carbon atoms are lost.[4]
Therefore, the entry of acetyl-CoA into the cycle does not involve any net gain in carbon.[6]
The key factor is therefore the entry point of the carbon units, upstream or downstream of the oxidative decarboxylations of the citric acid cycle.[8]
Additionally, in animals, there is no metabolic pathway that allows the production of pyruvate from acetyl-CoA, due to the irreversibility of the reaction catalyzed by the pyruvate dehydrogenase complex, namely, the oxidative decarboxylation of pyruvate to acetyl-CoA.[8]
Ketogenic amino acids during prolonged fasting
Unlike glucogenic amino acids, ketogenic amino acids do not play a crucial role during prolonged fasting or in diets with severe carbohydrate restriction.
During prolonged fasting, the primary source of acetyl-CoA for ketogenesis is the β-oxidation of fatty acids, while the contribution of ketogenic amino acids is marginal. Under these conditions, glucogenic amino acids play a central role, as they ensure the maintenance of glucose homeostasis through gluconeogenesis when hepatic glycogen stores are depleted.[5]
Glyoxylate cycle
Plants, yeasts, and many bacteria can use acetyl-CoA for glucose synthesis because they have the glyoxylate cycle.
This cycle shares several reactions with the citric acid cycle but features two unique steps, catalyzed by isocitrate lyase (EC 4.1.3.1) and malate synthase (EC 2.3.3.9), while lacking any decarboxylation reactions. Consequently, organisms possessing the glyoxylate cycle can convert fatty acids and ketogenic amino acids into glucose.[9]
References
^ D’Andrea G. Classifying amino acids as gluco(glyco)genic, ketogenic, or both. Biochem Educ 2000;28(1):27-28. doi:10.1016/s0307-4412(98)00271-4
^ Brosnan J.T. Interorgan amino acid transport and its regulation. J Nutr 2003;133(6 Suppl 1):2068S-2072S. doi:10.1093/jn/133.6.2068S
^ Litwack G. Human biochemistry. 2nd Edition. Academic Pr, 2021.
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ abc Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009.
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
^ abc Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
^ Kondrashov F.A., Koonin E.V., Morgunov I.G., Finogenova T.V., Kondrashova M.N. Evolution of glyoxylate cycle enzymes in Metazoa: evidence of multiple horizontal transfer events and pseudogene formation. Biol Direct 2006;1:31. doi:10.1186/1745-6150-1-31
Glucogenic amino acids are defined as amino acids whose carbon skeletons can be fully or partially catabolized into precursors for glucose synthesis or gluconeogenesis, hence the name “glucogenic”.[1]
Amino acids are the building blocks of proteins. When their pool meets the cellular needs for protein synthesis, since there are no amino acid stores in the body comparable to glycogen for glucose or triglycerides for fatty acids and glycerol, the excess amino acids are catabolized into precursors of the citric acid cycle and used for energy production.[2]
However, when the energy needs of the cell are met, the carbon skeletons resulting from amino acid catabolism are used for the synthesis of fatty acids, ketone bodies, or glucose.[3]
Glucogenic and ketogenic amino acids are classified based on the metabolic fate of their carbon skeletons. However, this classification is not clear-cut because, among the twenty standard amino acids found in proteins, five are both glucogenic and ketogenic.[4]
Glucose synthesis from glucogenic amino acids is possible because their carbon skeletons are fully or partially catabolized into pyruvate, oxaloacetate, α-ketoglutarate, succinyl-CoA, or fumarate.[5]
Pyruvate and oxaloacetate are intermediates of gluconeogenesis, while the entry of the other three metabolites into the citric acid cycle results in a net gain of carbon units, which can subsequently enter gluconeogenesis as oxaloacetate.[6]
Glucogenic amino acids play a crucial role during prolonged fasting, serving as a major source of precursors for gluconeogenesis, second only to glycerol derived from adipose tissue lipolysis.[7]
Of the twenty amino acids that make up proteins, thirteen are exclusively glucogenic, meaning that the catabolism of their carbon skeletons yields only glucose precursors.[4]
Classification of amino acids based on their catabolites: glucogenic and mixed (glucogenic and ketogenic)
Amino acid
Ketogenic
Glucogenic
Acetyl-CoA
Acetoacetyl-CoA
Alanine
✔ Pyruvate
Arginine
✔ α-Ketoglutarate
Asparagine
✔ Fumarate
Aspartate
✔ Oxaloacetate
Cysteine
✔ Pyruvate
Phenylalanine *
✔
✔ Fumarate
Glycine
✔ Pyruvate
Glutamate
✔ α-Ketoglutarate
Glutamine
✔ α-Ketoglutarate
Isoleucine *
✔
✔ Succinyl-CoA
Histidine
✔ α-Ketoglutarate
Methionine
✔ Succinyl-CoA
Proline
✔ α-Ketoglutarate
Serine
✔ Pyruvate
Tyrosine *
✔
✔ Fumarate
Threonine *
✔
✔ Succinyl-CoA
Tryptophan *
✔
✔
✔ Pyruvate
Valine
✔ Succinyl-CoA
* Amino acids that are both glucogenic and ketogenic.
Five amino acids, namely isoleucine, phenylalanine, threonine, tryptophan, and tyrosine, are both glucogenic and ketogenic, as a portion of their carbon skeleton is catabolized into glucogenic precursors, while another portion is converted into acetyl-CoA and/or acetoacetyl-CoA.[8]
Before amino acid carbon skeletons can be utilized, their amino groups must first be removed. Alanine and glutamate (or glutamine), are two of the primary molecules responsible for transporting amino groups from extrahepatic tissues to the liver, are particularly important glucogenic amino acids in mammals. Alanine is the primary gluconeogenic substrate for the liver, reaching it from muscle and other peripheral tissues via the glucose-alanine cycle.[9]
Biochemical basis
As with ketogenic amino acids, analyzing the stoichiometry of the citric acid cycle helps explain why the carbon skeletons of glucogenic amino acids act as precursors for glucose synthesis. The key factor lies in the specific point at which these carbons enter the cycle.[6]
Metabolic Map of Glucogenic Amino Acid Entry Points into the Krebs Cycle
When carbon atoms derived from amino acids enter the cycle as α-ketoglutarate, succinyl-CoA, or fumarate, they contribute to a net gain of carbon units. Except for α-ketoglutarate, these intermediates enter the cycle downstream of the two oxidative decarboxylation steps catalyzed by isocitrate dehydrogenase (EC 1.1.1.42) and the α-ketoglutarate dehydrogenase multienzyme complex.[5]
This results in a net gain of one carbon unit when entry occurs at α-ketoglutarate, or two when it occurs at succinyl-CoA or fumarate. These carbon atoms can then feed into gluconeogenesis via oxaloacetate.[4]
Furthermore, because the reaction catalyzed by the pyruvate dehydrogenase complex, the oxidative decarboxylation of pyruvate to acetyl-CoA, is irreversible, and since animals lack a pathway to convert acetyl-CoA back to pyruvate, acetyl-CoA cannot serve as a glucogenic substrate.[6]
Glucogenic amino acids during prolonged fasting
Glucogenic amino acids play a crucial role during prolonged fasting and in diets with severe carbohydrate restriction. Under these conditions, they are among the primary precursors for glucose synthesis.[6] The utilization of their carbon skeletons, along with glycerol and propionate, a short-chain fatty acid, helps maintain glycemic homeostasis via gluconeogenesis when liver glycogen stores are depleted.[7]
However, even under physiological conditions, such as during cellular protein turnover or after a protein-rich meal, amino acids exceeding the requirements for protein synthesis are catabolized. Depending on the metabolic state and the specific amino acid, they may be used for energy production or anabolic processes, including glucose synthesis. The glucose produced can subsequently be utilized for glycogen synthesis, converted into ketone bodies or fatty acids.[2]
References
^ D’Andrea G. Classifying amino acids as gluco(glyco)genic, ketogenic, or both. Biochem Educ 2000;28(1):27-28. doi:10.1016/s0307-4412(98)00271-4
^ ab Brosnan J.T. Interorgan amino acid transport and its regulation. J Nutr 2003;133(6 Suppl 1):2068S-2072S. doi:10.1093/jn/133.6.2068S
^ Litwack G. Human biochemistry. 2nd Edition. Academic Pr, 2021.
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ abcd Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
^ ab Kuriyama H., Shimomura I., Kishida K., Kondo H., Furuyama N., Nishizawa H., Maeda N., Matsuda M., Nagaretani H., Kihara S., Nakamura T., Tochino Y., Funahashi T., Matsuzawa Y. Coordinated regulation of fat-specific and liver-specific glycerol channels, aquaporin adipose and aquaporin 9. Diabetes 2002;51(10):2915-2921. 10.2337/diabetes.51.10.2915
^ Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009.
^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
2,3-Bisphosphoglycerate (2,3-BPG) is the conjugated base of 2,3-bisphosphoglyceric acid.[1]
In red blood cells, at sea level, 2,3-BPG is found in concentrations close to that of hemoglobin (Hb), about 5 mM. In contrast, in other cell types, it is found only in trace amounts.[2]
In erythrocytes, it is synthesized from 1,3-bisphosphoglycerate (1,3-BPG) in the first reaction of the Rapoport-Luebering shunt.[3][4] Since the shunt is a detour from the glycolytic pathway, occurring upstream of the reactions leading to ATP synthesis, the production of 2,3-bisphosphoglycerate incurs an energy cost of one ATP molecule per molecule produced.[5]
2,3-BPG is one of the regulators of the binding affinity of hemoglobin for oxygen.[6] It is also required for the catalytic activity of phosphoglycerate mutase (EC 5.4.2.1), and is an allosteric activator of ribose phosphate pyrophosphokinase (EC 2.7.6.1).[2][7]
By binding to hemoglobin, it lowers its oxygen affinity, thereby facilitating oxygen release in peripheral tissues.[6]
In red blood cells, the concentration of 2,3-bisphosphoglycerate can vary in response to changes in the flow of carbon through the glycolytic pathway. These variations may result from changes in altitude, diseases that impair blood oxygenation, or defects in glycolytic enzymes, and they affect hemoglobin binding affinity for oxygen.[8][9][10]
Summary: Key Points
Hemoglobin modulator: 2,3-BPG is an allosteric effector that lowers hemoglobin’s oxygen affinity, promoting oxygen release to tissues.
Rapoport-Luebering shunt: it is synthesized only in red blood cells via a glycolytic bypass, bypassing one ATP-generating step.
Structural selectivity: it binds tightly to adult hemoglobin (HbA) but less to fetal hemoglobin (HbF), facilitating maternal-fetal oxygen transfer.
Physiological adaptation: erythrocyte levels rise during hypoxia, anemia, and high-altitude exposure to optimize systemic oxygen delivery.
2,3-Bisphosphoglycerate is the conjugate base of 2,3-bisphosphoglyceric acid. It has a molecular weight of 261.00 g/mol, and molecular formula C3H3O10P2-5.[1]
According to the IUPAC nomenclature, its systematic name is (2R)-2,3-bis(phosphonooxy)propanoate.[11]
The pKa of 2,3-bisphosphoglyceric acid is 0.48. Therefore, at physiological pH, it exists almost exclusively in its ionized form, 2,3-BPG.[11]
It is soluble in water, with a solubility of 9.69 g/L.[1]
2,3-Bisphosphoglycerate is an isomer of 1,3-bisphosphoglycerate.
Metabolism
2,3-bisphosphoglycerate is produced from the glycolytic intermediate 1,3-bisphosphoglycerate in the first of the two steps of the Rapoport-Luebering shunt.[3][4]
Both reactions of the shunt are catalyzed by bisphosphoglycerate mutase (EC 5.4.2.4), a multifunctional enzyme with three main activities, 2,3-BPG synthase, its main activity, 2,3-BPG phosphatase, and 2,3-BPG mutase.[12]
In the first step of the Rapoport-Luebering shunt, bisphosphoglycerate mutase acts as a synthase and catalyzes the isomerization of 1,3-bisphosphoglycerate to 2,3-bisphosphoglycerate. The enzyme catalyzes the intermolecular transfer of a phosphate group from C-1 of 1,3-BPG to C-2 of 3-phosphoglycerate, which means that 3-phosphoglycerate must be present in the active site. During the reaction, 3-phosphoglycerate is converted into 2,3-bisphosphoglycerate, while 1,3-BPG is transformed into 3-phosphoglycerate.[6]
Biosynthetic Pathway and Functions of 2,3-BPG
In the second step, 2,3-bisphosphoglycerate is dephosphorylated into 3-phosphoglycerate. The reaction is catalyzed by the phosphatase activity of bisphosphoglycerate mutase. 3-Phosphoglycerate then re-enters the glycolytic pathway at the eighth step, which is catalyzed by phosphoglycerate mutase (EC 5.4.2.1).[5]
Energy cost of 2,3-bisphosphoglycerate synthesis
2,3-Bisphosphoglycerate synthesis incurs an energy cost for the red blood cell, equivalent to one ATP molecule per molecule produced.[5]
1,3-BPG is the substrate of the seventh reaction of glycolysis, in which phosphoglycerate kinase (EC 2.7.2.3) catalyzes the transfer of the high-energy phosphate group at the C-1 position to ADP, with the formation of 3-phosphoglycerate and ATP. Phosphoglycerate kinase catalyzes the first of two substrate-level phosphorylations that, along the glycolytic pathway, lead to the conservation of part of the chemical energy contained in glucose in the form of ATP, the cell’s energy currency.[6]
Since, under physiological conditions, the Rapoport-Luebering shunt intercepts approximately 20% of glycolytic carbon flux, the energy cost of 2,3-bisphosphoglycerate synthesis requires a finely tuned balance between the cell’s energy needs and the requirement to maintain hemoglobin in an optimal balance between deoxygenation and oxygenation.[13][14]
Role of 2,3-bisphosphoglycerate
2,3-bisphosphoglycerate has at least three biological functions.
It is required for the catalytic activity of phosphoglycerate mutase. Consequently, the molecule also contributes to the regulation of the levels of glycolytic intermediates.[7]
It is one of the allosteric activators of ribose phosphate pyrophosphokinase or PRPP synthase. The enzyme catalyzes the conversion of ribose-5-phosphate, one of the two products of the pentose phosphate pathway, into 5-phosphoribosyl-1-pyrophosphate (PRPP), an intermediate in the de novo synthesis of purines.[2]
2,3-BPG, along with other allosteric effectors, plays a role in the allosteric regulation of hemoglobin-oxygen affinity, facilitating oxygen release in peripheral tissues.[15] In humans, most primates, and many other mammals, 2,3-BPG reduces hemoglobin-oxygen affinity. In this way, 2,3-BPG promotes the dissociation of oxygen from hemoglobin, thus enhancing oxygen delivery to tissues.[6]
The action of 2,3-BPG is an example of heterotropic allosteric regulation, i.e., allosteric regulation in which the effector is different from the normal ligand of the protein.[5]
Hemoglobin
Hemoglobin is found in red blood cells and, in almost all vertebrates, it is responsible for carrying oxygen to tissues.
The protein has a quaternary structure formed by four subunits and contains four heme prosthetic groups, one per subunit, each of which can reversibly bind an oxygen molecule.[13]
Under physiological conditions, two types of hemoglobin are found in humans: hemoglobin A and hemoglobin F, which is found in the fetus.
Hemoglobin A is composed of two α chains of 141 amino acid residues and two β chains of 146 amino acid residues. Therefore, it is an α2β2 tetramer.
In hemoglobin F, the β chains are replaced by γ chains, which are very similar, but not identical, to the β chains. The result is an α2γ2 stoichiometry.[13] This difference has a major influence on the protein’s affinity for oxygen.[5]
Hemoglobin can exist in two conformations known as the T (tense) and R (relaxed) states.
Although oxygen binds to hemoglobin in both the T and R states, it has a greater affinity for the R state, which is stabilized by the binding. The binding of oxygen by hemoglobin is modulated by several factors, some exerting short-term effects and others, such as 2,3-bisphosphoglycerate, exerting long-term effects.[6]
Cooperativity of hemoglobin-oxygen binding
Hemoglobin in the deoxygenated state is in the T conformation. Oxygen binding to hemoglobin in the T state can occur only on the heme group of one α subunit, since the heme groups of the β subunits in the T state are virtually inaccessible.[6]
The binding of the first oxygen molecule triggers a conformational change in the α subunit, causing its conversion to the R state. This change is transmitted to adjacent subunits via subunit-subunit interactions, which then cause the second α subunit to switch to the R state. This triggers sequential conformational changes that lead to the transition of hemoglobin’s quaternary structure, which binds two oxygen molecules, Hb(O2)2, from the T state to the R state. At this point, the β subunits are now able to bind oxygen.[6]
This type of binding is called cooperative binding and is the basis of the sigmoidal shape of the hemoglobin-oxygen binding curve.[16]
Hemoglobin affinity for oxygen
In the lung capillaries, oxygen binds to hemoglobin and through the bloodstream reaches the peripheral tissues, where it is released.
The affinity between hemoglobin and oxygen is mainly determined by the structure of hemoglobin. However, it is modulated by various factors such as:
Changes in the affinity of the Hb-O2 bond that occur in the circulatory system due to the above factors optimize oxygen uptake in the lungs and its release in peripheral tissues.[10]
Graphically, changes in binding affinity induced by allosteric effectors and temperature translate into shifts of the hemoglobin-oxygen binding curve to the right, in the case of a decrease in binding affinity, or to the left, when binding affinity increases.[5]
Temperature, pH, and CO2 exert short-term effects.[10] For example, in skeletal muscle during physical activity, temperature, the concentration of hydrogen ions (H⁺) and partial pressure of CO2 increase. High concentrations of H⁺ and CO2 lower hemoglobin affinity for oxygen, an effect known as the Bohr effect. An increase in temperature also decreases affinity. These factors collectively favor the release of oxygen to the muscle tissue.[17]
Cl⁻ and 2,3-bisphosphoglycerate also decrease hemoglobin affinity for oxygen, but they are involved in long-term modulation.[10]
2,3-Bisphosphoglycerate and hemoglobin A
2,3-Bisphosphoglycerate binds to hemoglobin A through ionic bonds with positively charged residues of the β chains.
The bonds between 2,3-bisphosphoglycerate and hemoglobin involve the side chains of lysine 82, histidine 143, and the N-terminal amino group of each of the two β chains. These residues form an electrostatic pocket that is complementary to both the conformation and the charge distribution of 2,3-BPG.[5]
2,3-BPG binds to deoxygenated hemoglobin and stabilizes the T state. H⁺, Cl–, and CO2 also stabilize the T state.[10]
During the transition from the T to the R state, among the conformational changes occurring in the quaternary structure of hemoglobin, the binding pocket for 2,3-BPG between the β subunits narrows, preventing its binding. Notably, while hemoglobin has four binding sites for molecular oxygen, it has only one for 2,3-BPG.[6]
Furthermore, the binding site for 2,3-BPG is located far from that of oxygen.[5]
2,3-Bisphosphoglycerate and hemoglobin F
The γ chains of hemoglobin F, like the β chains of hemoglobin A, form the binding pocket for 2,3-BPG.[5]
However, there are two fewer positively charged residues in the binding pocket than in hemoglobin A. In fact, at position 143 of the γ chains, a histidine is replaced by a serine.[2]
This small difference in the primary structure means that 2,3-bisphosphoglycerate binds with lower affinity, and consequently, hemoglobin F has a higher oxygen affinity than maternal hemoglobin. This helps ensure the transfer of oxygen from hemoglobin A to hemoglobin F, and thus from the maternal circulation to the fetal circulation.[6]
Changes in 2,3-bisphosphoglycerate concentration
The concentration of 2,3-bisphosphoglycerate in red blood cells can be influenced by various factors, both physiological and pathological.[2]
Physiological conditions include the adaptation to high altitude, while pathological conditions include genetic defects affecting glycolytic enzymes, both downstream and upstream of the reaction catalyzed by glyceraldehyde 3-phosphate dehydrogenase (EC 1.2.1.12), and diseases that cause hypoxia.[18][19]
In all cases, changes are due to variations in the rate of 2,3-BPG synthesis and are reflected in hemoglobin-oxygen affinity.
Adaptation to high altitudes
Adaptation to high altitude is a rather complex process that also involves an increase in the number of red blood cells and their hemoglobin content. These changes require weeks to complete. However, after just one day at high altitude, some degree of adaptation is perceived. This effect is a consequence of the increase in 2,3-bisphosphoglycerate concentration in red blood cells.[2]
What happens is that the low partial pressure of oxygen leads to an increase in blood pH (alkalosis) which stimulates glycolysis. This, in turn, leads to an increased carbon flow through the Rapoport-Luebering shunt. This causes an increase in the synthesis of 2,3-bisphosphoglycerate, whose concentration can reach up to 8 mM. Such increase leads to a decrease in hemoglobin-oxygen affinity, facilitating oxygen release to the peripheral tissues and thus contributing to adaptation to high altitudes.[5]
Anemia
Variations in erythrocyte 2,3-bisphosphoglycerate concentration also occur in two autosomal recessive diseases: non-spherocytic hemolytic anemia due to hexokinase deficiency and hemolytic anemia due to red cell pyruvate kinase deficiency.[18][19]
In hemolytic anemia due to red cell pyruvate kinase (EC 2.7.1.40) deficiency, enzyme deficiency causes an increase in the synthesis of 2,3-BPG and, consequently, a reduction in hemoglobin-oxygen affinity.[8] An increase in 2,3-BPG concentration also occurs in individuals affected by conditions that limit blood oxygenation, such as hypoxia-inducing conditions. An example is cardiopulmonary failure.[2]
In non-spherocytic hemolytic anemia due to hexokinase deficiency (EC 2.7.1.1), the enzyme deficiency reduces 2,3-bisphosphoglycerate concentration in red blood cells, leading to an increase in hemoglobin-oxygen affinity.[9]
^ abcdefg Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
^ ab Rapoport S., Luebering J. Glycerate-2,3-diphosphatase. J Biol Chem 1951;189(2):683-694.
^ ab Rapoport S., Luebering J. The formation of 2,3-diphosphoglycerate in rabbit erythrocytes: the existence of a diphosphoglycerate mutase. J Biol Chem 1950;183(2):507-516.
^ abcdefghij Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ ab Oslund R.C., Su X., Haugbro M., Kee J-M., Esposito M., David Y., Wang B., Ge E., Perlman D.H., Kang Y., Muir T.W., Rabinowitz J.D. Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat Chem Biol 2017;13:1081-1087. doi:10.1038/nchembio.2453
^ ab Enegela O.A., Anjum F. Pyruvate Kinase Deficiency. [Updated 2023 Apr 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560581/
^ ab Paglia D.E., Shende A., Lanzkowsky P., Valentine W.N. Hexokinase “New Hyde Park”: a low activity erythrocyte isozyme in a Chinese kindred. Am J Hematol 1981;10(2):107-17. doi:10.1002/ajh.2830100202
^ abcde Webb K.L., Dominelli P.B., Baker S.E., Klassen S.A., Joyner M.J., Senefeld J.W., Wiggins C.C. Influence of high hemoglobin-oxygen affinity on humans during hypoxia. Front Physiol 2022;12:763933. doi:10.3389/fphys.2021.763933
^ Aljahdali A.S., Musayev F.N., Burgner J.W. 2nd, Ghatge M.S., Shekar V., Zhang Y., Omar A.M., Safo M.K. Molecular insight into 2-phosphoglycolate activation of the phosphatase activity of bisphosphoglycerate mutase. Acta Crystallogr D Struct Biol 2022;78(Pt 4):472-482. doi:10.1107/S2059798322001802
^ abc Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ Hodgkins S.R. 6 – Erythrocyte metabolism and membrane structure and function. Editor(s): Keohane E.M., Otto C.N., Walenga J.M. Rodak’s Hematology (Sixth Edition), Elsevier. 2020:78-90. doi:10.1016/B978-0-323-53045-3.00015-5
^ ab Mairbäurl H., Oelz O., Bärtsch P. Interactions between Hb, Mg, DPG, ATP, and Cl determine the change in Hb-O2 affinity at high altitude. J Appl Physiol 1993;74(1):40-8. doi:10.1152/jappl.1993.74.1.40
^ Henry E.R., Bettati S., Hofrichter J., Eaton W.A. A tertiary two-state allosteric model for hemoglobin. Biophys Chem 2002;98(1-2):149-64. doi:10.1016/s0301-4622(02)00091-1
^ Böning D., Schwiegart U., Tibes U., Hemmer B. Influences of exercise and endurance training on the oxygen dissociation curve of blood under in vivo and in vitro conditions. Eur J Appl Physiol Occup Physiol 1975;34(1):1-10. doi:10.1007/BF00999910
The Rapoport-Luebering shunt is a metabolic pathway predominantly present in red blood cells and is a side glycolytic pathway that branches off at the level of 1,3-bisphosphoglycerate (1,3-BPG).[1][2]
The two reactions of the shunt are catalyzed by a multifunctional enzyme, bisphosphoglycerate mutase (EC 5.4.2.4). However, a second enzyme involved in the shunt, 2,3-bisphosphoglycerate 3-phosphatase (EC 3.1.3.80), has recently been identified.[3]
The most important product is 2,3-bisphosphoglycerate (2,3-BPG), an allosteric effector of hemoglobin that reduces its affinity for molecular oxygen, thereby facilitating its release at the tissue level.[4][5]
As it branches off from glycolysis upstream of the reactions leading to ATP synthesis, the Rapoport-Luebering shunt imposes an energy cost on the cell.[3]
Defects in glycolytic enzymes in red blood cells lead to abnormalities in 2,3-BPG synthesis and, consequently, in the affinity of hemoglobin for molecular oxygen.[6]
In Summary: Key Points
Biological function: the Rapoport-Luebering shunt is a side pathway of glycolysis exclusive to erythrocytes, essential for regulating oxygen delivery to body tissues.
Crucial metabolite: it produces 2,3-bisphosphoglycerate, an allosteric effector that binds to deoxyhemoglobin, stabilizing it and thereby promoting oxygen release to peripheral tissues.
Enzymatic control: metabolic flux is primarily governed by the trifunctional enzyme bisphosphoglycerate mutase, alongside the phosphatase activity of MIPP1.
Energy trade-off and pathology: Bypassing glycolysis costs the cell 2 ATP per glucose molecule; imbalances caused by enzyme defects (e.g., pyruvate kinase or hexokinase deficiency) lead to hemolytic anemia.
Schematic Representation of the Rapoport-Luebering Shunt
Bisphosphoglycerate mutase is a central enzyme in the Rapoport-Luebering shunt, and is found in red blood cells.[4]
It is a multifunctional enzyme, as it functions as a 2,3-bisphosphoglycerate synthase and a 2,3-bisphosphoglycerate phosphatase. It is also a 2,3-bisphosphoglycerate mutase, an activity similar to that of glycolytic phosphoglycerate mutase (EC 5.4.2.1), but does not participate in the shunt. All three catalytic activities take place at the same active site, with synthase being the primary activity, which is inhibited by free 2,3-bisphosphoglycerate.[7][8]
The phosphatase activity is low, approximately 1000 times lower than the synthase activity, and is stimulated by various anions, including chloride, phosphate, sulfite, and, more markedly, 2-phosphoglycolate.[9]
Finally, it appears that the mutase activity is physiologically insignificant, catalyzing about 5 percent of the reversible conversion of 3-phosphoglycerate to 2-phosphoglycerate.[10][11]
Bisphosphoglycerate mutase is one of the three isoforms of phosphoglycerate mutase found in mammals. The other two isoforms are phosphoglycerate mutase 1 or type M, present in muscle, and phosphoglycerate mutase 2 or type B, present in all other tissues. These two isoforms are also trifunctional enzymes.[4]
Reactions
The Rapoport-Luebering shunt involves two reactions.
In the first reaction, bisphosphoglycerate mutase acts as a synthase, catalyzing the isomerization of 1,3-BPG to 2,3-bisphosphoglycerate. The enzyme requires 3-phosphoglycerate, as it catalyzes the intermolecular transfer of a phosphate group from C-1 of 1,3-bisphosphoglycerate to C-2 of 3-phosphoglycerate. In the reaction, therefore, the starting 3-phosphoglycerate becomes 2,3-BPG, while 1,3-BPG becomes the new 3-phosphoglycerate.[12]
In the second step, the phosphatase activity of bisphosphoglycerate mutase catalyzes the synthesis of 3-phosphoglycerate, a glycolytic intermediate, following the hydrolysis of the phosphate group at position C-2 of 2,3-BPG. The 3-phosphoglycerate produced can then re-enter the glycolytic pathway at the reaction catalyzed by phosphoglycerate mutase, i.e., the eighth step of the pathway.[5]
2,3-Bisphosphoglycerate 3-phosphatase
A study has identifying a second enzyme involved in the Rapoport-Luebering shunt. In addition to bisphosphoglycerate mutase, researchers have discovered a distinct 2,3-bisphosphoglycerate 3-phosphatase activity catalyzed by an evolutionarily conserved multiple inositol polyphosphate phosphatase, known as MIPP1, which has been found in Dictyostelium, birds, and mammals.
This discovery reveals that the formation of 3-phosphoglycerate, a precursor for serine biosynthesis and an activator of AMP-activated protein kinase, can be bypassed. By genetically manipulating MIPP1 expression in Dictyostelium, scientists demonstrated the enzyme’s physiologically relevant role in regulating cellular 2,3-BPG content.[3]
Role of the Rapoport-Luebering shunt
The Rapoport-Luebering shunt leads to the formation of 2,3-BPG, an allosteric effector of hemoglobin, along with carbon dioxide and hydrogen ions.[5]
2,3-Bisphosphoglycerate is found in high concentrations in human red blood cells, whereas in other cells, it is present only in trace amounts.[6] In most primates and many other mammals, it decreases hemoglobin’s affinity for molecular oxygen, thereby facilitating its release to the tissues.[3]
2,3-BPG binds to deoxyhemoglobin by means of ionic bonds with the β chains in the T state, a state in which these subunits form an electrostatic pocket complementary to both the conformation and the charge distribution of the molecule. The bond stabilizes the T state, hence deoxyhemoglobin, and is primarily responsible for hemoglobin’s cooperative binding of molecular oxygen.[12]
In trace amounts, 2,3-BPG is also required for the catalytic activity of phosphoglycerate mutase. It follows that another function of bisphosphoglycerate mutase, and hence of the Rapoport-Luebering shunt, is to control the level of glycolytic intermediates.[13]
Energy cost of the Rapoport-Luebering shunt
1,3-bisphosphoglycerate is an intermediate of the glycolytic pathway. In the seventh step of glycolysis, phosphoglycerate kinase (EC 2.7.2.3) catalyzes the transfer of the high-energy phosphoryl group at the C-1 position of 1,3-BPG to ADP, with the formation of ATP and 3-phosphoglycerate. The reaction, a substrate-level phosphorylation, is the first of two reactions in the glycolytic pathway in which part of the chemical energy contained in glucose is conserved in the form of ATP.[5]
Therefore, the entry of 1,3-BPG into the Rapoport-Luebering cycle incurs an energy cost equivalent to two ATP molecules per 1,3-bisphosphoglycerate molecule diverted from glycolysis.[3] As a result, a delicate balance exists between the energy demands of red blood cells and the requirement to maintain hemoglobin in an optimal state of oxygenation and deoxygenation.[14]
Relationship between glycolysis and the Rapoport-Luebering shunt
Under physiological conditions, the Rapoport-Luebering shunt intercepts approximately 20% of the glycolytic flux in red blood cells.[8] It follows that defects in erythrocyte glycolytic enzymes influence the shunt and, consequently, the affinity of hemoglobin for molecular oxygen.[6]
Two conditions that illustrate this connection are nonspherocytic hemolytic anemia due to hexokinase deficiency and hemolytic anemia due to pyruvate kinase deficiency in erythrocytes, both of which are autosomal recessive diseases.[15][16]
In nonspherocytic hemolytic anemia due to hexokinase deficiency there is a deficiency of erythrocyte hexokinase (EC 2.7.1.1).[17] The enzyme catalyzes the phosphorylation of glucose to glucose-6-phosphate. Its deficiency leads to a decrease in the erythrocyte concentration of 2,3-BPG and, consequently, an increase in the affinity of hemoglobin for molecular oxygen.[6]
In contrast, hemolytic anemia due to pyruvate kinase deficiency in erythrocytes is characterized by a deficiency of pyruvate kinase (EC 2.7.1.40), leading to increased synthesis of 2,3-BPG and a decreased affinity of hemoglobin for molecular oxygen.[18]
Reference
^ Rapoport S., Luebering J. Glycerate-2,3-diphosphatase. J Biol Chem 1951;189(2):683-694.
^ Rapoport S., Luebering J. The formation of 2,3-diphosphoglycerate in rabbit erythrocytes: the existence of a diphosphoglycerate mutase. J Biol Chem 1950;183(2):507-516.
^ abcde Cho J., King J.S., Qian X., Harwood A.J., Shears S.B. Dephosphorylation of 2,3-bisphosphoglycerate by MIPP expands the regulatory capacity of the Rapoport-Luebering glycolytic shunt. Proc Natl Acad Sci U S A 2008;105(16):5998-6003. doi:10.1073/pnas.0710980105
^ abc DiMauro S., Miranda A.F., Khan S., Gitlin K., Friedman R. Human muscle phosphoglycerate mutase deficiency: newly discovered metabolic myopathy. Science 1981;212(4500):1277-9. doi:10.1126/science.6262916
^ abcd Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ abcd Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
^ Aljahdali A.S., Musayev F.N., Burgner J.W. 2nd, Ghatge M.S., Shekar V., Zhang Y., Omar A.M., Safo M.K. Molecular insight into 2-phosphoglycolate activation of the phosphatase activity of bisphosphoglycerate mutase. Acta Crystallogr D Struct Biol 2022;78(Pt 4):472-482. doi:10.1107/S2059798322001802
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ Reynolds C.H. Activation of human erythrocyte 2,3-bisphosphoglycerate phosphatase at physiological concentrations of substrate. Arch Biochem Biophys 1986;250(1):106-11. doi:10.1016/0003-9861(86)90706-x
^ Lemarchandel V., Joulin V., Valentin C., Rosa R., Galactéros F., Rosa J., Cohen-Solal M. Compound heterozygosity in a complete erythrocyte bisphosphoglycerate mutase deficiency. Blood 1992;80(10):2643-9. doi:10.1182/blood.V80.10.2643.2643
^ Sasaki R., Chiba H. Role and induction of 2,3-bisphosphoglycerate synthase. Mol Cell Biochem 1983;53-54(1-2):247-56. doi:10.1007/BF00225257
^ Oslund R.C., Su X., Haugbro M., Kee J-M., Esposito M., David Y., Wang B., Ge E., Perlman D.H., Kang Y., Muir T.W., Rabinowitz J.D. Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat Chem Biol 2017;13:1081-1087. doi:10.1038/nchembio.2453
^ Hodgkins S.R. 6 – Erythrocyte metabolism and membrane structure and function. Editor(s): Keohane E.M., Otto C.N., Walenga J.M. Rodak’s Hematology (Sixth Edition), Elsevier. 2020:78-90. doi:10.1016/B978-0-323-53045-3.00015-5
^ Paglia D.E., Shende A., Lanzkowsky P., Valentine W.N. Hexokinase “New Hyde Park”: a low activity erythrocyte isozyme in a Chinese kindred. Am J Hematol 1981;10(2):107-17. doi:10.1002/ajh.2830100202
^ Enegela O.A., Anjum F. Pyruvate Kinase Deficiency. [Updated 2023 Apr 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560581/
Pyruvic acid, an alpha-ketoacid, is a molecule with a central role in cellular metabolism.[1][2]
It can be produced through various metabolic pathways, mostly cytosolic, among which glycolysis is usually the most important pathway, while its fate depends on the cell type and the availability of oxygen, as it can be used for energy, biosynthetic, and anaplerotic purposes.[3]
Given its central role in cellular metabolism, mutations in the genes that encode the proteins involved in its metabolism cause, in humans, mild to severe diseases.[4]
Summary: Key Points
Metabolic hub: pyruvic acid is a central molecule in cellular metabolism; at physiological pH, it exists almost entirely in its anionic form as pyruvate.
Cytosolic origin: under physiological conditions, it is mainly derived from glycolysis, but it can also be generated via lactate oxidation, alanine transamination, or the malic enzyme.
Oxygen crossroads: under hypoxic conditions (or in cells without mitochondria), it is reduced to lactate to regenerate NAD+; in the presence of oxygen, it enters the mitochondrion via MPC transporters.
Mitochondrial fates: once in the matrix, it is converted into acetyl-CoA by the pyruvate dehydrogenase complex for energy production, or carboxylated into oxaloacetate for anaplerotic purposes.
Pyruvic acid or, according to the IUPAC nomenclature, 2-oxopropanoic acid, has a molecular formula C3H4O3, condensed formula CH3COCOOH, and molecular weight of 88.06 g/mol.
It belongs to the group of alpha-ketoacids, meaning keto acids that have the carbonyl group adjacent to the carboxylic acid. It has the simplest chemical structure among the alpha-ketoacids.[1]
In purified form it appears as a colorless liquid, with a smell similar to that of acetic acid.
Its acid dissociation constant (pKa), at 25 °C, is equal to 2.45. It is therefore a very strong acid, and, at physiological pH, both in cells and in extracellular fluids, is present almost entirely in its anionic form, pyruvate.
Its melting point is 13.8 °C (56.84 °F; 286.95 K), while its boiling point is 163.5 °C (329 °F; 438 K).[1]
Pyruvate metabolism
The biosynthetic pathways leading to pyruvic acid production, as well as its subsequent utilization, depend on the cell type and/or the availability of oxygen.[3]
Cytosolic and Mitochondrial Metabolic Pathways of Pyruvic Acid
In cells with mitochondria, pyruvate metabolism consists of a cytosolic and a mitochondrial phase.
In the cytosol, several metabolic pathways lead to its formation, namely, glycolysis, the oxidation of lactate, the reaction catalyzed by the cytosolic malic enzyme, and the catabolism of at least six amino acids, the most important of which is alanine.
Pyruvic acid production from alanine and lactate can also occur in the mitochondrial matrix.[5]
The pyruvic acid produced in the cytosol, by means of specific transporters, enters the mitochondrion where it can be used for energy, entering the citric acid cycle, and/or biosynthetic/anaplerotic purposes, depending on the needs of the cell.[6]
If, instead, we consider cells without mitochondria, such as red blood cells, and, under hypoxic conditions, cells with mitochondria too, pyruvate is reduced to lactate and/or leaves the cell as such to be metabolized in other tissues, for example cardiac muscle.[7]
Glycolysis
Under physiological conditions, in most cells pyruvic acid is mainly derived from glycolysis, of which it is one of the three products, with ATP and NADH.
In the last step of the glycolytic pathway, pyruvate kinase (EC 2.7.1.40) catalyzes a substrate-level phosphorylation that leads to the transfer of the phosphoryl group from phosphoenolpyruvate to ADP. The reaction, essentially irreversible, produces one molecule of ATP and one of pyruvate.
Phosphoenolpyruvate + ADP + H+ → Pyruvate + ATP
Therefore, during the glycolytic pathway, two molecules of pyruvate are produced from a glucose molecule.[2][8]
Lactate dehydrogenase
In the cytosol, pyruvic acid can be produced from lactic acid.
The enzyme lactate dehydrogenase (LDH) (EC 1.1.1.27) catalyzes the reversible conversion of pyruvate to lactate and of NADH to NAD+.
Pyruvate + NADH + H+ ⇄ Lactate + NAD+
The direction of the reaction depends on lactate dehydrogenase isozymes present and the NADH/NAD+ ratio in the cytosol.[3][5]
Isoenzymes in which the H subunit predominates, such as LDH-1, predominate in cardiac muscle, an exclusively aerobic tissue, where they catalyze the oxidation of lactate (formed in other tissues) to pyruvate, which is then used for energy.
The oxidation of lactate produced for example by red blood cells or skeletal muscle under hypoxic conditions, can also occur in hepatocytes, favored by the low NADH/NAD+ ratio in the cytosol, although LDH-5 is the main isoenzyme. In the hepatocyte, pyruvic acid can be enter gluconeogenesis, which in this case is part of the Cori cycle, or be oxidized for energy.[9]
Conversely, in skeletal muscle fiber under hypoxic conditions, in which the pyruvate dehydrogenase complex is inhibited and oxidative phosphorylation is blocked, in order for glycolysis to proceed pyruvate is reduced to lactate with the concomitant oxidation of NADH to NAD+.[10] Note that the conversion of glucose into lactate is defined as lactic fermentation. This reduction is favored by the fact that in skeletal muscle fiber isoenzymes with a prevalence of the M subunit predominate, such as LDH-4 and LDH-5.[11]
Alanine aminotransferase
Another source of pyruvic acid is alanine, an amino acid particularly abundant in muscle proteins.
The utilization of the carbon skeleton of amino acids for energy and/or anabolic purposes involves the removal of the basic amino group, which occurs in reactions catalyzed by enzymes called transaminases (EC 2.6.1-), and the subsequent disposal of nitrogen in a non-toxic form through the urea cycle.[2][8]
The removal of the amino group of alanine is catalyzed by alanine aminotransferase (ALT; EC 2.6.1.2). The reaction, reversible, leads to the formation of pyruvate and glutamate.
Two forms of ALT have been identified: ALT1, localized in the cytosol, and ALT2, localized in the mitochondrial matrix.[4] Hence, pyruvic acid can be produced by transamination of alanine also in the mitochondrial matrix.
Alanine is one of the main gluconeogenic precursors, and, through the glucose-alanine cycle, represents a link between the metabolism of carbohydrates and amino acids.[12]
In the cytosol, the carbon skeletons of five other amino acids, namely cysteine, glycine, serine, threonine and tryptophan, can be converted partly or entirely to pyruvate.[2]
Malic enzyme
Pyruvate can also be produced from malate.
In the reaction catalyzed by the cytosolic malic enzyme (EC 1.1.1.40), malate undergoes an oxidative decarboxylation to yield pyruvate.[3]
Malate + NADP+ → Pyruvate + CO2 + NADPH + H+
Malic enzyme plays an important role in the transport of intermediates of the citric acid cycle, such as, in addition to malate, oxaloacetate and citrate, between the cytosol and the mitochondrial matrix.[7]
Mitochondrial pyruvate carriers
In cells with mitochondria, most of the pyruvate produced in the cytosol enters the mitochondrial matrix passing through the outer and the inner mitochondrial membranes. In the mitochondrial matrix pyruvate can then be used for both anabolic and catabolic purposes.
The passage through the outer mitochondrial membrane occurs by free diffusion through non-specific voltage-dependent anion channels (VDACs) or porins, the most abundant proteins of the outer mitochondrial membrane, whose function is to mediate the exchange of ions and small molecules, including, in addition to pyruvate, also ATP, NADH and others, between the cytosol and the intermembrane space of mitochondria.[13][14]
Conversely, the inner mitochondrial membrane is impermeable to charged molecules, which allows the maintenance of the proton gradient needed for oxidative phosphorylation to occur. The passage of pyruvate therefore occurs through specific transporters, the mitochondrial pyruvate carriers (MPC), a hetero-oligomeric complex of two subunits, indicated as MPC1 and MPC2. This transport is coupled to a symport of protons. MPC therefore links the cytosolic and mitochondrial metabolism of pyruvate.[15][16]
Pyruvate dehydrogenase complex
Once in the mitochondrial matrix, pyruvate is mostly oxidized to carbon dioxide in order to support ATP production.
The first step in this oxidation process involves the pyruvate dehydrogenase complex (PDC), one of the most important multienzyme complexes present in cells. The complex catalyzes the irreversible oxidative decarboxylation of pyruvate to form acetyl-coenzyme A (acetyl-CoA) and a carbon dioxide molecule, with the release of two electrons, which are carried by NAD.
Pyruvate + CoA + NAD+ → Acetyl-CoA + NADH + H+ + CO2
The acetyl group can enter the citric acid cycle to be completely oxidized to carbon dioxide, with the production of a molecule of GTP, 3 molecules of NADH, one of FADH2. The two reduced coenzymes, through oxidative phosphorylation, will allow the production of further ATP molecules.[2]
Alternatively, the acetyl group derived from pyruvic acid can be used for anabolic purposes, among which the synthesis of some lipids, such as of fatty acids, phospholipids and cholesterol, or for regulatory purposes through histone acetylation.
Since lipid biosynthesis occurs in the cytosol and histone acetylation in the nucleus, acetyl-CoA must leave the mitochondrial matrix, a process that requires the formation of citrate through the condensation between the acetyl group and carbonyl of oxaloacetate, reaction catalyzed by citrate synthase (EC 2.3.3.1).
Once citrate has reached the cytosol, through a citrate carrier, an integral protein of the inner mitochondrial membrane, it is cleaved to acetyl-CoA and oxaloacetate in the reaction catalyzed by citrate lyase (EC 4.1.3.6).[17]
Pyruvate carboxylase
In the mitochondrial matrix, pyruvic acid can be carboxylated to oxaloacetate in an irreversible reaction catalyzed by pyruvate carboxylase (PC) (EC 6.4.1.1).[8]
Pyruvate + HCO3– + ATP → Oxaloacetate + ADP + Pi
Several intermediates of the citric acid cycle are precursors for the synthesis of various molecules. It follows that each of these intermediates removed from the citric acid cycle must be reintegrated for the cycle to continue. Reactions that reintegrate the cycle are defined as anaplerotic. In this perspective, the reaction catalyzed by pyruvate carboxylase plays an anaplerotic function, catalyzing the formation of oxaloacetate.[6]
^ abcde Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ abcd Berg J.M., Tymoczko J.L., Gregory J.G. Jr, Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
^ ab Gray L.R., Tompkins S.C., Taylor E.B. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-604. doi:10.1007/s00018-013-1539-2
^ ab Markert C.L., Shaklee J.B., Whitt G.S. Evolution of a gene. Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science 1975;189(4197):102-14. doi:10.1126/science.1138367
^ ab Owen O.E., Kalhan S.C., Hanson R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 2002;277(34):30409-12. doi:10.1074/jbc.R200006200
^ ab McCommis K.S. and Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J 2015;466(3):443-454. doi:10.1042/BJ20141171
^ abc Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ Gleeson T.T. Post-exercise lactate metabolism: a comparative review of sites, pathways, and regulation. Annu Rev Physiol 1996;58:565-81. doi:10.1146/annurev.ph.58.030196.003025
^ Li X., Yang Y., Zhang B., Lin X., Fu X., An Y., Zou Y., Wang J.X., Wang Z., Yu T. Lactate metabolism in human health and disease. Signal Transduct Target Ther 2022;7(1):305. doi:10.1038/s41392-022-01151-3
^ Farhana A., Lappin S.L. Biochemistry, lactate dehydrogenase. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557536/
^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
^ Colombini M. The VDAC channel: molecular basis for selectivity. Biochim Biophys Acta 2016;1863(10):2498-502. doi:10.1016/j.bbamcr.2016.01.019
^ Young M.J., Bay D.C., Hausner G., Court D.A. The evolutionary history of mitochondrial porins. BMC Evol Biol 2007;7:31. doi:10.1186/1471-2148-7-31
^ Bricker D.K., Taylor E.B., Schell J.C., Orsak T., Boutron A., Chen Y.C., Cox J.E., Cardon C.M., Van Vranken J.G., Dephoure N., Redin C., Boudina S., Gygi S.P., Brivet M., Thummel C.S., Rutter J. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 2012;337(6090):96-100. doi:10.1126/science.1218099
^ Herzig S., Raemy E., Montessuit S., Veuthey J.L., Zamboni N., Westermann B., Kunji E.R., Martinou J.C. Identification and functional expression of the mitochondrial pyruvate carrier. Science 2012;337(6090):93-6. doi:10.1126/science.1218530
^ Zara V., Assalve G., Ferramosca A. Multiple roles played by the mitochondrial citrate carrier in cellular metabolism and physiology. Cell Mol Life Sci 2022;79(8):428. doi:10.1007/s00018-022-04466-0
Lactate dehydrogenase (LDH; EC 1.1.1.27) is a family of oxidoreductases that catalyze the reversible conversion of pyruvate, the conjugate base of pyruvic acid, to lactate, with the concomitant interconversion of NADH and NAD+, which act as cofactors.
They are tetrameric enzymes, where each subunit has catalytic activity. The subunits, encoded by distinct genes, can be assembled in different combination to form isozymes with specific kinetic and regulatory properties.[1]
Lactate dehydrogenase is found in almost all animal tissues, plants, but also in microorganisms. Although it is mostly present in the cytosol, its presence has also been demonstrated in mitochondria, where it catalyzes the oxidation of lactate to pyruvate, and in peroxisomes.[2][3]
In humans, different isoenzymes have preferential tissue localizations, based on the specific metabolic phenotype of the tissue.
Lactate dehydrogenase is a important enzyme in cellular metabolism, as it is involved in energy production from carbohydrates under anaerobic conditions, in the synthesis of glucose from lactate, and utilization of the carbon skeleton of lactate for energy under aerobic conditions.[1][4]
Summary: Key Points
Core reaction: lactate dehydrogenase (LDH) catalyzes the reversible interconversion between pyruvate and lactate, accompanied by the oxidation/reduction of NADH and NAD+.
Isoenzymes: human LDH exists as five main tetrameric isoforms (LDH-1 to LDH-5) formed by combinations of H and M subunits with tissue-specific distribution.
Genetic basis: distinct genes (LDHA, LDHB, LDHC, LDHx) encode L-lactate specific subunits, while the LDHD gene encodes a separate D-lactate dehydrogenase using FAD.
Metabolic role: essential for anaerobic glycolysis, allowing the regeneration of NAD+ to sustain cellular ATP production under hypoxic conditions.
Tissue specificity: heart tissue favors H-rich isoforms adapted for aerobic oxidative metabolism, while skeletal muscle and liver favor M-rich isoforms geared toward glycolysis.
In mammals, several genes encode the subunits of lactate dehydrogenase and are designated LDHA, LDHB, LDHC, LDHx, and LDHD.
The first four genes encode enzymes that recognize as substrate the L-isomers of lactic acid, a molecule with a chirality center, the major enantiomeric form of the molecule present in vertebrates, and have NAD as a cofactor, whereas LDHD encodes an enzyme that recognizes as substrate the D-isomer of lactic acid and has FAD as a cofactor.[5][6]
LDHA, located on chromosome 11p15.4, encodes the M subunit, named for its discovery in muscle, whereas LDHB, located on chromosome 12p12.2-p12.1, encodes the H subunit, named for its discovery in heart tissue.[2][7]
LDHC, located on chromosome 11p15.5-p15.3 and probably derived from the duplication of the LDHA, encodes the C subunit.[8]
Finally, LDHx encodes the peroxisomal form of LDH.[2] LDHx is the readthrough form of the LDHB gene; in fact what happens is that when the ribosome reaches the stop codon of the mRNA for the H subunit, reads it as encoding an amino acid. Then the translation proceeds by adding another seven amino acids that constitute the peroxisomal targeting signal by which the proteins are imported into the peroxisome.[3]
Structure
The enzymes belonging to the lactate dehydrogenase family have an oligomeric structure, specifically they are tetramers resulting from the assembly of subunits, of approximately 35 kD, which can be of the same type or two different types. Each subunit has a catalytic site, hence, the tetramer has four active sites. However, the subunits, taken individually, are catalytically inactive.[7][9]
Considering the H and M subunits, their primary structures are approximately 75% identical, and consequently, their three-dimensional structure is also very similar, but with small differences at the at the substrate binding site that lead to significant differences in the kinetic properties of the proteins.[1][10] Another consequence of the differences in the primary structure concerns the net surface charge, which is -6 for the M subunit and +1 for the H subunit.[11]
The secondary structure is made up of approximately 40 percent alpha-helices and 23% beta sheets, forming beta-alpha-beta structures, with the parallel beta sheets as the main component.[12]
In humans, the prevalent isozymes are those formed by the H and M subunits. The assembly, in different combinations, of the two subunits leads to the formation of five isozymes, namely:
From Gene Expression to the Tetrameric Structure of LDH
The C subunit forms a sixth isoform, a homotetramer, named as LDH-6 or C4.[8]
A seventh isoform derives from the assembly of the LDHx subunit.[3][8]
Reaction
Lactate dehydrogenase catalyzes the reversible conversion of pyruvate to lactate, with the concomitant interconversion of NADH and NAD+. In both cases, the removal of two hydrogen atoms from the reducing agent occurs, followed by the transfer of a hydride ion, namely, a proton and two electrons, to the oxidizing agent, while the remaining proton is released into the aqueous medium as free H+ ion. The enzyme is able to increase the reaction rate by 14 times.[5]
When the reaction proceeds from pyruvate to lactate, the first step is the binding of NADH, the reducing agent, to the enzyme, which is followed by conformational changes that facilitate the formation of hydrogen bonds between specific residues surrounding the active site and the carbonyl carbon of pyruvate, namely C2, and the subsequent interaction between NADH and pyruvate.[13] At this point, a hydride ion is transferred from the nicotinamide ring of NADH to the C2 of pyruvate, which is therefore reduced to lactate. This causes the oxidation of the coenzyme and neutralization of the positive charge carried by the nitrogen of the nicotinamide ring, and reduction of pyruvate to lactate. Therefore, in this case the C2 of pyruvate is reduced from ketone to alcohol.
In the opposite direction, the hydride ion is transferred from the C2 of lactate, which in this case is the reducing agent, to the to the nicotinamide atom C-4. In this case the C2 of lactate is oxidized from alcohol to ketone.[14][15]
Active site
The active site of the H and M subunits has a highly conserved structure in different species, with the same amino acids participating in the reaction.[16]His-193, which is found near the binding site for the coenzyme, is among these, not only for human lactate dehydrogenase, but also for that of many other species.
However, the small differences in their primary structure lead to different biochemical properties. Among the differences, one is crucial for the catalytic properties: an alanine of the M subunit is replaced by a glutamine in the H subunit, namely, a non-polar amino acid is replaced by a polar one.[11] Below is a brief overview of the catalytic differences between the two subunits.
The H subunit binds substrate faster than the M subunit, but has about five times less catalytic activity than the M subunit.
The M subunit has a higher affinity for pyruvate, thus favoring the formation of lactate and NAD+, and is not inhibited by high concentrations of pyruvate. Conversely, the H subunit has a higher affinity for lactate, which favors the formation of pyruvate and NADH, and is inhibited by high concentrations of pyruvate.[17][18]
From all this it follows that the kinetic/catalytic properties of the different isozymes depend on the prevalence of one of the two subunits.[19]
Regulation by pyruvate and lactate
Lactate dehydrogenase isozymes are subject to inhibition by pyruvate and lactate.
The isozymes where the H subunit predominates are inhibited by lower pyruvate concentrations than those where the M subunit predominates. For example, H4 is inhibited by pyruvate concentrations of about 0.2 mM, while M4 is weakly inhibited by pyruvate concentrations up to 5 mM.
Conversely, H4 is inhibited by lactate concentrations greater than 20-40 mM, whereas M4 is inhibited to a lesser extent by high lactate concentrations.[20]
Tissue distribution
In humans, the different lactate dehydrogenase isozymes have preferential tissue localizations, which generally reflect the metabolic phenotype of the tissue.
In fact, tissues with a predominantly or exclusively aerobic metabolic phenotype, such as the heart, produce mainly isozymes in which the H subunit predominate.[21]
By contrast, tissues where anaerobic metabolism is important, such as muscle fibers during vigorous exercise, or hypoxic regions in solid tumors, produce mainly isozymes in which the M subunit predominate.[5]
However, there are exceptions, such as the liver, an organ with an aerobic metabolic phenotype where LDH-5 predominates, but where the oxidation of lactate to pyruvate, which can also be considered as a part of the hepatic branch of the Cori cycle, is favored by the low NADH/NAD+ ratio in the hepatocyte cytosol.[13]
Below is a brief overview.
LDH-1 or H4: cardiac muscle, kidney and red blood cells.
LDH-2 or M1H3 has a distribution similar to LDH-1.
LDH-3 or M2H2: spleen, brain, white blood cells, kidney and lung.
LDH-4 or M3H1: spleen, lung, skeletal muscle, red blood cells and kidney.
LDH-5 or M4: liver, skeletal muscle and lung.
LDH-6: sperm and testis.
In individual organs, LDH-1 and LDH-2 predominates in the heart, kidney and red blood cells, LDH-3 in the lung, LDH-4 and LDH-5 in skeletal muscle and LDH-5 in the liver.
The isozymes present in serum, whose dosage is used for diagnostic purposes, are always of tissue origin.[5]
Finally, in the same tissue/organ, different isozymes can be present in significant quantities in different cell types, as for example occurs in skeletal muscle, in the brain, but also in solid tumors.
Role
Lactate dehydrogenase is involved both in the synthesis and utilization/removal of lactate.[4]
Under hypoxic conditions, the cell obtains ATP from the anaerobic oxidation of glucose. Through the glycolytic pathway, the monosaccharide is oxidized to two molecules of pyruvate, yielding 2 ATP and two NADH. However, for glycolysis to proceed, the NADH produced must be reoxidized to NAD+, as the oxidized coenzyme is involved in the reaction catalyzed by glyceraldehyde-3-phosphate dehydrogenase (EC 1.2.1.12). As the pyruvate dehydrogenase complex is inhibited and oxidative phosphorylation is blocked, the oxidation of NADH occurs in the reaction catalyzed by lactate dehydrogenase, which then allows glycolysis to proceed even in hypoxic conditions. This metabolic pathway produces lactate from glucose, and is known as lactic acid fermentation.[13][22]
Lactic acid is often considered an end product of glucose metabolism, and its accumulation in the body is harmful as it may potentially causelactic acidosis.[22] Therefore, it must be rapidly removed from tissues and circulation. Considering for example the lactose produced by red blood cells or muscle fibers working in under hypoxic conditions, such as during vigorous exercise, or continuously by the red blood cell, through bloodstream, it can reach, among others, the liver and the heart. In the liver, although LDH-5 is the major isozyme, the low cytosolic NADH/NAD+ ratio shifts the equilibrium of the reaction toward pyruvate synthesis, which can be used for energy or enter into gluconeogenesis.[23] In the cardiomyocyte LDH-1 oxidizes lactate to pyruvate, that will be used for energy.
References
^ abc Berg J.M., Tymoczko J.L., Gregory J.G. Jr, Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
^ abc Markert C.L., Shaklee J.B., Whitt G.S. Evolution of a gene. Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science 1975;189(4197):102-14. doi:10.1126/science.1138367
^ abc Schueren F., Lingner T., George R., Hofhuis J., Dickel C., Gärtner J., Thoms S. Peroxisomal lactate dehydrogenase is generated by translational readthrough in mammals. Elife 2014;3:e03640. doi:10.7554/eLife.03640
^ abc Laughton J.D., Charnay Y., Belloir B., Pellerin L., Magistretti P.J., Bouras C. Differential messenger RNA distribution of lactate dehydrogenase LDH-1 and LDH-5 isoforms in the rat brain. Neuroscience 2000;96(3):619-25. doi:10.1016/s0306-4522(99)00580-1
^ abcd Farhana A., Lappin S.L. Biochemistry, lactate dehydrogenase. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557536/
^ Jin S., Chen X., Yang J., Ding J. Lactate dehydrogenase D is a general dehydrogenase for D-2-hydroxyacids and is associated with D-lactic acidosis. Nat Commun 2023;14(1):6638. doi:10.1038/s41467-023-42456-3
^ ab Krieg A.F., Rosenblum L.J., Henry J.B. Lactate dehydrogenase isozymes a comparison of pyruvate-to-lactate and lactate-to-pyruvate assays. Clin Chem 1967;13(3):196-203. doi:10.1093/clinchem/13.3.196
^ abc Zinkham W.H., Blanco A., Clowry L.J. Jr. An unusual isozyme of lactic dehydrogenase in mature testes: localization, ontogeny, and kinetic properties. Ann N Y Acad Sci 1964;121:571-88. doi:10.1111/j.1749-6632.1964.tb14227.x
^ Bellamacina C.R. The nicotinamide dinucleotide binding motif: a comparison of nucleotide binding proteins. FASEB J 1996;10(11):1257-69. doi:10.1096/fasebj.10.11.8836039
^ ab Read J.A., Winter V.J., Eszes C.M., Sessions R.B., Brady R.L. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins 2001;43(2):175-85.
^ Auerbach G., Ostendorp R., Prade L., Korndörfer I., Dams T., Huber R., Jaenicke R. Lactate dehydrogenase from the hyperthermophilic bacterium thermotoga maritima: the crystal structure at 2.1 A resolution reveals strategies for intrinsic protein stabilization. Structure 1998;6(6):769-81. doi:10.1016/s0969-2126(98)00078-1
^ abc Forkasiewicz A., Dorociak M., Stach K., Szelachowski P., Tabola R., Augoff K. The usefulness of lactate dehydrogenase measurements in current oncological practice. Cell Mol Biol Lett 2020;25:35. doi:10.1186/s11658-020-00228-7
^ Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ Holmes R.S., Goldberg E. Computational analyses of mammalian lactate dehydrogenases: human, mouse, opossum and platypus LDHs. Comput Biol Chem 2009;33(5):379-85. doi:10.1016/j.compbiolchem.2009.07.006
^ Feng Y., Xiong Y., Qiao T., Li X., Jia L., Han Y. Lactate dehydrogenase A: a key player in carcinogenesis and potential target in cancer therapy. Cancer Med 2018;7(12):6124-6136. doi:10.1002/cam4.1820
^ Gray L.R., Tompkins S.C., Taylor E.B. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-604. doi:10.1007/s00018-013-1539-2
^ Rogatzki M.J., Ferguson B.S., Goodwin M.L., Gladden L.B. Lactate is always the end product of glycolysis. Front Neurosci 2015;9:22. doi:10.3389/fnins.2015.00022
^ Stambaugh R., Post D. Substrate and product inhibition of rabbit muscle lactic dehydrogenase heart (H4) and muscle (M4) isozymes. J Biol Chem 1966;241(7):1462-7. doi:10.1016/S0021-9258(18)96733-5
^ Wroblewski F., Gregory K.F. Lactic dehydrogenase isozymes and their distribution in normal tissues and plasma and in disease states. Ann N Y Acad Sci 1961;94:912-32. doi:10.1111/j.1749-6632.1961.tb35584.x
^ ab Li X., Yang Y., Zhang B., Lin X., Fu X., An Y., Zou Y., Wang J.X., Wang Z., Yu T. Lactate metabolism in human health and disease. Signal Transduct Target Ther 2022;7(1):305. doi:10.1038/s41392-022-01151-3
^ Gleeson T.T. Post-exercise lactate metabolism: a comparative review of sites, pathways, and regulation. Annu Rev Physiol 1996;58:565-81. doi:10.1146/annurev.ph.58.030196.003025
Keto acids or ketoacids are organic compounds containing two functional groups: a carboxyl acid group (−COOH) and a carbonyl group (˂C=O).
Based on the position of the carbonyl group relative to the carboxylic acid group, to which IUPAC nomenclature rules assign the highest priority, ketoacids are classified as α-keto acids, β-keto acids, and γ-keto acids.[1]
General Chemical Structure of Keto Acids (α, β e γ)
Keto acids, and in particular α-keto acids, are very important in biochemistry, being involved in many metabolic pathways.[2]
In Summary: Key Points
Definition and structure: keto acids are organic compounds containing both a carboxylic acid group and a carbonyl group within their molecular structure.
IUPAC classification: they are classified into alpha (α), beta (β), and gamma (γ) keto acids based on the position of the carbonyl group relative to the carboxylic acid.
Role of alpha-keto acids: pyruvic acid (the end product of glycolysis), oxaloacetic acid, and α-ketoglutaric acid are vital metabolic intermediates driving the citric acid cycle, gluconeogenesis, and amino acid metabolism.
Significance of beta and gamma: beta-ketoacids (such as acetoacetic acid) are key to ketone body production and gut microbiota pathways, while gamma-keto acids (such as levulinic acid) originate from cellulose catabolism.
α-Keto acids
They have the carbonyl group adjacent to the carboxylic acid. Many of these compounds, in the form of their conjugate bases, have important biological functions. Below are some examples.[2]
Pyruvic acid, the simplest α-keto acid, is the end metabolic product of glycolysis.
Oxaloacetic acid and α-ketoglutaric acid are intermediates of the citric acid cycle.
α-Ketoacids can arise from transamination and oxidative deamination reactions of amino acids. In transamination reactions, the α amino group of the amino acid is transferred to an α-keto acid, usually α-ketoglutarate, with the formation of a new amino acid and a new α-keto acid. These reactions are catalyzed by enzymes called aminotransferases or transaminases (EC 2.6.1.-).
α-Keto acid + Amino acid ⇄ New amino acid + New α-keto acid
In oxidative deaminations, amino acids are converted into the corresponding α-keto acids by removing the amino group, which is converted to ammonia and replaced by a carbonyl group. Since the reaction is reversible, ketoacids are also precursors of amino acids.
Note: ammonia is a toxic compound, and is converted into the safer compound urea via the urea cycle in the liver.[2]
Pyruvate, oxaloacetate and α-ketoglutarate, the latter via oxaloacetate, are the entry points into gluconeogenesis of the carbon skeleton of many glucogenic amino acids.[3]
It has also been observed that, in vitro, murine and human tumor cell lines secrete 2-keto acids into the tumor microenvironment, such as α-ketoisocaproate, α-keto-β-methylvalerate and α-ketoisovalerate, which are capable to influencing the anti-tumor activity of macrophages.[4]
β-Keto acids
They have the carbonyl group at the second carbon from the carboxylic acid.
Examples of β-ketoacids are acetoacetic acid, the simplest one, and β-hydroxybutyric acid, which are two of the three ketone bodies, together with acetone, produced by the hepatocyte when acetyl-CoA is produced in excess of the capacity of citrate synthase (EC 2.3.3.1), namely, of citric acid cycle to oxidize it fully, as during prolonged fasting or diets very low in carbohydrates.
Note that acetoacetyl-CoA and β-hydroxybutyryl-CoA, namely, the activated forms of these β-keto acids, are also intermediates in the butyric acid synthesis pathway which occurs in most butyrate-producing bacteria of the gut microbiota.[5][6][7]
γ-Keto acids
They have the carbonyl group at the third carbon from the carboxylic acid.
An example is levulinic acid, the simplest one, which arises from the catabolism of cellulose.
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ D’Andrea G. Classifying amino acids as gluco(glyco)genic, ketogenic, or both. Biochem Educ 2000;28(1):27-28. doi:10.1016/s0307-4412(98)00271-4
^ Cai Z., Li W., Brenner M., Bahiraii S., Heiss E.H., Weckwerth W. Branched-chain ketoacids derived from cancer cells modulate macrophage polarization and metabolic reprogramming. Front Immunol 2022;13:966158. doi:10.3389/fimmu.2022.966158.
^ Miller T.L., Wolin M.J. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl Environ Microbiol 1996;62(5):1589-92. doi:10.1128/aem.62.5.1589-1592
^ Portincasa P., Bonfrate L.,Vacca M., De Angelis M., Farella I., Lanza E., Khalil M.,Wang D.Q.-H., Sperandio M., Di Ciaula A. Gut microbiota and short chain fatty acids: implications in glucose homeostasis. Int J Mol Sci 2022;23:1105. doi:10.3390/ijms23031105
^ Pryde S.E., Duncan S.H., Hold G.L., Stewart C.S., Flint H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett 2002;217(2):133-9. doi:10.1111/j.1574-6968.2002.tb11467.x
A futile cycle, or substrate cycle, occurs when two non-equilibrium opposing reactions, catalyzed by different enzymes, or two opposite metabolic pathways run simultaneously with no other overall effect than the dissipation of energy.[1]
The term “futile cycle” was coined because apparently these cycles seemed to confer no benefit to the cell, a sort of metabolic imperfection leading to energy expenditure.[2]
However, recently, they have been recognized as important for the generation of heat, the amplification of metabolic signals, the redistribution of the energy, in the form of triglycerides, between adipocytes and hepatocytes, and the modification of fatty acids stored as triglycerides in adipose tissue.
In order to avoid uncontrolled dissipation of energy, futile cycles are strictly regulated.[3][4][5]
Examples of futile cycles are glycolysis and gluconeogenesis when they proceed simultaneously at a high rate in the same cell, the Cori cycle, and the triglyceride/fatty acid cycle.[6]
Summary: Key Points
Definition and nature: a futile (or substrate) cycle occurs when two opposing, irreversible reactions run simultaneously, resulting only in net ATP consumption and energy dissipation.
Control mechanisms: to prevent uncontrolled energy waste, cells tightly and independently regulate the enzymes of opposing pathways using allosteric mechanisms, covalent modifications, or nuclear compartmentalization.
Biological functions: although initially considered metabolic imperfections, they are now recognized as vital for heat generation (thermogenesis), metabolic signal amplification, and fatty acid remodeling.
Primary examples: the most prominent biological models include the coupling of glycolysis and gluconeogenesis (PFK-1/FBPase), the Cori cycle between muscle and liver, and the triglyceride/fatty acid cycle in adipose tissue.
Let us consider the conversion of A into B, which proceeds at a rate of 100, and B into A, which proceeds at a rate of 90. This results in a net flow of 10. Suppose that an effector increases the rate of conversion of A into B by 30 percent, to 130 percent, and reduces the rate of conversion of B into A by 30 percent, to 63 percent. The resulting net flux is equal to 130 − 63 = 67, namely, a 30 percent change in the rates of the opposing reactions has led to a 570 percent increase in the net flux.
A mechanism of this type could, at least in part, explain the even 1000-fold increase in carbon flux down glycolysis in the initial phase of intense exercise.[2]
Regulation
In the course of evolution, the selection of different enzymes to catalyze irreversible and opposing reactions has made it possible to avoid or put under strict control futile cycles. How? The selection of one enzyme that catalyzes the conversion of A into B, and another enzyme that catalyzes the opposing reaction, whose activities are regulated separately, allows the control of the net flux.[7]
Enzymatic activities are controlled by:
allosteric mechanisms;
covalent modifications;
modifications in the concentration of the enzymes, due to variations in the ratio between their synthesis and/or degradation. A different mechanism regulates hepatic glucokinase (EC 2.7.1.2). During fasting, the enzyme is reversibly bound to GKPR, one of the liver-specific proteins, which anchors it inside the nucleus, separating it from the other glycolytic enzymes, and thus preventing the futile cycle between glycolysis and gluconeogenesis.
In this way, it is possible to obtain a coordinated regulation of the two opposing pathways, thus avoiding an uncontrolled futile cycle. Obviously, such a fine regulation could not be achieved if a single enzyme would operate in both directions.[8]
Glycolysis and gluconeogenesis
If glycolysis, which converts glucose into pyruvate, the conjugate base of pyruvic acid, with the production of ATP, and gluconeogenesis, which converts pyruvate into glucose with the consumption of ATP, run simultaneously at a high rate in the same cell, the net result would be a net consumption of ATP, therefore a futile cycle. This is avoided by the control of the irreversible steps of the two metabolic pathways, in particular the reactions catalyzed by phosphofructokinase-1 (PFK-1; EC 2.7.1.11), and by fructose-1,6-bisphosphatase (FBPase; EC 3.1.3.11), mainly by the allosteric effector fructose 2,6-bisphosphate.[7]
Example of a Futile Cycle Between Glycolysis and Gluconeogenesis.
It should be noted that in glycolysis, the control involves all the irreversible reactions, whereas in gluconeogenesis, the key regulatory points are the reactions catalyzed by pyruvate carboxylase (EC 6.4.1.1) and fructose 1,6-bisphosphatase.[8]
Cori Cycle
In the Cori cycle, lactic acid produced from glucose in the muscle and other extrahepatic tissues reaches the liver, where it is converted back into glucose, which, released into the circulation, returns to the muscle and other extrahepatic tissues, thereby closing the cycle. From an energetic point of view, the Cori cycle can be considered a futile cycle because it results in a net consumption of 4 ATP with no other overall effect. However, it allows many different types of extrahepatic cells to work at the expense of the liver.[6]
Triglyceride/fatty acid cycle
In the triglyceride/fatty acid cycle, triglycerides in adipose tissue are partially or completely hydrolyzed to free fatty acids and glycerol, in a process called lipolysis; the released fatty acids are then used to resynthesize new molecules of triglycerides.[3][6][7] Four moles of ATP are consumed for every mole of triglycerides that completes the cycle.
This futile cycle can take place:
between adipose tissue, which releases fatty acids, and the liver, which re-esterifies them to triglycerides, leading to a redistribution of stored energy;[4]
in adipocytes, where it may contribute to thermogenesis and modifications of the stored fatty acids.
Regarding the modifications, the cycle renders fatty acids accessible for re-arrangements such as elongations and desaturations, which allow saturated fatty acids to be converted to unsaturated fatty acids. However, the efficiency of the process seems to depend on the type of fatty acids.[7] For example, the metabolism of the released medium-chain fatty acids is faster than the conversion of palmitic acid, one of the long-chain fatty acids, to palmitoleic acid, oleic acid, and then, in hepatocytes, to arachidonic acid.
Note that the conversion of medium-chain fatty acids and palmitic acid to long-chain unsaturated fatty acids reduces the health risk associated to their accumulation in stored triglycerides.[5]
Generation of heat
In some cases a futile cycle has the only function of producing heat through the hydrolysis of ATP. This occurs, for example, in the flight muscles of bumblebees, which, in order to fly, must maintain a thoracic temperature of about 30 °C, even when the external temperature is 10 °C. The thoracic temperature is maintained at the optimal levels for flight thanks to the futile cycle between the reactions catalyzed by PFK-1 and FBPase. In fact, flight muscle FBPase is not inhibited by AMP, which suggests that, during evolution, this protein has been selected for the generation of heat.
Unlike bumblebees, flight muscles of honeybees contain almost no FBPase, and therefore these insects cannot fly when the external temperature is low.[2]
References
^ Newsholme E.A. Substrate cycles: their metabolic, energetic and thermic consequences in man. Biochem Soc Symp. 1978;43:183–205.
^ abc Berg J.M., Tymoczko J.L., Gatto J.G., Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
^ ab Reshef L., Olswang Y., Cassuto H., Blum B., Croniger C.M., Kalhan S.C., Tilghman S.M., Hanson R.W. Glyceroneogenesis and the triglyceride/fatty acid cycle. J Biol Chem 2003;278(33):30413-6. doi:10.1074/jbc.R300017200.
^ ab Sharma A.K., Wolfrum C. Lipid cycling isn’t all futile. Nat Metab 2023;5(4):540-541. doi:10.1038/s42255-023-00779-x.
^ ab Wunderling K., Zurkovic J., Zink F., Kuerschner L., Thiele C. Triglyceride cycling enables modification of stored fatty acids. Nat Metab 2023;5(4):699-709. doi:10.1038/s42255-023-00769-z.
^ abc Brownstein A.J., Veliova M., Acin-Perez R., Liesa M., Shirihai O.S. ATP-consuming futile cycles as energy dissipating mechanisms to counteract obesity. Rev Endocr Metab Disord 2022;23(1):121-131. doi:10.1007/s11154-021-09690-w.
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
Short-chain fatty acids (SCFAs) are saturated fatty acids with a straight or branched carbon chain made of 2–5 carbon atoms including acetic, propionic, butyric, isobutyric, valeric, isovaleric, and 2-methylbutyric acids.[1]
In humans, they are, along with secondary bile salts, the main metabolites produced by bacteria in the gut microbiota in the cecum and colon, and derive almost entirely from the anaerobic fermentation of non-digestible carbohydrates.[2] The most abundant are acetic, propionic, and butyric acids, which represent 90–95% of the produced SCFAs. The remaining percentage consists of branched SCFAs.[3]
They are the major anions present in the colon.[4] Their concentration is higher in the cecum and in the proximal colon, where the substrates for their synthesis are more abundant.[5][6][7] They reduce the colonic pH and thus acidify the stool.
About 90–95% of the SCFAs are absorbed in the cecum and colon, whereas 5–10% are excreted with the feces, and are thought to provide about 70% of the energy needs of colonocytes.[8][9]
Short-chain fatty acids modulate the physiology and composition of the gut microbiota. Furthermore, a growing body of research suggests that they play an important role in maintaining human health.[4]
Summary: Key Points
Microbiota origin: short-chain fatty acids derive almost entirely from the anaerobic fermentation of dietary fibers and resistant starches by gut bacteria.
The three pillars: acetic, propionic, and butyric acids are the most abundant metabolites, representing 90–95% of total SCFAs in the cecum and colon.
Cellular fuel: butyric acid holds paramount biological importance, providing roughly 70% of the energy needs required by colonocytes.
Regulation and health: they modulate host physiology by inhibiting histone deacetylases and binding to GPR membrane receptors, regulating glucose homeostasis and the gut-brain axis.
Like medium-chain fatty acids and long-chain fatty acids, short-chain fatty acids are present in animal and plant tissues mostly in the form of triglycerides, although in much lower amounts than their long-chain counterparts.[10]
In adults, the main food source is milk and dairy products, where butyric acid is the SCFA present with the highest concentration. Other sources are some vegetable oils, such as palm kernel oil and coconut oil.
In breastfed infants, the main source is breast milk.[11]
However, for humans, and most mammals, the most important source is the anaerobic fermentation of fibers and resistant starch, namely, indigestible carbohydrates, by the bacteria of the gut microbiota.[12] Approximately 500–600 mM of SCFAs are produced through this pathway per day. Acetic, propionic, and butyric acids are present in a molar ratio of about 60:20:20, respectively, although the relative proportion of each depends on the microbiota composition, the substrate, and the intestinal transit time.[2][4]
Properties
Short-chain fatty acids have carbon chains made of 2–5 carbon atoms, a characteristic that strongly affects their physical properties.
Acetic, propionic, butyric, and valeric acids are straight-chain fatty acids, whereas isobutyric, isovaleric, and 2-methylbutyric acid are branched-chain fatty acids.[13]
Physicochemical Properties of the Seven Short-Chain Fatty Acids
They are small molecules, and are the smallest among all lipids.
They are liquid at room temperature, and are soluble in polar solvents such as water. Unlike straight-chain saturated fatty acids with longer carbon chains, whose solubility decreases as the chain lengthens, SCFAs remain soluble because their hydrophobic carbon chain is short, while the carboxyl group remains highly polar.[11]
Finally, butyric acid and isobutyric acid, which have the molecular formula C4H8O2, are an example of chain isomerism; the same applies to are valeric, isovaleric, and 2-methylbutyric acids, which share the molecular formula C5H10O2.
Health effects
Short-chain fatty acids appear to play a crucial role in maintaining human health.[7] Their activity seems to occur through direct and/or indirect effects on cellular processes such as proliferation, differentiation, and gene expression, thus contributing to the regulation of processes such as glucose homeostasis, intestinal and immune function, and the regulation of the gut-brain axis.[14]
Their health effects seem to be confirmed by studies showing that intestinal dysbiosis appears to be implicated in metabolic pathologies, such as disorders involving glucose homeostasis, and behavioral and neurological pathologies, such as depression, and Alzheimer’s and Parkinson’s.[15]
Synthesis
In humans, the enzyme equipment carrying out carbohydrate digestion lacks the enzymes capable of digesting fiber and resistant starch, the latter being so-called precisely because it resists the action of alpha-amylase.
On the contrary, the bacteria of the gut microbiota encode a large number of different glycoside hydrolases, more than 260, which also hydrolyze fibers and resistant starch, releasing the constituent monosaccharides.[16]
Hexoses and deoxyhexoses enter glycolysis, and pentoses enter the pentose phosphate pathway, to give pyruvate, which is the main precursor for the synthesis of short-chain fatty acids.[17][18][19]
The synthesis of SCFAs is affected by several factors; below are some examples.
The fiber content of the diet. For example, a diet rich in fibers, such as the Mediterranean diet, may influence their synthesis.[2]
The pH of the intestinal lumen, as the bacteria that produce butyric acid dominate at a pH value around 5.5, while the bacteria that produce acetic and propionic acids dominate at a pH value around 6.5.[21]
Acetic acid and propionic acid are mainly produced by species of the phylum Bacteroidetes, while butyric acid, for whose synthesis resistant starch is particularly important, by species of the phylum Firmicutes.[23][24]
Synthesis of acetic acid
Acetic acid, the most abundant SCFA in the colon, can be synthesized via the Wood-Ljungdahl pathway in the reductive direction, through the reduction of CO2 to acetate, or from acetyl-CoA, which is the most important metabolic pathway, responsible for the production of about two thirds of the butyric acid present in the intestinal lumen.[25]
Synthesis of propionic acid
Propionic acid can be synthesized through three different metabolic pathways: the acrylate and succinate pathways, which use lactic acid produced by other bacteria, and the propanediol pathway, in which the precursors are deoxyhexoses.[6]
The acrylate pathway converts lactic acid to propionyl-CoA, via lactoyl-CoA. In the final step, propionyl-CoA is hydrolyzed to propionic acid.[26]
In the succinate pathway, lactate is reduced to pyruvate, which is carboxylated to oxaloacetate, which, through a pathway that has malate, fumarate, succinate, and methylmalonyl-CoA as intermediates, is converted to propionyl-CoA, which in turn is hydrolyzed to propionic acid. The succinate pathway is thought to be the dominant pathway for propionic acid synthesis in the gut.[27]
In the propanediol pathway, some deoxyhexoses, such as fucose and rhamnose, are converted via 1,2-propanediol to propionyl-CoA and then propionic acid.[28]
Synthesis of butyric acid
The synthesis of butyric acid can follow two routes.[4][11]
In most butyric acid-producing bacteria, the short-chain fatty acid is synthesized through a pathway that begins with the condensation of two acetyl-CoA molecules to acetoacetyl-CoA, which, through a pathway that has β-hydroxybutyryl-CoA and crotonyl-CoA as intermediates, is converted to butyryl-CoA. The final step is the release of butyric acid from butyryl-CoA.[29]
In a small number of bacterial species, butyryl-CoA is converted to butyryl phosphate, from which butyric acid is released.[6]
Synthesis from amino acids
Acetic acid, propionic acid, and butyric acid can also be produced from amino acids obtained from peptide and protein degradation, although the amount produced by these pathways is small.[24]
These syntheses occur in the distal part of the colon, often by non-commensal bacteria, as in the case of glutamate and lysine fermentation.[27] Different short-chain fatty acids are produced by the metabolism of different amino acids; below are some examples.[30]
Glutamic acid mainly produces acetic acid and butyric acid.
Aspartic acid mainly produces acetic acid and propionic acid.
The basic amino acids lysine, arginine, and histidine produce acetic acid and butyric acid.
Cysteine produces acetic, propionic, and butyric acids.
Methionine mainly produces propionic acid and butyric acid.
Branched-chain fatty acids derive from the branched-chain amino acids leucine, isoleucine, and valine.
The pH of the intestinal lumen influences the metabolism of proteins by the gut microbiota; for example, their breakdown into amino acids is more likely at neutral or weakly alkaline pH.[31]
It should be noted that potentially toxic compounds, such as ammonia, sulfites and phenols, are also produced from the intestinal metabolism of amino acids.[30]
Endogenous synthesis
Mammals, and therefore humans, have the enzymatic equipment for the endogenous synthesis of short-chain fatty acids. The synthesis occurs mainly in the liver, by β-oxidation cycles which lead to the formation of acyl-CoA with a shorter carbon chain than the starting fatty acid. The acyl-CoA is then hydrolyzed to fatty acid and CoA by an acyl-CoA thioesterase (EC 3.1.2.20).[11][32]
Membrane receptors
Short-chain fatty acids can bind to specific receptors on the plasma membrane, namely G protein-coupled receptors, including GPR41, GPR43, and GPR109A.[33]
The effects produced by the binding of SCFAs on the receptors depend on the cell type. For example, binding to receptors on intestinal L cells is associated with the release of glucagon-like peptide-1 (GLP-1), and peptide YY, hormones that affect appetite and food intake.[4] Binding to enterochromaffin cells induces the release of serotonin, which may affect intestinal motility.[34] Finally, binding to receptors on pancreatic beta-cells increases insulin release.[35]
The different short-chain fatty acids have different abilities to activate the receptors: GPR43 is more likely to be activated by acetic and propionic acids, GPR41 by propionic and butyric acids, while GPR109A is activated by butyric acid.[36]
Absorption
About 90% of the short-chain fatty acids present in the intestinal lumen are absorbed by colonocytes.[37] The passage across the plasma membrane can occur by passive diffusion or by active transport mediated by two types of membrane transporters: the H+-dependent and Na+-dependent monocarboxylate transporters, or MCTs and SMCTs, respectively.[38]
Passive transport affects protonated forms of SCFAs, so it is influenced by the colonic pH value. A weak acidification of the intestinal lumen, which can be due to the metabolic activity of the microorganisms, increases the prevalence of the protonated form and therefore enhances passive transport.[11]
Role in colonocytes
In colonocytes, short-chain fatty acids have both energy and regulatory functions.[33]
When used for energy purposes, acetic acid and butyric acid are converted to acetyl-CoA, and propionic acid to propionyl-CoA. Through the production of ATP, SCFAs contribute to the maintenance of cellular homeostasis, but also, for example, to the maintenance of the integrity of the tight junctions at the cell apex, and therefore of the integrity of the intestinal barrier.[39] Of the three major short-chain fatty acids produced by the gut microbiota, butyric acid is the major source of energy for colonocytes, while acetic and propionic acids are poorly metabolized and mostly drained by the portal vein.[27][40]
Considering the regulatory role, SCFAs are, for example, capable of inhibiting histone deacetylases (EC 3.5.1.98), enzymes that catalyze the removal of acetyl groups from lysine residues of histone proteins, acetyl groups previously inserted by histone acetyltransferase (EC 2.3.1.48).[41] The R groups of deacetylated lysines have positive charges, which allows histone proteins to wrap the DNA more tightly, making the nucleosome more compact and thereby hindering transcription and gene expression. The different short-chain fatty acids have different abilities to inhibit histone deacetylases:
This mode of action on histone deacetylases has been observed not only in the gut and associated immune tissue, but also in the central and peripheral nervous systems.[42]
Transport
Short-chain fatty acids that have not been utilized by colonocytes leave the cell by passive diffusion and active transport across the basolateral membrane, and enter the portal circulation where acetic acid reaches the highest concentration, about 260 mM/L, while propionic and butyric acids reach concentrations of about 30 mM/L.[4]
In the rectum, a small amount of these lipids can pass directly into the systemic circulation, thus bypassing the liver, via the internal iliac vein.[40]
Unlike long-chain fatty acids, short- and medium-chain fatty acids are present in the circulation in free form, namely, as non-esterified fatty acids, and, bound to albumin, reach the liver. Subsequent cell uptake and intracellular transport do not require fatty acid transport proteins, plasma membrane fatty acid translocases, or cytosolic fatty acid binding proteins. Therefore, their oxidation may be much faster than that of long-chain fatty acids and the longest members of medium-chain fatty acids, namely, fatty acid with carbon chains longer than 8 carbon atoms.[11]
Hepatic and extrahepatic metabolism
The liver is an important site for short-chain fatty acid metabolism.[37]
It can absorb about 40% of the acetic acid and 80% of the propionic acid from the portal vein.[43] Propionic acid is mostly metabolized in the liver, where it can also be used as a substrate for gluconeogenesis.[44]
A small amount of the gut-derived SCFAs, about 36% for acetic acid, 9% for propionic acid, and only 2% for butyric acid, reach, through the systemic circulation, the peripheral tissues. In muscle, acetic acid can be used for lipid synthesis or be oxidized for energy production. Furthermore, it is thought that SCFA concentrations in the systemic circulation, even if small, are capable of influencing the metabolism and physiology of peripheral cells and tissues.[4][12]
References
^ Cummings J.H. Short chain fatty acids in the human colon. Gut 1981;22(9):763-79. doi:10.1136/gut.22.9.763
^ abc Silva Y.P., Bernardi A., Frozza R.L. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front Endocrinol (Lausanne) 2020;11:25. doi:10.3389/fendo.2020.00025
^ abcdefg Portincasa P., Bonfrate L., Vacca M., De Angelis M., Farella I., Lanza E., Khalil M., Wang D.Q.-H., Sperandio M., Di Ciaula A. Gut microbiota and short chain fatty acids: implications in glucose homeostasis. Int J Mol Sci 2022;23:1105. doi:10.3390/ijms23031105
^ Abdul Rahim M.B.H., Chilloux J., Martinez-Gili L., Neves A.L., Myridakis A., Gooderham N., Dumas M.-E. Diet-induced metabolic changes of the human gut microbiome: importance of short-chain fatty acids, methylamines and indoles. Acta Diabetol 2019;56:493-500. doi:10.1007/s00592-019-01312-x
^ abcd Deleu S., Machiels K., Raes J., Verbeke K., Vermeire S. Short chain fatty acids and its producing organisms: an overlooked therapy for IBD? EBioMedicine 2021;66:103293. doi:10.1016/j.ebiom.2021.103293
^ abc Liu X.F., Shao J.H., Liao Y.T., Wang L.N., Jia Y., Dong P.J., Liu Z.Z., He D.D., Li C., Zhang X. Regulation of short-chain fatty acids in the immune system. Front Immunol 2023;14:1186892. doi:10.3389/fimmu.2023.1186892
^ McLoughlin R.F., Berthon B.S., Jensen M.E., Baines K.J., Wood L.G. Short-chain fatty acids, prebiotics, synbiotics, and systemic inflammation: a systematic review and meta-analysis. Am J Clin Nutr 2017;106(3):930-945. doi:10.3945/ajcn.117.156265
^ Brahe L.K., Astrup A., Larsen L.H. Is butyrate the link between diet, intestinal microbiota and obesity-related metabolic diseases? Obes Rev 2013;14(12):950-9. doi:10.1111/obr.12068
^ Marten B., Pfeuffer M., Schrezenmeir J. Medium-chain triglycerides. Int Dairy J 2006;16:11, Pages 1374-1382. doi:10.1016/j.idairyj.2006.06.015
^ abcdef Schönfeld P., Wojtczak L. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res 2016;57(6):943-54. doi:10.1194/jlr.R067629
^ ab Cummings J.H., Pomare E.W., Branch W.J., Naylor C.P., Macfarlane G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987;28(10):1221-7. doi:10.1136/gut.28.10.1221
^ Dalile B., Van Oudenhove L., Vervliet B., Verbeke K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat Rev Gastroenterol Hepatol 2019;16(8):461-478. doi:10.1038/s41575-019-0157-3
^ Borre Y.E., O’Keeffe G.W., Clarke G., Stanton C., Dinan T.G., Cryan J.F. Microbiota and neurodevelopmental windows: implications for brain disorders. Trends Mol Med 2014;20(9):509-18. doi:10.1016/j.molmed.2014.05.002
^ Usuda H., Okamoto T., Wada K. Leaky gut: effect of dietary fiber and fats on microbiome and intestinal barrier. Int J Mol Sci 2021;22(14):7613. doi:10.3390/ijms22147613
^ Hugenholtz F., Mullaney J. A., Kleerebezem M., Smidt H., Rosendale D. I. Modulation of the microbial fermentation in the gut by fermentable carbohydrates. Bioact Carbohydr Diet Fibre 2013;2(2)-133-142. doi:10.1016/j.bcdf.2013.09.008
^ Krautkramer K.A., Fan J., Bäckhed F. Gut microbial metabolites as multi-kingdom intermediates. Nat Rev Microbiol 2021;19(2):77-94. doi:10.1038/s41579-020-0438-4
^ Miller T.L., Wolin M.J. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl Environ Microbiol 1996;62(5):1589-92. doi:10.1128/aem.62.5.1589-1592
^ Bergman E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev 1990;70(2):567-90. doi: 10.1152/physrev.1990.70.2.567
^ Topping D.L., Clifton P.M. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev 2001;81(3):1031-64. doi:10.1152/physrev.2001.81.3.1031
^ Louis P., Flint H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett 2009;294(1):1-8. doi: 10.1111/j.1574-6968.2009.01514.x
^ ab Louis P., Flint H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol 2017;19(1):29-41. doi:10.1111/1462-2920.13589
^ Ragsdale S.W., Pierce E. Acetogenesis and the Wood-Ljungdahl pathway of CO2, fixation. Biochim Biophys Acta 2008;1784(12):1873-98. doi:10.1016/j.bbapap.2008.08.012
^ Hetzel M., Brock M., Selmer T., Pierik A.J., Golding B.T., Buckel W. Acryloyl-CoA reductase from Clostridium propionicum. An enzyme complex of propionyl-CoA dehydrogenase and electron-transferring flavoprotein. Eur J Biochem 2003;270(5):902-10. doi:10.1046/j.1432-1033.2003.03450.x
^ abc Koh A., De Vadder F., Kovatcheva-Datchary P., Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 2016;165(6):1332-1345. doi:10.1016/j.cell.2016.05.041
^ Scott K.P., Martin J.C., Campbell G., Mayer C.D., Flint H.J. Whole-genome transcription profiling reveals genes up-regulated by growth on fucose in the human gut bacterium Roseburia inulinivorans. J Bacteriol 2006;188(12):4340-9. doi:10.1128/JB.00137-06
^ Pryde S.E., Duncan S.H., Hold G.L., Stewart C.S., Flint H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett 2002;217(2):133-9. doi:10.1111/j.1574-6968.2002.tb11467.x
^ ab Smith E.A., Macfarlane G.T. Dissimilatory amino acid metabolism in human colonic bacteria. Anaerobe 1997;3(5):327-37. doi:
^ Windey K., De Preter V., Verbeke K. Relevance of protein fermentation to gut health. Mol Nutr Food Res 2012;56(1):184-96. doi:10.1002/mnfr.201100542
^ ab Parada Venegas D., De la Fuente M.K., Landskron G., González M.J., Quera R., Dijkstra G., Harmsen H.J.M., Faber K.N., Hermoso M.A. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol 2019;10:1486. doi:10.3389/fimmu.2019.00277
^ Fukumoto S., Tatewaki M., Yamada T., Fujimiya M., Mantyh C., Voss M., Eubanks S., Harris M., Pappas T.N., Takahashi T. Short-chain fatty acids stimulate colonic transit via intraluminal 5-HT release in rats. Am J Physiol Regul Integr Comp Physiol 2003;284(5):R1269-76. doi:10.1152/ajpregu.00442.2002
^ Puddu A., Sanguineti R., Montecucco F., Viviani G.L. Evidence for the gut microbiota short-chain fatty acids as key pathophysiological molecules improving diabetes. Mediators Inflamm 2014;2014:162021. doi:10.1155/2014/162021
^ Brown A.J., Goldsworthy S.M., Barnes A.A., Eilert M.M., Tcheang L., Daniels D., Muir A.I., Wigglesworth M.J., Kinghorn I., Fraser N.J., Pike N.B., Strum J.C., Steplewski K.M., Murdock P.R., Holder J.C., Marshall F.H., Szekeres P.G., Wilson S., Ignar D.M., Foord S.M., Wise A., Dowell S.J. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 2003;278(13):11312-9. doi:10.1074/jbc.M211609200
^ ab Xiong R.G., Zhou D.D., Wu S.X., Huang S.Y., Saimaiti A., Yang Z.J., Shang A., Zhao C.N., Gan R.Y., Li H.B. Health benefits and side effects of short-chain fatty acids. Foods 2022;11(18):2863. doi:10.3390/foods11182863
^ Kim C.H., Park J., Kim M. Gut microbiota-derived short-chain Fatty acids, T cells, and inflammation. Immune Netw 2014;14(6):277-88. doi:10.4110/in.2014.14.6.277
^ Chambers E.S., Preston T., Frost G., Morrison D.J. Role of gut microbiota-generated short-chain fatty acids in metabolic and cardiovascular health. Curr Nutr Rep 2018;7(4):198-206. doi:10.1007/s13668-018-0248-8
^ ab van der Beek C.M., Dejong C.H.C., Troost F.J., Masclee A.A.M., Lenaerts K. Role of short-chain fatty acids in colonic inflammation, carcinogenesis, and mucosal protection and healing. Nutr Rev 2017;75(4):286-305. doi:10.1093/nutrit/nuw067
^ Marchix J., Goddard G., Helmrath M.A. Host-gut microbiota crosstalk in intestinal adaptation. Cell Mol Gastroenterol Hepatol 2018;6(2):149-162. doi:10.1016/j.jcmgh.2018.01.024
^ Stilling R.M., van de Wouw M., Clarke G., Stanton C., Dinan T.G., Cryan J.F. The neuropharmacology of butyrate: the bread and butter of the microbiota-gut-brain axis? Neurochem Int 2016;99:110-132. doi:10.1016/j.neuint.2016.06.011
^ Chambers E.S. Gut-derived short-chain fatty acids: a friend or foe for hepatic lipid metabolism? Nutr Bull 2019;44:154-159. doi:10.1111/nbu.12377
^ Leung C., Rivera L., Furness J.B., Angus P.W. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol 2016;13(7):412-25. doi:10.1038/nrgastro.2016.85
Starch phosphorylase, also known as alpha-glucan phosphorylase (EC 2.4.1.1), is a multimeric protein both with enzymatic and regulatory activity.
It plays an important role in carbohydrate metabolism in both prokaryotes and eukaryotes.[1]
The enzyme catalyzes the transfer of a glucosyl unit from glucose 1-phosphate to the non-reducing end of a nascent α-(1→4)-glucan, forming an α-(1→4) glycosidic linkage.[2] This reaction is reversible, and its direction depends on the phosphate/glucose 1-phosphate ratio present in vivo.[3] The enzyme belongs to the family of glucosyltransferases (EC 2.4), along with other enzymes such as starch synthase (EC 2.4.1.21), involved in the synthesis of amylose and amylopectin, the polysaccharides that compose starch granules, glycogen phosphorylase (EC 2.4.1.1), involved in glycogenolysis, and glycogen synthase (EC 2.4.1.11), which participates in glycogen synthesis.[1]
It is important to note that while starch synthase uses ADP-glucose as the glucosyl donor and glycogen synthase uses UDP-glucose, starch phosphorylase utilizes glucose 1-phosphate.[4]
Additionally, starch phosphorylase appears to be involved in both the synthesis and degradation of amylose and amylopectin.[2]
In industrial applications, the phosphorolytic action of starch phosphorylase is employed in the production of glucose 1-phosphate and carbohydrates such as glucans and modified starches.[5]
At least two isoforms of starch phosphorylase are present in plants: one located in the stroma of plastids, named Pho1, and another with cytosolic localization, known as Pho2. Both isoforms play a critical role in the synthesis and degradation of starch.[6]
Starch degradation
Although alpha-amylase (EC 3.2.1.1) is the first enzyme to act in the polysaccharide degradation during the early stages of germination, and beta-amylase (EC 3.2.1.2) initiates transient starch degradation in chloroplasts, other enzymatic activities are also involved. These include alpha-glucan water dikinase (EC 2.7.9.4), phospho-glucan water dikinase (EC 2.7.9.5), and debranching enzymes.[7]
Of the two isoenzymes of starch phosphorylase, Pho1 appears to have an indirect or regulatory role, influencing the activity of the other enzymes involved in starch degradation.[6] In contrast, Pho2 is capable of directly degrading starch granules and other branched glucans.[8]
Starch synthesis
During starch biosynthesis, starch phosphorylase, particularly Pho1, appears to play both enzymatic and regulatory roles.
Pho1 seems to be involved in the early stages of starch synthesis, contributing to the elongation of the nascent glucan chain.[4]
Starch synthesis involves at least five classes of enzymes: ADP-glucose pyrophosphorylase (EC 2.7.7.27), starch synthases, starch branching enzymes (EC 2.4.1.18), starch debranching enzymes (EC 3.2.1.41), and starch phosphorylases.[9] In addition to these, there are also non-catalytic proteins essential for the correct assembly of the starch granule.[10]
In the endosperm of cereals, the formation of multienzyme complexes between starch synthases and branching enzymes depends not only on specific phosphorylation enents but also on the presence of Pho1.[11]
Starch phosphorylase is capable of forming a complex with disproportionating enzyme (EC 2.4.1.25).[11] This complex appears to synthesize short malto-oligosaccharides (MOS), which are α-(1→4)-glucans with a degree of polymerization between 2 and 7.[12][13]
MOS serve as primers for starch synthase IV and granule-bound starch synthase in the initial steps of amylopectin and amylose synthesis, respectively, role analogous to that of glycogenin in glycogen biosynthesis.[14]
MOS can also originate from the activity of starch debranching enzymes during the trimming of amylopectin molecules.[13]
References
^ ab Rathore R.S., Garg N., Garg S., Kumar A. Starch phosphorylase: role in starch metabolism and biotechnological applications. Crit Rev Biotechnol 2009;29(3):214-24. doi:10.1080/07388550902926063
^ ab Tickle P., Burrell M.M., Coates S.A., Emes M.J., Tetlow I.J., Bowsher C.G. Characterization of plastidial starch phosphorylase in Triticum aestivum L. endosperm. J Plant Physiol 2009;166(14):1465-78. doi:10.1016/j.jplph.2009.05.004
^ Newgard C.B., Hwang P.K., Fletterick R.J. The family of glycogen phosphorylases: structure and function. Crit Rev Biochem Mol Biol 1989;24(1):69-99. doi:10.3109/10409238909082552
^ ab Cuesta-Seijo J.A., Ruzanski C., Krucewicz K., Meier S., Hägglund P., Svensson B., Palcic M.M. Functional and structural characterization of plastidic starch phosphorylase during barley endosperm development. PLoS One 2017;12(4):e0175488. doi:10.1371/journal.pone
^ Kadokawa J.I. α-Glucan phosphorylase-catalyzed enzymatic reactions using analog substrates to synthesize non-natural oligo- and polysaccharides. Catalysts 2018;8(10):473. doi:10.3390/catal8100473
^ ab Yu G., Shoaib N., Xie Y., Liu L., Mughal N., Li Y., Huang H., Zhang N., Zhang J., Liu Y., Hu Y., Liu H., Huang Y. Comparative study of starch phosphorylase genes and encoded proteins in various Monocots and Dicots with emphasis on maize. Int J Mol Sci 2022;23:4518. doi:10.3390/ijms23094518
^ Steup M., Robenek H., Melkonian M. In-vitro degradation of starch granules isolated from spinach chloroplasts. Planta 1983;158(5):428-36. doi:10.1007/BF00397736
^ Crofts N., Nakamura Y., Fujita N. Critical and speculative review of the roles of multi-protein complexes in starch biosynthesis in cereals. Plant Sci 2017;262:1-8. doi:10.1016/j.plantsci.2017.05.007
^ Seung D., Soyk S., Coiro M., Maier B.A., Eicke S., Zeeman S.C. PROTEIN TARGETING TO STARCH is required for localising GRANULE-BOUND STARCH SYNTHASE to starch granules and for normal amylose synthesis in Arabidopsis. PLOS Biol 2015;13(2):e1002080. doi:10.1371/journal.pbio.1002080
^ ab Crofts N., Abe N., Oitome N.F., Matsushima R., Hayashi M., Tetlow I.J., Emes M.J., Nakamura Y., Fujita N. Amylopectin biosynthetic enzymes from developing rice seed form enzymatically active protein complexes. J Exp Bot 2015;66(15):4469-82. doi:10.1093/jxb/erv212
^ Hwang S.K., Koper K., Satoh H., Okita T.W. Rice endosperm starch phosphorylase (Pho1) assembles with disproportionating enzyme (Dpe1) to form a protein complex that enhances synthesis of malto-oligosaccharides. J Biol Chem 2016;291(38):19994-20007. doi:10.1074/jbc.M116.735449
^ ab Tetlow I.J., Bertoft E. A review of starch biosynthesis in relation to the building block-backbone model. Int J Mol Sci 2020;21(19):7011. doi:10.3390/ijms21197011
^ Pfister B., Zeeman S.C., Rugen M.D., Field R.A., Ebenhöh O., Raguin A. Theoretical and experimental approaches to understand the biosynthesis of starch granules in a physiological context. Photosynth Res 2020;145:55-70. doi:10.1007/s11120-019-00704-y
Amylopectin is a highly branched polysaccharide composed of α-D-glucose units. Together with amylose, it is one of the two main constituents of starch granules, the primary form of energy storage in plants and the most widespread and abundant form of carbohydrate reserve on Earth.[1]
Glucose monomers are connected by α-(1→4) glycosidic bonds to form linear chains, while branches are introduced through α-(1→6) glycosidic bonds.[2]
Its biosynthesis requires the coordinated activity of at least four distinct classes of enzymes: starch synthases (EC 2.4.1.21), starch branching enzymes (EC 2.4.1.18), starch debranching enzymes, and starch phosphorylase (EC 2.4.1.1).[3]
Within starch granules, amylopectin is more abundant than amylose and forms a semi-crystalline matrix in which amylose appears to be embedded.
The amylose/amylopectin ratio significantly affects the physicochemical properties of starch, thereby influencing both its industrial applications, such as the production of food additives, and its potential health effects.[4][5]
Amylopectin is a highly branched polysaccharide with a molecular weight on the order of 107-108 Daltons, making it much larger than amylose. It is composed of 104-105 glucose residues linked by α-1,4 glycosidic bonds to form numerous relatively short chains, with a degree of polymerization ranging from 18 to 25 units.[6] The chain length varies depending on the source of starch as well as the environmental and nutritional conditions during plant growth and seed development.[2]
The chains are interconnected by α-1,6 glycosidic bonds, giving rise to a tree-like architecture in which neighboring chains assemble into cluster-like structure.[7]
Amylopectin Structure
In most starches, α-1,6 linkages account for about 5% of the total glycosidic bonds, a lower proportion than in glycogen (≈9%), where the branches are more evenly distributed. The length and distribution of branches strongly influence the physicochemical properties of amylopectin, including solubility, viscosity, retrogradation behavior, and the gelatinization and pasting temperatures. For example, glycogen is water-soluble, whereas amylopectin and starch are not.[5]
Amylopectin chains
Amylopectin chains can be classified either by their length or by the presence or absence of branching.
Length-based classification
Two main types of chains are distinguished: short and long. Short chains have a degree of polymerization (DP) ranging from 6 to 36 glucosyl units, although the upper limit depends on the amylopectin source. Long chains are defined as those with a DP ≥ 36. In most starches, the molar ratio of long to short chains is approximately 19:6, and is generally higher in A-type crystalline starches (e.g., cereal endosperm starches) than in B-type crystalline starches (e.g., potato starch).[6]
Branching-based classification
According to their connections to other chains, amylopectin chains are divided into three categories: A-chains, B-chains, and C-chains.[8]
A-chains are unbranched, short chains with an average DP of about 13; they represent the outer chains of the molecule.
B-chains contain at least one branch (either an A- or another B-chain), are longer than A-chains, and are located in the inner regions of the molecule. They are further subclassified into B1-chains (DP ≈ 22), B2-chains (DP ≈ 42), B3-chains (DP ≈ 69), B4-chains, and so forth.
The C-chain is a special type of B-chain that carries the sole reducing end of the amylopectin molecule.
A-chains and B1-chains participate in the formation of clusters, while B2-, B3-, and B4-chains are thought to span two, three, and four clusters, respectively.[8][9]
A-type, B-type and C-type polymorphs
Within the clusters, neighboring linear chain segments form double helices that run parallel to each other, with a periodicity of 2.1 nm, where each turn comprises six glucose residues per chain.[10] These double helices assemble into two distinct crystalline structures: the A-type polymorph, which is denser and typical of cereal grains, and the B-type polymorph, which has a more open hexagonal structure, is less dense and more hydrated, and is characteristic of tuber and high-amylose starches. The C-type polymorph represents a mixture of A- and B-type structures and is found in the starches of roots, legumes, and certain fruits.
This organization underlies the semi-crystalline nature of starch granules.[2]
Growth rings
Although starch granules vary in shape, their internal architecture is remarkably conserved across species. When observed under a microscope, most starches display a regular pattern of alternating light and dark rings, known as growth rings because of their resemblance to tree rings.[10]
The growth rings surround the hilum, the central region of the granule, whose precise structure remains unclear, although it appears to consist of a relatively disordered α-glucan arrangement. Each ring has a thickness of 200–400 nm and reflects the alternation of less dense amorphous regions and denser semi-crystalline regions.[10]
According to the cluster model, the semi-crystalline regions arise from the alternation of crystalline and amorphous lamellae, stacked with a periodicity of about 9–10 nm.[9] The crystalline lamellae, composed of amylopectin linear chains arranged in A-, B-, or C-type polymorphs, extend about 6–7 nm, whereas the amorphous lamellae, which contain most of the branch points, extend about 3 nm.[11][12]
Amylopectin synthesis
Amylopectin synthesis is thought to initiate at the hilum of the starch granule.[13]
It requires the coordinated activities of at least four distinct classes of enzymes: starch synthases, starch phosphorylase, starch branching enzymes, and starch debranching enzymes.[1] Each class comprises several isoforms with distinct biochemical properties.
Like amylose biosynthesis, amylopectin synthesis depends on short malto-oligosaccharides (MOS), α-(1→4)-glucans with a DP of 2 to 7, which act as primers and are elongated by starch synthase, in a manner similar to the role of glycogenin during the early stages of glycogen synthesis.[14]
MOS appear to originate from multiple sources, all of which require the action of enzymes involved in starch biosynthesis, including:
starch synthase III and starch phosphorylase (the latter in cooperation with the disproportionating enzyme, EC 2.4.1.25), which use ADP-glucose and glucose-1-phosphate as substrates, respectively;
starch debranching enzymes, which generate MOS during the trimming of other amylopectin molecules.[14]
Because MOS exhibit low solubility in aqueous environments, they seem to evade hydrolysis by alpha-amylase (EC 3.2.1.1) and beta-amylase (EC 3.2.1.2).[6]
The spatial and temporal coordination of the enzymes involved, many of which physically interact to form multienzyme complexes, is essential for converting the products of photosynthesis into the highly organized and insoluble polysaccharide structure.[5] Moreover, as in amylose and glycogen synthesis, the polymerization of glucose into amylopectin, and more generally of osmotically active monosaccharides into osmotically inert polysaccharides, enables the storage of large amounts of carbohydrates within the cell without causing a substantial increase in osmotic pressure.[15]
Starch synthase
Six isoforms of starch synthase have been identified, all structurally related. Five of these, starch synthase I, II, III, IV, and V (SSI, SSII, SSIII, SSIV, and SSV; EC 2.4.1.21), are involved in amylopectin biosynthesis. They are localized either in the plastid stroma or partitioned between the stroma and starch granules. The sixth isoform, granule-bound starch synthase (GBSS; EC 2.4.1.242), is almost exclusively associated with granules and is responsible for amylose synthesis.[10][16]
The first four isoforms are catalytically active and, like GBSS, glycogen phosphorylase (EC 2.4.1.1), and glycogen synthase (EC 2.4.1.11), enzymes involved in glycogenolysis and glycogen synthesis, belong to the glycosyltransferase family (EC 2.4). In contrast, starch synthase V lacks catalytic activity.
During amylopectin synthesis, starch synthase catalyzes the addition of one glucose residue to the non-reducing end of a pre-existing α-(1→4)-linked glucan chain, forming a new α-(1→4) glycosidic bond.[12]
Unlike glycogen synthase, which uses UDP-glucose, starch synthase employs ADP-glucose as the glucosyl donor.[5]
The mode of action of starch synthases I–IV differs from that of GBSS. While SS isoforms catalyze the incorporation of only one glucose unit per substrate encounter, a distributive mode of action, GBSS is capable of adding multiple glucose units per encounter, a processive mode of action.[6]
Roles of starch synthases
The initial steps of amylopectin biosynthesis, as well as the formation of normal starch granules, require the presence of SSIV, although SSIII also appears to contribute, with partially overlapping functions.[14]
Like GBSS, SSIV depends on a member of the PTST protein family, PTST2, which lacks catalytic activity but facilitates enzyme binding to α-glucans through a specific carbohydrate-binding domain. SSIV can also dimerize, a feature important both for its catalytic activity and for protein–protein interactions.
According to a proposed model of PTST2 function, the protein recognizes malto-oligosaccharides with a specific three-dimensional helical conformation via its carbohydrate-binding domain. PTST2 then forms a complex with MOS, which subsequently interacts with an SSIV dimer. The dimer catalyzes the elongation of the α-glucan, while PTST2 is released, enabling it to bind another MOS and mediate further interactions with SSIV dimers.[17]
Following the action of SSIV, the other starch synthase isoforms extend the glucan chains. SSI elongates malto-oligosaccharides with a DP of 6–7 to form oligosaccharides with a DP of 8–12. These, in turn, are excellent substrates for SSII, which extends them to chains with a DP of 12–30. SSIII further elongates the glucans, producing linear chains with DP values greater than 30. Thus, SSIII appears to function not only in the early stages of granule initiation but also during later phases of starch biosynthesis.[1]
Finally, SSIV and SSV seem to be required for the production of a regular number of starch granules with normal morphology.[7][14]
Starch branching enzymes
Starch branching enzymes (SBEs) catalyze the formation of α-(1→6) glycosidic bonds, thereby introducing branch points into linear α-glucan chains, such as those in glycogen and amylopectin.[18][19] Their action increase the number of non-reducing ends, which serve as acceptors of glucose units during elongation reactions.[20]
SBEs function by cleaving an internal α-(1→4) bond within an α-glucan chain, releasing an oligosaccharide whose reducing end is subsequently attached to the hydroxyl group at the C6 position of a glucosyl residue on an α-glucan chain. The result is a new α-(1→6) linkage. The acceptor chain can be either the same chain from which the oligosaccharide was removed (intra-chain transfer) or a different chain (inter-chain transfer).
One of the factors influencing the type of transfer appears to be the relative concentration of linear α-(1→4) chains. In particular, closely associated chains, such as those forming double helices within clusters, tend to favor inter-chain transfer.[21] Moreover, the interaction between starch synthase I and starch branching enzymes seems to play a key role in determining the bimodal chain-length distribution characteristic of plant starches.[14]
Starch branching enzyme isoforms
Plants possess two isoforms of starch branching enzyme, referred to as SBEI and SBEII. Encoded by different genes, they exhibit distinct biochemical properties, suggesting that they play different roles in determining the structure of amylopectin and amylose.[6]
SBEI is expressed predominantly in storage tissues, indicating an important role in defining the structural properties of storage starches. It shows a substrate preference for amylose and can transfer oligosaccharides with a DP greater than 30, although most transfers involve chains of DP 10–13. SBEI also appears to be involved in the synthesis of super-long (or extra-long) amylopectin chains, while its other contributions to amylopectin structure seem less significant. Not all plants express SBEI; for example, Arabidopsis and canola (Brassica napus L.), both oil-storing plants, possess only SBEII, and starch is found exclusively in their photosynthetic tissues.[22][23]
SBEII is widely expressed in grasses, cereals, and many other plants. Loss of SBEII activity results in pronounced alterations to amylopectin architecture and a reduction in starch content. The enzyme has a substrate preference for amylopectin and transfers oligosaccharides with a DP of 6–14. In cereals and grasses, two tissue-specific isoforms are encoded by distinct genes: SBEIIa, expressed mainly in leaves, and SBEIIb, expressed mainly in the endosperm.[6]
Starch debranching enzymes
Starch debranching enzymes catalyze the hydrolysis of α-(1→6) glycosidic bonds and are members of the α-amylase superfamily.
Two types of debranching enzymes are present in plants: isoamylases (EC 3.2.1.68) and pullulanases (EC 3.2.1.411).[24] Isoamylases act on amylopectin and other polyglucans, whereas pullulanases debranch both amylopectin and pullulan, a fungal polysaccharide. Isoamylases and pullulanases also differ in substrate specificity: isoamylases hydrolyze branches composed of at least three glycosidic residues, while pullulanases can act on branches of just two.[25]
During starch granule formation, starch debranching enzymes play a crucial role in defining the water-insoluble properties and fine structure of amylopectin. Enzyme activity is thought to promote clustering of the remaining branches, thereby enhancing interactions between adjacent chains and facilitating α-helix formation. This process, in turn, appears essential for the development of the semi-crystalline structures of amylopectin and, consequently, of starch. In these semi-crystalline regions, branches are presumably inaccessible to starch debranching enzymes, α-amylase, and β-amylase.[6]
Starch debranching enzymes are also employed industrially, particularly in the production of resistant starch and cyclodextrins, which are cyclic oligosaccharides.[25]
Starch phosphorylase
Like starch synthases, starch phosphorylase belongs to the family of glycosyltransferases and closely resembles glycogen phosphorylase. In plants, it occurs in at least two isoenzymatic forms: Pho1, located in the stroma of plastids and considered the true starch phosphorylase involved in starch synthesis, and Pho2, which is cytosolic.[26]
Starch phosphorylase is thought to participate in the initial steps of starch synthesis by catalyzing the reversible transfer of glucosyl units to an α-glucan, where they are linked by an α-(1→4) glycosidic bond.[5] Unlike starch synthases, starch phosphorylase uses glucose-1-phosphate, not ADP-glucose, as the glucosyl donor.[26]
Phosphorylation of amylopectin
Amylopectin, like glycogen, carries phosphate groups in variable amounts depending on the botanical source of the starch. For example, potato starch contains relatively high levels of phosphate groups, with a degree of substitution of about 0.1–0.3%, whereas cereal endosperm starches generally have phosphate contents below 0.01%.[27]
Phosphorylation of amylopectin is catalyzed by two dikinases present in plastids: α-glucan water dikinase (EC 2.7.9.4) and phospho-glucan water dikinase (EC 2.7.9.5). These enzymes transfer the β-phosphate group of ATP to a glucosyl unit of an α-glucan chain, while the γ-phosphate group is transferred to water. Specifically, α-glucan water dikinase phosphorylates the hydroxyl group at the C6 position, whereas phospho-glucan water dikinase phosphorylates the hydroxyl group at the C3 position, usually on a prephosphorylated glucan chain. About two-thirds of phosphate groups are bound at the C6 position and 20–30% at the C3 position. Phosphate groups are also detected at the C2 position, but in much smaller amounts. The enzyme responsible for this phosphorylation is not known.[28][29]
With regard to substrate specificity, phosphorylation appears to accumulate more readily on longer chains. Furthermore, there seems to be an inverse correlation between total phosphate content and the frequency of amylopectin branching. The negative charges carried by phosphate groups cause mutual repulsion between neighboring phosphorylated oligosaccharides. These repulsions seem to promote chain opening and hydration, thereby influencing the activity of biosynthetic enzymes and making the chains more susceptible to attack by amylases.[30][31]
Amylose/amylopectin ratio
Starch granules are composed primarily of amylopectin and amylose.[1] The relative proportions of the two polysaccharides vary, with amylose typically accounting for no more than 35% of the granule’s dry weight.[12] However, some plants produce starch granules composed mostly, or almost entirely, of amylopectin, starches known as waxy starches, while others produce starch granules composed predominantly, or almost exclusively, of amylose.[21]
The amylose/amylopectin ratio strongly influences the physicochemical properties of starch, including water absorption, gelatinization, retrogradation, and resistance to enzymatic hydrolysis. The latter property is especially relevant during carbohydrate digestion, as it determines the rate at which amylose and amylopectin are hydrolyzed by α-amylase to maltose and maltotriose. Consequently, the amylose/amylopectin ratio affects both the health impacts of different starch types and their industrial applications.[5][32][33]
References
^ abcd Qu J., Xu S., Zhang Z., Chen G., Zhong Y., Liu L., Zhang R., Xue J., Guo D. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci Rep 2018;8(1):12736. doi:10.1038/s41598-018-30411-y
^ abc Cornejo-Ramírez Y.I., Martínez-Cruz O., Del Toro-Sánchez C.L., Wong-Corral F.J., Borboa-Flores J. & Cinco-Moroyoqui F.J. The structural characteristics of starches and their functional properties. CYTA J Food 2018;16(1):1003-1017. doi:10.1080/19476337.2018.1518343
^ Smith A.M., Zeeman S.C. Starch: a flexible, adaptable carbon store coupled to plant growth. Annu Rev Plant Biol 2020;71:217-245. doi:10.1146/annurev-arplant-050718-100241
^ Junejo S.A., Flanagan B.M., Zhang B., Dhital S. Starch structure and nutritional functionality – Past revelations and future prospects. Carbohydr Polym 2022;277:118837. doi:10.1016/j.carbpol.2021.118837
^ abcdef Crofts N., Abe N., Oitome N.F., Matsushima R., Hayashi M., Tetlow I.J., Emes M.J., Nakamura Y., Fujita N. Amylopectin biosynthetic enzymes from developing rice seed form enzymatically active protein complexes. J Exp Bot 2015;66(15):4469-82. doi:10.1093/jxb/erv212
^ abcdefg Tetlow I.J., Bertoft E. A review of starch biosynthesis in relation to the building block-backbone model. Int J Mol Sci 2020;21(19):7011. doi:10.3390/ijms21197011
^ ab Abt M.R., Pfister B., Sharma M., Eicke S., Bürgy L., Neale I., Seung D., Zeeman S.C. STARCH SYNTHASE5, a noncanonical starch synthase-like protein, promotes starch granule initiation in Arabidopsis. Plant Cell 2020;32(8):2543-2565. doi:10.1105/tpc.19.00946
^ ab Li G., Yacine Y., Zhu F. Relationships between supramolecular organization and amylopectin fine structure of quinoa starch. Food Hydrocoll 2021;117:106685. doi:10.1016/j.foodhyd.2021.106685
^ ab Pfister B., Zeeman S.C., Rugen M.D., Field R.A., Ebenhöh O., Raguin A. Theoretical and experimental approaches to understand the biosynthesis of starch granules in a physiological context. Photosynth Res 2020;145:55-70. doi:10.1007/s11120-019-00704-y
^ abcd Pfister B., Zeeman S.C. Formation of starch in plant cells. Cell Mol Life Sci 2016;73(14):2781-807. doi:10.1007/s00018-016-2250-x
^ Jenkins P.J., Donald A.M. The influence of amylose on starch granule structure. Int J Biol Macromol 1995;17(6):315-21. doi:10.1016/0141-8130(96)81838-1
^ abc Gous P.W., Fox G.P. Review: Amylopectin synthesis and hydrolysis – Understanding isoamylase and limit dextrinase and their impact on starch structure on barley (Hordeum vulgare) quality. Trends Food Sci Technol 2017;62:23-32. doi:10.1016/j.tifs.2016.11.013
^ Ziegler G.R., Creek J.A., Runt J. Spherulitic crystallization in starch as a model for starch granule initiation. Biomacromolecules 2005;6(3):1547-54. doi:10.1021/bm049214p
^ abcde Szydlowski N., Ragel P., Raynaud S., Lucas M.M., Roldán I., Montero M., Muñoz F.J., Ovecka M., Bahaji A., Planchot V., Pozueta-Romero J., D’Hulst C., Mérida A. Starch granule initiation in Arabidopsis requires the presence of either class IV or class III starch synthases. Plant Cell 2009;21(8):2443-57. doi:10.1105/tpc.109.066522
^ Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ Zeeman S.C., Kossmann J., Smith A.M. Starch: its metabolism, evolution, and biotechnological modification in plants. Annu Rev Plant Biol 2010;61:209-34. doi:10.1146/annurev-arplant-042809-112301
^ Seung D., Boudet J., Monroe J., Schreier T.B., David L.C., Abt M., Lu K.J., Zanella M., Zeeman S.C. Homologs of PROTEIN TARGETING TO STARCH control starch granule initiation in Arabidopsis leaves. Plant Cell 2017;29(7):1657-1677. doi:10.1105/tpc.17.00222
^ Ball S.G., Morell M.K. From bacterial glycogen to starch: understanding the biogenesis of the plant starch granule. Annu Rev Plant Biol. 2003;54:207-33. doi:10.1146/annurev.arplant.54.031902.134927
^ Wilkens C., Svensson B., Møller M.S. Functional roles of starch binding domains and surface binding sites in enzymes involved in starch biosynthesis. Front Plant Sci 2018;9:1652. doi:10.3389/fpls.2018.01652
^ Sawada T., Nakamura Y., Ohdan T., Saitoh A., Francisco P.B. Jr, Suzuki E., Fujita N., Shimonaga T., Fujiwara S., Tsuzuki M., Colleoni C., Ball S. Diversity of reaction characteristics of glucan branching enzymes and the fine structure of α-glucan from various sources. Arch Biochem Biophys 2014;562:9-21. doi:10.1016/j.abb.2014.07.032
^ ab Wang J., Hu P., Lin L., Chen Z., Liu Q., Wei C. Gradually decreasing starch branching enzyme expression is responsible for the formation of heterogeneous starch granules. Plant Physiol 2018;176(1):582-595. doi:10.1104/pp.17.01013
^ Guan H.P., Preiss J. Differentiation of the properties of the branching isozymes from maize (Zea mays). Plant Physiol 1993;102(4):1269-1273. doi:10.1104/pp.102.4.1269
^ Dumez S., Wattebled F., Dauvillee D., Delvalle D., Planchot V., Ball S.G., D’Hulst C. Mutants of Arabidopsis lacking starch branching enzyme II substitute plastidial starch synthesis by cytoplasmic maltose accumulation. Plant Cell 2006;18(10):2694-709. doi:10.1105/tpc.105.037671
^ Møller M.S., Henriksen A., Svensson B. Structure and function of α-glucan debranching enzymes. Cell Mol Life Sci 2016;73(14):2619-41. doi:10.1007/s00018-016-2241-y
^ ab Xia W., Zhang K., Su L., Wu J. Microbial starch debranching enzymes: developments and applications. Biotechnol Adv 2021;50(3):107786. doi:10.1016/j.biotechadv.2021.107786
^ ab Cuesta-Seijo J.A., Ruzanski C., Krucewicz K., Meier S., Hägglund P., Svensson B., Palcic M.M. Functional and structural characterization of plastidic starch phosphorylase during barley endosperm development. PLoS One 2017;12(4):e0175488. doi:10.1371/journal.pone
^ Nitschke F., Wang P., Schmieder P., Girard J.M., Awrey D.E., Wang T., Israelian J., Zhao X., Turnbull J., Heydenreich M., Kleinpeter E., Steup M., Minassian B.A. Hyperphosphorylation of glucosyl C6 carbons and altered structure of glycogen in the neurodegenerative epilepsy Lafora disease. Cell Metab 2013;17(5):756-67. doi:10.1016/j.cmet.2013.04.006
^ Hizukuri S., Tabata S., Kagoshima Nikuni Z. Studies on starch phosphate Part 1. Estimation of glucose-6-phosphate residues in starch and the presence of other bound phosphate(s). Starch/Stärke 1970;22:338-343. doi:https://doi.org/10.1002/star.19700221004
^ Ritte G., Heydenreich M., Mahlow S., Haebel S., Kötting O., Steup M. Phosphorylation of C6- and C3-positions of glucosyl residues in starch is catalysed by distinct dikinases. FEBS Lett 2006;580(20):4872-6. doi:10.1016/j.febslet.2006.07.085
^ Zhou W., He S., Naconsie M., Ma Q., Zeeman S.C., Gruissem W. & Zhang P. Alpha-glucan, water dikinase 1 affects starch metabolism and storage root growth in Cassava (Manihot esculenta Crantz). Sci Rep 2017;7:9863 doi:10.1038/s41598-017-10594-6
^ Baunsgaard L., Lütken H., Mikkelsen R., Glaring M.A., Pham T.T., Blennow A. A novel isoform of glucan, water dikinase phosphorylates pre-phosphorylated alpha-glucans and is involved in starch degradation in Arabidopsis. Plant J 2005;41(4):595-605. doi:10.1111/j.1365-313X.2004.02322.x
^ Tester R.F., Morrison W.R. Swelling and gelatinization of cereal starches. I. Effects of amylopectin, amylose and lipids. Cereal Chem 1990;67:551-557.
^ Hoover R. Composition, molecular structure, and physicochemical properties of tuber and root starches: a review. Carbohydr Polym 2001;45(3):253-267 doi:10.1016/S0144-8617(00)00260-5
Starch synthase (EC 2.4.1.21) is an enzyme that catalyzes the transfer of glucose molecules from ADP-glucose to the non-reducing end of a pre-existing α-(1→4) linked glucan chain, to which the monosaccharides are attached via an α-(1→4) glycosidic bond.[1]
This enzymatic activity is involved in the synthesis of amylose and amylopectin, the two major constituents of starch, which is the main storage form of carbohydrates in plants.[2]
Starch synthase belongs to the family of glycosyltransferases (EC 2.4), like starch phosphorylase (EC 2.4.1.1), another enzyme involved in starch metabolism, and glycogen synthase (EC 2.4.1.11) and glycogen phosphorylase (EC 2.4.1.1), which are involved in glycogen synthesis and glycogen degradation (glycogenolysis), respectively.[3]
However, whereas glycogen synthase uses UDP-glucose as the glucosyl donor, and starch phosphorylase uses glucose 1-phosphate, starch synthase uses ADP-glucose.[4][5]
Six isoforms of starch synthase are known in plants. They are structurally related proteins, with five involved in the synthesis of amylopectin, referred to as starch synthase I, II, III, IV and V (SSI, SSII, SSIII, SSIV and SSV, respectively), and one involved in the synthesis of amylose, the granule-bound starch synthase (GBSS; EC 2.4.1.242).[3]
GBSS, SSI, SSII, SSIII, and SSIV possess catalytic activity, whereas SSV lacks it.[6]
GBSS is almost exclusively bound to starch granules and is located mainly within them, as evidenced by protease treatment of the granule surface. The other isoforms of starch synthase are found either only in the stroma of plastids or partitioned between starch granules and the stroma, and are referred to as soluble starch synthases.[7]
Starch synthases I, II, III, and IV catalyze the addition of a single glucose residue per substrate encounter, a distributive mode of action, whereas GBSS catalyzes the addition of more than one glucosidic unit per substrate encounter, a processive mode of action.[8]
Processive: adds multiple glucose units per encounter
Primarily bound to starch granules
Catalyzes the elongation of amylose chains
SSI–IV
Amylopectin
Distributive: adds one glucose unit per encounter
Stroma and/or soluble in starch granules
Exhibit catalytic activity; contribute to amylopectin branching
SSV
Amylopectin
Not specified
Not specified
Lacks catalytic activity; function not fully understood
Starch synthase and MOS
Starch synthases involved in the early steps of amylose and amylopectin synthesis require short malto-oligosaccharides (MOS) to initiate the de novo synthesis of these two polysaccharides.
These small oligosaccharides, α-(1→4)-glucans with a degree of polymerization from 2 to 7, act as primers and are elongated, a function analogous to that performed by glycogenin in glycogen synthesis.
MOS can originate from the activity of starch synthase III, starch phosphorylase, or starch debranching enzymes.
Being poorly water soluble, MOS seem able to evade the hydrolytic action of alpha-amylase (EC 3.2.1.1) and beta-amylase.[8]
Starch synthase and amylopectin synthesis
The synthesis of amylopectin requires the temporally coordinated action of at least four classes of enzymes: starch synthase isoenzymes, starch phosphorylase, starch branching enzymes (EC 2.4.1.18), and starch debranching enzymes.[2][4] It is also thought that, in many cases, these enzymes physically interact with each other to form multienzyme complexes, structures capable of increasing the efficiency of the metabolic pathway.[8]
It is generally accepted that starch granule growth occurs from a central core called the hilum, whose precise structure is unknown, although it appears to consist of a disordered arrangement of α-glucans.[9] The initiation of the hilum, the formation of normal starch granule morphology, and the degree of starch accumulation require the presence of SSIV, although SSIII and SSV are also thought to contribute, overlapping in function with SSIV.[10]
GBSS and amylose synthesis
Granule-bound starch synthase is involved in the synthesis of amylose. This enzyme was first described by Luis Federico Leloir in the early 1960s, the same researcher who, in 1948, had discovered the main pathway for galactose metabolism, known as the Leloir pathway.[11]
Its catalytic action is not entirely simultaneous with that of the other starch synthases, as it requires the presence of an amylopectin matrix.[3]
In grasses, granule-bound starch synthase is present in two isoforms, encoded by distinct genes, referred to as GBSSI and GBSSII.[12]
GBSS requires, for its catalytic activity, the presence of a protein from the PTST family, PTST1, which enables its binding to the starch granule and whose action appears to be more important in chloroplasts than in amyloplasts.
Mechanism of GBSS and PTST1 Interaction
PTST1 appears to associate, in the plastid stroma, with GBSS. The complex binds to the nascent starch granule; the protein then dissociates from the enzyme, which begins catalyzing the elongation of malto-oligosaccharides, while the protein returns to the stroma to recruit another GBSS molecule.[13]
References
^ Zeeman S.C., Kossmann J., Smith A.M. Starch: its metabolism, evolution, and biotechnological modification in plants. Annu Rev Plant Biol 2010;61:209-34. doi:10.1146/annurev-arplant-042809-112301
^ ab Qu J., Xu S., Zhang Z., Chen G., Zhong Y., Liu L., Zhang R., Xue J., Guo D. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci Rep 2018;8(1):12736. doi:10.1038/s41598-018-30411-y
^ abc Pfister B., Zeeman S.C. Formation of starch in plant cells. Cell Mol Life Sci 2016;73(14):2781-807. doi: 10.1007/s00018-016-2250-x
^ ab Crofts N., Abe N., Oitome N.F., Matsushima R., Hayashi M., Tetlow I.J., Emes M.J., Nakamura Y., Fujita N. Amylopectin biosynthetic enzymes from developing rice seed form enzymatically active protein complexes. J Exp Bot 2015;66(15):4469-82. doi:10.1093/jxb/erv212
^ Cuesta-Seijo J.A., Ruzanski C., Krucewicz K., Meier S., Hägglund P., Svensson B., Palcic M.M. Functional and structural characterization of plastidic starch phosphorylase during barley endosperm development. PLoS One 2017;12(4):e0175488. doi:10.1371/journal.pone
^ Abt M.R., Pfister B., Sharma M., Eicke S., Bürgy L., Neale I., Seung D., Zeeman S.C. STARCH SYNTHASE5, a noncanonical starch synthase-like protein, promotes starch granule initiation in Arabidopsis. Plant Cell 2020;32(8):2543-2565. doi:10.1105/tpc.19.00946
^ Seung D., Boudet J., Monroe J., Schreier T.B., David L.C., Abt M., Lu K.J., Zanella M., Zeeman S.C. Homologs of PROTEIN TARGETING TO STARCH control starch granule initiation in Arabidopsis leaves. Plant Cell 2017;29(7):1657-1677. doi:10.1105/tpc.17.00222
^ abc Tetlow I.J., Bertoft E. A review of starch biosynthesis in relation to the building block-backbone model. Int J Mol Sci 2020;21(19):7011. doi:10.3390/ijms21197011
^ Ziegler G.R., Creek J.A., Runt J. Spherulitic crystallization in starch as a model for starch granule initiation. Biomacromolecules 2005;6(3):1547-54. doi:10.1021/bm049214p
^ Szydlowski N., Ragel P., Raynaud S., Lucas M.M., Roldán I., Montero M., Muñoz F.J., Ovecka M., Bahaji A., Planchot V., Pozueta-Romero J., D’Hulst C., Mérida A. Starch granule initiation in Arabidopsis requires the presence of either class IV or class III starch synthases. Plant Cell 2009;21(8):2443-57. doi:10.1105/tpc.109.066522
^ Leloir L.F., de Fekete M.A., Cardini C.E. Starch and oligosaccharide synthesis from uridine diphosphate glucose. J Biol Chem 1961;236:636-41. doi:10.1016/S0021-9258(18)64280-2
^ Vrinten P.L., Nakamura T. Wheat granule-bound starch synthase I and II are encoded by separate genes that are expressed in different tissues. Plant Physiol 2000;122(1):255-64. doi:10.1104/pp.122.1.255
^ Seung D., Soyk S., Coiro M., Maier B.A., Eicke S., and Zeeman S.C. PROTEIN TARGETING TO STARCH is required for localising GRANULE-BOUND STARCH SYNTHASE to starch granules and for normal amylose synthesis in Arabidopsis. PLoS Biol 2015;13:e1002080. doi:10.1371/journal.pbio.1002080
Amylose is a polysaccharide composed of α-D-glucose units linked by α-(1→4) glycosidic bonds, with a few branches connected to the main chain by α-(1→6) glycosidic bonds.[1]
Together with amylopectin, it is one of the two main constituents of starch, the major storage form of energy and carbohydrates in the biosphere.[2][3]
Its synthesis is catalyzed by the enzyme granule-bound starch synthase (GBSS; EC 2.4.1.242) and requires the presence of a second protein, known as protein targeting to starch 1 (PTST1), which has no catalytic activity.[4]
Within the starch granule, amylose is embedded in the semi-crystalline matrix formed by amylopectin.[5] Unlike amylopectin, amylose is not essential for the formation of starch granules, is present in smaller amounts, but exerts a strong influence on the physicochemical properties of starch.[6][7]
Plant varieties have been developed whose starch granules contain either negligible amounts or, conversely, very high amounts of amylose. These phenotypes have both industrial applications and potential health benefits.[8][9][10]
Amylose molecules have a molecular weight of about 106 daltons. They are mostly linear and composed of α-D-glucose units (hereinafter referred to as glucose) linked by α-(1→4) glycosidic bonds, that is, covalent bonds between the C-1 atom of one unit and the hydroxyl group on the C-4 atom of the next unit.[11] The linear chains are composed of several hundred to several thousand monosaccharides; therefore, they are much longer than amylopectin chains.[1]
The few branches are connected to the linear chain by α-(1→6) glycosidic bonds, as in amylopectin and glycogen, the storage form of carbohydrates in animals. An α-(1→6) glycosidic bond is a covalent bond between the C-1 atom of one unit and the hydroxyl group on the C-6 atom of another glucose unit. The number of branches ranges from 5 to 20, depending on the botanical origin of the starch. Unlike those of amylopectin, the branches are not clustered.[12][13]
Studies on the length of amylose branches have shown a bimodal distribution, with two fractions termed:
AM1, comprising shorter chains, with a degree of polymerization between 100 and 700;
AM2, comprising longer chains, with a degree of polymerization between 700 and 40,000.[14]
A similar bimodal distribution of branch length is also observed in amylopectin, whose fractions are termed AP1 (shorter and more abundant) and AP2. The intraspecies variation in the distribution of AM1 and AM2 fractions is relatively small, whereas the variation among different species is considerable and has a genetic basis.[14]
Amylose location in starch granules
The precise location of amylose within starch granules is not fully known, although it is generally believed to be concentrated in the amorphous regions. However, some studies suggest that its distribution is not limited to these regions, but that amylose can also be found interspersed between amylopectin chains and on the surface of the granules. Thus, amylose may occupy multiple locations within the granule.[5][15]
Synthesis
In plants, the synthesis of amylose is catalyzed by GBSS, one of the six isoforms of starch synthase.[16] This enzyme, whose activity is the major determinant of amylose content in starch granules, requires the presence of the PTST1 protein.[4] Amylose branching appears to be carried out by the starch branching enzyme (SBE; EC 2.4.1.18).[17]
As in the case of amylopectin biosynthesis, it is believed that the enzymes involved physically interact to form multienzyme complexes, which optimize the efficiency of the process.[1] Because amylose synthesis requires a pre-existing amylopectin matrix to target GBSS to the starch granules, the synthesis of the two polysaccharides is not entirely simultaneous.[16]
Priming and initiation of amylose synthesis
In the initial steps, GBSS, like other starch synthases, requires short malto-oligosaccharides (MOS), α-(1→4) glucans with a degree of polymerization of 2 to 7, which act as primers for chain elongation.[17] MOS can originate from various sources, such as the trimming of nascent amylopectin molecules by starch debranching enzymes, or the activity of starch phosphorylase (EC 2.4.1.1), another enzyme involved in starch metabolism.[18] Because MOS are poorly soluble in water, they appear able to evade the hydrolytic activity of α-amylase (EC 3.2.1.1) and β-amylase (EC 3.2.1.2), and diffuse within the starch granule matrix, where they are elongated by GBSS.[19] Once malto-oligosaccharides are elongated beyond seven glucose residues, they can no longer diffuse out of the granule and are further extended in situ.[20]
Since a primer is required, the early stages of amylose and amylopectin biosynthesis resemble those of glycogen. However, in glycogen synthesis the primer is the self-glucosylating protein glycogenin.[21]
Importantly, the polymerization of glucose into amylopectin, amylose, glycogen, and, more generally, the conversion of osmotically active monosaccharides into osmotically inactive polysaccharides, enables the storage of large amounts of carbohydrate within the cell without increasing osmotic pressure.[21][22]
Granule-bound starch synthase
GBSS, together with the other isoforms of starch synthase and starch phosphorylase, belongs to the glycosyltransferase family (EC 2.4).[16]
GBSS was discovered by Louis Federico Leloir’s group, which had previously identified the main pathway for galactose, known as the Leloir pathway. It is the most abundant protein associated with starch granules.[23] Unlike the other isoforms of starch synthase, which are mostly located in the plastid stroma or partitioned between starch granules and the stroma, GBSS is found almost exclusively bound to granules. Protease treatment of the starch granule surface has shown that most GBSS is located within the granule rather than on its surface, a position consistent with amylose synthesis inside nascent granules.[4]
In grasses, GBSS is present in two isoforms encoded by distinct genes. These isoforms are:
GBSSI, present in the amyloplasts of storage tissues (non-photosynthetic tissues);
GBSSII, present in chloroplasts (photosynthetic tissues), where it participates in the synthesis of transient starch.[24]
GBSS catalyzes the transfer of a glucose residue from ADP-glucose to the non-reducing end of an α-(1→4)-glucan, forming a new α-(1→4) glycosidic bond.[21][25]
Notably, starch synthases use ADP-glucose as the glucosyl donor, whereas glycogen synthase, the enzyme responsible for glycogen synthesis, uses UDP-glucose.[22][26]
Granule-bound starch synthase can add more than one glucose monomer per substrate encounter, a processive mode of action. This contrasts with the other isoforms of starch synthase, which add only a single glucose unit per encounter, a distributive mode of action. The processive activity of GBSS enables the biosynthesis of long linear chains and appears to be strongly enhanced by the presence of amylopectin.[1]
PTST1
Amylose synthesis requires the presence of a protein from the PTST family, namely PTST1, which was discovered more than fifty years after GBSS.
PTST1 has no catalytic activity but enables the binding of GBSS to the starch granule. This function appears to be more important in chloroplasts, and therefore in the synthesis of transient starch, than in amyloplasts.
It has been proposed that PTST1 associates with GBSS in the plastid stroma. The complex then binds to the nascent starch granule, after which PTST1 dissociates, allowing GBSS to initiate amylose synthesis. PTST1 subsequently returns to the plastid stroma to recruit another GBSS molecule. The importance of PTST1 is highlighted by its conservation throughout the plant kingdom. Loss of PTST1 causes GBSS to detach from the nascent starch granule, thereby preventing amylose synthesis.[4][19]
Starch branching enzymes
The enzyme responsible for generating the few α-(1→6) linkages present in amylose molecules is not definitively known, although an isoform of starch branching enzyme, SBEI, may be involved.[5]
SBEI is located primarily in the plastid stroma, with only a small proportion bound to starch granules, and is mainly expressed in storage tissues. The low frequency of branching in amylose may result from its biosynthesis inside the granules, where SBEI is scarce, thereby protecting the nascent molecule from the enzyme’s action.[27]
Amylose/amylopectin ratio
In the starch granules of land plants, amylose is almost always present in variable amounts, generally between 5% and 35%. Variability occurs not only among species but also within the same species, depending on the organ or tissue examined. In tubers and seeds, the amylose content also varies with the stage of development: it is generally low in the early stages and increases until the final value is reached, a pattern consistent with the synthesis of amylose within an amylopectin matrix.[5][25]
However, some plants have starch with very low, or even no, amylose content. This type of starch is referred to as waxy, owing to the raw endosperm’s appearance, which resembles wax. Conversely, certain plants produce starch granules composed mostly, or even entirely, of amylose.[9]
Although its precise role is not yet fully understood, the near-constant presence of amylose suggests that this polysaccharide plays an important structural role in starch granules and confers some advantage to plants during growth and development.[5]
The amylose/amylopectin ratio strongly influences the physicochemical properties of starch. For example, it affects water absorption, which in turn influences processes such as starch retrogradation and gelatinization, as well as resistance to enzymatic hydrolysis. This determines, for instance, the rate at which maltose and maltotriose are released during starch degradation by α-amylase or β-amylase.[28] These properties, in turn, influence both the industrial applications of starch and its effects on human health.[10]
High-amylose starches
High-amylose cereals can be obtained by enhancing GBSSI gene expression or by suppressing or eliminating the genes encoding starch branching enzymes, SSIIa, or other enzymes and proteins involved in amylopectin biosynthesis. However, in cereal endosperm, the most effective method is the suppression or elimination of one or more starch branching enzyme isoforms.[9]
High-amylose starches exhibit distinctive physicochemical properties, such as high gelling strength, easy retrogradation, and excellent film-forming ability. These properties make them suitable for industrial applications, including the production of biodegradable plastics, paper, and adhesives.[6][28]
High-amylose starches also contain elevated levels of resistant starch, a form of starch that escapes digestion in the small intestine by α-amylase, one of the key hydrolases involved in carbohydrate digestion. Studies with resistant starch–enriched foods have shown improvements in insulin sensitivity and glycemic response, as well as a reduced risk of cardiovascular disease, obesity, and type II diabetes mellitus.[29]
Mechanism of action
Resistant starch, by escaping intestinal digestion, lowers the glycemic index of foods in which it is present, thereby helping regulate blood glucose levels. Once it reaches the colon, it is fermented by the gut microbiota, which is part of the human microbiota, producing short-chain fatty acids (mainly butyric acid, acetic acid, and propionic acid), which play an essential role in maintaining intestinal health.[30][31]
References
^ abcd Tetlow I.J., Bertoft E. A review of starch biosynthesis in relation to the building block-backbone model. Int J Mol Sci 2020;21(19):7011. doi:10.3390/ijms21197011
^ Qu J., Xu S., Zhang Z., Chen G., Zhong Y., Liu L., Zhang R., Xue J., Guo D. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci Rep 2018;8(1):12736. doi:10.1038/s41598-018-30411-y
^ Apriyanto A., Compart J., Fettke J. A review of starch, a unique biopolymer – Structure, metabolism and in planta modifications. Plant Sci 2022;318:111223. doi:10.1016/j.plantsci.2022.111223
^ abcd Seung D., Soyk S., Coiro M., Maier B.A., Eicke S., Zeeman S.C. PROTEIN TARGETING TO STARCH is required for localising GRANULE-BOUND STARCH SYNTHASE to starch granules and for normal amylose synthesis in Arabidopsis. PLOS Biol 2015;13(2):e1002080. doi:10.1371/journal.pbio.1002080
^ abcde Seung D. Amylose in starch: towards an understanding of biosynthesis, structure and function. New Phytol 2020;228:1490-1504. doi:10.1111/nph.16858
^ ab Jobling S. Improving starch for food and industrial applications. Curr Opin Plant Biol 2004;7(2):210-8. doi:10.1016/j.pbi.2003.12.001
^ Junejo S.A., Flanagan B.M., Zhang B., Dhital S. Starch structure and nutritional functionality – Past revelations and future prospects. Carbohydr Polym 2022;277:118837. doi:10.1016/j.carbpol.2021.118837
^ Funnell-Harris D.L., Sattler S.E., O’Neill P.M., Eskridge K.M., and Pedersen J.F. Effect of waxy (low amylose) on fungal infection of Sorghum grain. Phytopathology 2015;105(6):716-846. doi:10.1094/PHYTO-09-14-0255-R
^ abc Wang J., Hu P., Chen Z., Liu Q., Wei C. Progress in high-amylose cereal crops through inactivation of starch branching enzymes. Front Plant Sci 2017;8:469. doi:10.3389/fpls.2017.00469
^ ab Li H-T., Zhang W., Zhu H, Chao C., Guo Q. Unlocking the potential of high-amylose starch for gut health: not all function the same. Fermentation 2023;9(2):134. doi:https://doi.org/10.3390/fermentation9020134
^ Hizukuri S., Takeda Y., Yasuda M., Suzuki A. Multi-branched nature of amylose and the action of debranching enzymes. Carbohydr Res 1981;94:205-213. doi:10.1016/S0008-6215(00)80718-1
^ Zhiguang C., Haixia Z., Min C., Fayong G., Jing L. The fine structure of starch: a review. NPJ Sci Food 2025;9(1):50. doi:10.1038/s41538-025-00414-x
^ ab Wang K., Hasjim J., Wu A.C., Henry R.J., Gilbert R.G. Variation in amylose fine structure of starches from different botanical sources. J Agric Food Chem 2014;62(19):4443-53. doi:10.1021/jf5011676
^ Teobaldi A.G., Carrillo Parra E.J., Barrera G.N., Ribotta P.D. The properties of damaged starch granules: the relationship between granule structure and water–starch polymer interactions. Foods 2025;14(1):21. doi:https://doi.org/10.3390/foods14010021
^ abc Pfister B., Zeeman S.C. Formation of starch in plant cells. Cell Mol Life Sci 2016;73(14):2781-807. doi:10.1007/s00018-016-2250-x
^ Tetlow I.J., Emes M.J. A review of starch-branching enzymes and their role in amylopectin biosynthesis. IUBMB Life 2014;66(8):546-58. doi:10.1002/iub.1297
^ Szydlowski N., Ragel P., Raynaud S., Lucas M.M., Roldán I., Montero M., Muñoz F.J., Ovecka M., Bahaji A., Planchot V., Pozueta-Romero J., D’Hulst C., Mérida A. Starch granule initiation in Arabidopsis requires the presence of either class IV or class III starch synthases. Plant Cell 2009;21(8):2443-57. doi:10.1105/tpc.109.066522
^ ab Seung D., Boudet J., Monroe J., Schreier T.B., David L.C., Abt M., Lu K.J., Zanella M., Zeeman S.C. Homologs of PROTEIN TARGETING TO STARCH control starch granule initiation in Arabidopsis leaves. Plant Cell 2017;29(7):1657-1677. doi:10.1105/tpc.17.00222
^ Denyer K., Johnson P., Zeeman S., Smith A.M. The control of amylose synthesis. J Plant Physiol 2001;158: 479-487. doi:10.1078/0176-1617-00360
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ ab Heldt H-W. Plant biochemistry – 4th Edition. Elsevier Academic Press, 2010.
^ Leloir L.F., de Fekete M.A., Cardini C.E. Starch and oligosaccharide synthesis from uridine diphosphate glucose. J Biol Chem 1961;236:636-41. doi:10.1016/S0021-9258(18)64280-2
^ Vrinten P.L., Nakamura T. Wheat granule-bound starch synthase I and II are encoded by separate genes that are expressed in different tissues. Plant Physiol 2000;122(1):255-64. doi:10.1104/pp.122.1.255
^ ab Gous P.W., Fox G.P. Review: amylopectin synthesis and hydrolysis – Understanding isoamylase and limit dextrinase and their impact on starch structure on barley (Hordeum vulgare) quality. Trends Food Sci Technol 2016;62:23-32. doi:10.1016/j.tifs.2016.11.013
^ Crofts N., Abe N., Oitome N.F., Matsushima R., Hayashi M., Tetlow I.J., Emes M.J., Nakamura Y., Fujita N. Amylopectin biosynthetic enzymes from developing rice seed form enzymatically active protein complexes. J Exp Bot 2015;66(15):4469-82. doi:10.1093/jxb/erv212
^ Kram A.M., Oostergetel G.T., Van Bruggen E. Localization of branching enzyme in potato tuber cells with the use of immunoelectron microscopy. Plant Physiol 1993;101(1):237-243. doi:10.1104/pp.101.1.237
^ ab Magallanes-Cruz P.A., Flores-Silva P.C., Bello-Perez L.A. Starch structure influences its digestibility: a review. J Food Sci 2017;82(9):2016-2023. doi:10.1111/1750-3841.13809
^ Birt D.F., Boylston T., Hendrich S., Jane J.L., Hollis J., Li L., McClelland J., Moore S., Phillips G.J., Rowling M., Schalinske K., Scott M.P., Whitley E.M. Resistant starch: promise for improving human health. Adv Nutr 2013;4(6):587-601. doi:10.3945/an.113.004325
^ Usuda H., Okamoto T., Wada K. Leaky gut: effect of dietary fiber and fats on microbiome and intestinal iarrier. Int J Mol Sci 2021;22(14):7613. doi:10.3390/ijms22147613
^ Topping D.L., Clifton P.M. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev 2001;81(3):1031-64. doi:10.1152/physrev.2001.81.3.1031
The Leloir pathway is the primary route for galactose metabolism.
Discovered by Luis F. Leloir and colleagues in 1948, this pathway converts galactose into glucose 1-phosphate. Specifically, it involves the inversion of the configuration of the hydroxyl group at carbon 4 of galactose, one of the monosaccharide’s chiral centers.[1]
The metabolic intermediates involved in this isomerization serve as building blocks in various biosynthetic pathways, such as the glycosylation of proteins and lipids or glycogen synthesis. Their role depends on the developmental stage, the cell’s metabolic state, and the tissue type.[2]
With the exception of the first step, the reactions in the Leloir pathway are reversible, proceeding in either direction depending on substrate concentrations and the tissue’s metabolic needs. This enables the interconversion of galactose and glucose.[3]
The importance of the Leloir pathway, and of galactose itself, is highlighted by its high degree of evolutionary conservation, from bacteria to plants and animals.[4] In humans, its relevance is further underscored by the severe consequences of mutations in the genes encoding its enzymes, which lead to the inherited metabolic disorder known as galactosemia.[5]
Summary: Key Points
Primary function: The Leloir pathway is the essential metabolic route that converts dietary galactose into glucose 1-phosphate, making it available for energy production or glycogen storage.
Cellular trapping and irreversibility: The galactokinase reaction is the only irreversible step; it adds a negative charge to galactose, effectively preventing it from diffusing out of the cell.
Biochemical flexibility: Except for the second step, the reactions are fully reversible, enabling cells to interconvert sugar nucleotides based on developmental stages and specific tissue needs.
Clinical relevance: Inherited mutations in the pathway’s genes cause galactosemia. The resulting accumulation of galactitol triggers osmotic and oxidative stress, leading to early cataract formation.
Galactose, along with glucose and fructose, is one of the monosaccharides that can be absorbed in the intestine. The main dietary source of galactose is lactose, which, together with maltose, trehalose and sucrose, is one of the disaccharides commonly found in food.
Since there are no transporters for disaccharides, their glycosidic bonds are hydrolyzed in the final stage of carbohydrate digestion, releasing their constituent monosaccharides. In the case of lactose, these are glucose and galactose.[6]
The monosaccharides are then absorbed and transported via the portal circulation to the liver, the primary site of galactose metabolism. The liver absorbs most of the galactose (approximately 88%) through facilitated passive diffusion mediated by the GLUT2 transporter.[7]
The small remaining amount of circulating galactose reaches other organs and tissues, such as the mammary gland, which during lactation uses galactose for lactose synthesis and for the glycosylation of milk proteins and lipids.[4]
The steps of the Leloir pathway
The Leloir pathway consists of four enzymatic reactions, catalyzed respectively by:
galactose mutarotase (also known as aldose 1-epimerase; EC 5.1.3.3);
Detailed Schematic Representation of the Leloir Pathway
Step 1: galactose mutarotase
The hydrolysis of the β-(1→4) glycosidic bond in lactose results in the release of glucose and β-D-galactose.
Galactokinase, the enzyme responsible for the second step of the Leloir pathway, is specific for the α-anomer of galactose.[8] Therefore, the β-anomer must first be converted into the α-anomer. This conversion is catalyzed by galactose mutarotase.
In addition to galactose, this enzyme is also capable of interconverting the α- and β-anomeric forms of other sugars, including glucose, xylulose, maltose, and lactose, although with varying degrees of efficiency.[9]
Step 2: galactokinase
In the second step, α-D-galactose is phosphorylated to galactose 1-phosphate in a reaction catalyzed by galactokinase.[8] Phosphorylation of galactose, like that of glucose to glucose 6-phosphate, is metabolically important for several reasons.
Galactose 1-phosphate cannot diffuse out of the cell because it carries a negative charge, and there are no transporters for phosphorylated sugars in the plasma membrane.[10]
Phosphorylation increases the energy content of the monosaccharide, initiating destabilization and facilitating its subsequent metabolism.[11]
By converting galactose into galactose 1-phosphate, the reaction lowers the intracellular concentration of free galactose, promoting its continued uptake via facilitated transport.[10]
The reaction catalyzed by galactokinase represents the irreversible step of the Leloir pathway.[3] Unlike hexokinase and glucokinase (EC 2.7.1.1), which phosphorylate the hydroxyl group at carbon 6 of glucose, galactokinase and fructokinase (EC 2.7.1.4) phosphorylate the hydroxyl group at carbon 1 of galactose and fructose, respectively.[12]
The conversion of galactose 1-phosphate to glucose-1-phosphate requires two additional reactions: the third and fourth steps of the Leloir pathway.
Step 3: galactose 1-phosphate uridylyltransferase
In the third step, galactose 1-phosphate uridylyltransferase catalyzes the transfer of a uridine monophosphate (UMP) group from UDP-glucose to galactose 1-phosphate, yielding UDP-galactose and glucose 1-phosphate.
The reaction follows a ping-pong (double displacement) mechanism, involving the formation of a covalent intermediate between the enzyme and the UMP group.[10]
Step 4: UDP-galactose 4-epimerase
In the final step, UDP-galactose is converted into UDP-glucose in a reaction catalyzed by UDP-galactose 4-epimerase. This enzyme inverts the configuration of the hydroxyl group at carbon 4 and is responsible for the interconversion between UDP-galactose and UDP-glucose, and, by extension, between galactose and glucose.[13]
UDP-galactose 4-epimerase requires NAD+ as a cofactor, and the reaction proceeds through the formation of a ketonic intermediate at C4, accompanied by the reduction of NAD+ to NADH. The sugar then rotates within the active site, exposing the opposite face to NADH, which transfers a hydride ion back to carbon 4, but in the reversed configuration.[14]
Because NAD+ is first reduced and then re-oxidized, there is no net redox change for the coenzyme, which therefore does not appear in the overall reaction equation.
UDP-galactose 4-epimerase is thought to be the rate-limiting enzyme of the Leloir pathway.[4]
The UDP-glucose produced in this reaction is subsequently recycled by UDP-glucose pyrophosphorylase, releasing glucose 1-phosphate.[2]
In mammals, UDP-galactose 4-epimerase also catalyzes the interconversion between UDP-N-acetylgalactosamine and UDP-N-acetylglucosamine.[15]
It is important to note that UDP-galactose and UDP-glucose, like galactose and glucose in general, have the same molecular formula. They differ only in the configuration of the hydroxyl group at carbon 4, making them stereoisomers, and more specifically, epimers, a form of optical isomerism.
Role of the Leloir pathway
The Leloir pathway enables cells to utilize galactose or its derivative, glucose, in various metabolic pathways, both anabolic and catabolic, depending on the metabolic state of the cell or tissue.
Because the reaction catalyzed by UDP-galactose 4-epimerase is reversible, the conversion of glucose into galactose (and its nucleotide derivatives) is also possible.[2]
UDP-galactose can be used in:
the glycosylation of proteins and lipids, such as the formation of galactocerebrosides, which are major glycolipid components of myelin. For this reason, galactose was initially referred to as cerebrose;[4][16]
the lactating mammary gland, where it is used for lactose synthesis in a reaction catalyzed by the lactose synthase system.[17]
Furthermore, since the UDP-galactose 4-epimerase reaction is reversible, glucose, once activated to UDP-glucose by UDP-glucose pyrophosphorylase (EC 2.7.7.9), can be converted to UDP-galactose for lactose production.[18]
In the liver and skeletal muscle, UDP-glucose derived from UDP-galactose can be used:[3]
for protein and lipid glycosylation;
in glycogen synthesis, especially when the cell’s energy demand is low. Compared to glucose and fructose, galactose is preferentially incorporated into hepatic glycogen rather than entering oxidative pathways;
after conversion to glucose 1-phosphate (via UDP-glucose pyrophosphorylase), and subsequent isomerization to glucose 6-phosphate (via phosphoglucomutase, EC 5.4.2.2), it may enter various metabolic routes, including glycolysis, the pentose phosphate pathway or gluconeogenesis.[4]
Note: UDP-galactose, discovered during studies on the Leloir pathway, was the first nucleotide sugar to be identified.[19]
Leloir pathway and galactosemia
Glycosylations are post-translational modifications that play a crucial role in enabling and regulating a wide range of biological processes. Defects in glycosylation have been associated with numerous pathological conditions, including cancer, diabetes, and congenital metabolic disorders, particularly the congenital disorders of glycosylation (CDGs), which are primarily autosomal recessive monogenic conditions.[20] Among these, galactosemia is a well-known disorder, first described by von Reuss A. in 1908.[21] Galactosemia results from mutations in one of the genes encoding the enzymes of the Leloir pathway, and four clinical types have been identified.
Type I: due to galactose-1-phosphate uridylyltransferase deficiency. This is the most common and severe form and is referred to as classic galactosemia.[22]
Type III: due to UDP-galactose 4-epimerase deficiency.[24]
Type IV: due to galactose mutarotase deficiency.[25]
At present, the standard treatment for all forms of galactosemia consists of a galactose-restricted diet, aimed at preventing the toxic accumulation of galactose or its metabolites.[26]
Galactosemia and cataracts
The accumulation of galactose activates alternative metabolic pathways, including the synthesis of galactitol and galactonate.
One of the early clinical manifestations of galactosemia is the development of cataracts, typically within the first two years of life. In the most severe cases, the disease may also lead to brain, liver, and kidney damage.[9]
One proposed mechanism underlying cataract formation involves the reduction of galactose, accumulated in the lens, to galactitol in a reaction catalyzed by aldose reductase (EC 1.1.1.21).[27] Galactitol is poorly metabolized and does not readily diffuse across cell membranes due to its low lipophilicity. As an osmotically active compound, it increases intracellular osmotic pressure, leading to a net influx of water into the lens cells.[28]
Moreover, its synthesis consumes NADPH, potentially depleting cellular NADPH pools and impairing the activity of glutathione reductase (EC 1.8.1.7). This reduction in antioxidant capacity may contribute to the accumulation of free radicals.[29]
The combined effects of osmotic stress and oxidative damage can ultimately compromise cell integrity, resulting in cell death and the formation of lens opacities. Additionally, galactitol has been reported to act as an inhibitor of galactose mutarotase, which may exacerbate galactose accumulation by further hindering its metabolism.[30]
References
^ Leloir L.F., de Fekete M.A., Cardini C.E. Starch and oligosaccharide synthesis from uridine diphosphate glucose. J Biol Chem 1961;236:636-41. doi:10.1016/S0021-9258(18)64280-2
^ abc Frey P.A. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J 1996;10(4):461-70. doi:10.1096/fasebj.10.4.8647345
^ abc Conte F., van Buuringen N., Voermans N.C., Lefeber D.J. Galactose in human metabolism, glycosylation and congenital metabolic diseases: time for a closer look. Biochim Biophys Acta Gen Subj 2021;1865(8):129898. doi:10.1016/j.bbagen.2021.129898
^ abcde Coelho A.I., Berry G.T., Rubio-Gozalbo M.E. Galactose metabolism and health. Curr Opin Clin Nutr Metab Care 2015;18(4):422-427. doi:10.1097/MCO.0000000000000189
^ Petry K.G., Reichardt J.K. The fundamental importance of human galactose metabolism: lessons from genetics and biochemistry. Trends Genet 1998;14(3):98-102. doi:10.1016/s0168-9525(97)01379-6
^ Coelho A.I., Rubio-Gozalbo M.E., Vicente J.B., Rivera I. Sweet and sour: an update on classic galactosemia. J Inherit Metab Dis 2017;40(3):325-342. doi:10.1007/s10545-017-0029-3
^ abc Holden H.M., Rayment I., Thoden J.B. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem 2003;278(45):43885-43888. doi:10.1074/jbc.R300025200
^ ab Thoden J.B., Timson D.J., Reece R.J., and M. Holden H.M. Molecular structure of human galactose mutarotase. J Biol Chem 2004;279(22):23431-23437. doi:10.1074/jbc.M402347200
^ Berg J.M., Tymoczko J.L., Gatto J.G., Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
^ Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009.
^ Thoden J.B., Holden H.M. Dramatic differences in the binding of UDP-galactose and UDP-glucose to UDP-galactose 4-epimerase from Escherichia coli. Biochemistry 1998;37(33):11469-77. doi:10.1021/bi9808969
^ Thoden J.B., Wohlers T.M., Fridovich-Keil J.L., Holden H.M. Human UDP-galactose 4-epimerase. Accommodation of UDP-N-acetylglucosamine within the active site. J Biol Chem 2001;276(18):15131-6. doi:10.1074/jbc.M100220200
^ Helenius A., Aebi M. Intracellular functions of N-linked glycans. Science 2001;291(5512):2364-9. doi:10.1126/science.291.5512.2364
^ Lin Y., Sun X., Hou X., Qu B., Gao X., Li Q. Effects of glucose on lactose synthesis in mammary epithelial cells from dairy cow. BMC Vet Res 2016;12:81. doi:10.1186/s12917-016-0704-x
^ Sunehag A., Tigas S., Haymond M.W. Contribution of plasma galactose and glucose to milk lactose synthesis during galactose ingestion. J Clin Endocrinol Metab 2003;88(1):225-9. doi:10.1210/jc.2002-020768
^ Cardini C.E., Paladini A.C., Caputto R., Leloir L.F. Uridine diphosphate glucose: the coenzyme of the galactose-glucose phosphate isomerization. Nature 1950;165:191-192. doi:10.1038/165191a0
^ Los E., Ford G.A. Galactose-1-phosphate uridyltransferase deficiency. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441957/
^ Ramani P.K., Arya K. Galactokinase deficiency. [Updated 2023 Jul 31]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560683/
^ Fridovich-Keil J., Bean L., He M., et al. Epimerase deficiency galactosemia. 2011 Jan 25 [Updated 2021 Mar 4]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK51671/
^ Wada Y., Kikuchi A., Arai-Ichinoi N., Sakamoto O., Takezawa Y., Iwasawa S., Niihori T., Nyuzuki H., Nakajima Y., Ogawa E., Ishige M., Hirai H., Sasai H., Fujiki R., Shirota M., Funayama R., Yamamoto M., Ito T., Ohara O., Nakayama K., Aoki Y., Koshiba S., Fukao T., Kure S. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet Med 2019;21(6):1286-1294. doi:10.1038/s41436-018-0340-x
^ Succoio M., Sacchettini R., Rossi A., Parenti G., Ruoppolo M. Galactosemia: biochemistry, molecular genetics, newborn screening, and treatment. Biomolecules 2022;12(7):968. doi:10.3390/biom12070968
^ Pintor J. Sugars, the crystalline lens and the development of cataracts. Biochem Pharmacol 2012;1:4. doi:10.4172/2167-0501.1000e119
^ Tsakiris S., Marinou K., Schulpis K. H. The in vitro effects of galactose and its derivatives on rat brain Mg2+-ATPase activity. Pharmacol Toxicol 2022;91:254-257. doi:10.1034/j.1600-0773.2002.910506.x
^ Lai K., Elsas L.J., Wierenga K.J. Galactose toxicity in animals. IUBMB Life 2009;61(11):1063-74. doi:10.1002/iub.262
^ Keston A.S. Inhibition of action of lens mutarotase on glucose by cataractogenic sugars and corresponding polyols. Arch Biochem Biophys 1963;102(2):306-312. doi:10.1016/0003-9861(63)90184-X
In a solution, solvent molecules tend to move from a region of higher concentration to one of lower concentration. When two different solutions are separated by a semipermeable membrane, namely, a membrane that allows certain ions or molecules to pass, in this case the solvent molecules, a net flow of solvent molecules from the side with higher concentration to the side with lower concentration will occur.[1] This net flow through the semipermeable membrane produces a pressure called osmotic pressure, denoted as Π, which can be defined as the force that must be applied to prevent the movement of solvent molecules through a semipermeable membrane.[2][3]
Osmotic pressure, along with boiling point elevation, freezing point depression, and vapor pressure lowering, is one of the four colligative properties of solutions. These are properties that do not depend on the chemical nature of the solute particles, namely, ions, molecules, or supramolecular structures, but only on the number of solute particles present in the solution.[4] For a solution containing n solutes, the equation that describes osmotic pressure is the sum of the contributions of each solute:
Π = RT(i1c1 + i2c2 + … + incn)
This equation is known as the van ’t Hoff equation, and it relates osmotic pressure (Π) to the following variables:
The van ’t Hoff factor is a measure of the degree of dissociation of solutes in solution. It is described by the equation:
i = 1 + α(n-1)
where:
α is the degree of dissociation of the solute, defined as the ratio between the number of moles of solute that have dissociated and the initial number of moles. Its value ranges from 0, for substances that do not ionize or dissociate in solution, to 1, for substances that completely dissociate or ionize;
n is the number of ions formed from the dissociation of the solute moleculeis the number of ions formed per molecule of solute upon dissociation.[7]
Non-ionizable compounds, such as glucose, glycogen, or starch, have n = 1 and i = 1. In contrast, compounds that fully dissociate, like strong acids, strong bases, or salts, exhibit a van ’t Hoff factor greater than one, as their degree of dissociation is 1 and n is at least 2. A typical example can be found in dilute solutions.
In the first two cases, i = 2, while for calcium chloride, i = 3.
Finally, for substances that do not fully dissociate, such as weak acids and weak bases, i is not an integer.
The product of the van ’t Hoff factor and the molar concentration of solute particles (i·c) is called the osmolarity of the solution, namely, the concentration of osmotically active solute particles per liter of solution.[7]
Osmotic pressure, osmosis, and plasma membranes
Osmosis can be defined as the net movement of solvent molecules through a semipermeable membrane, driven by osmotic pressure differences across the membrane, in an attempt to equalize solute concentrations on both sides.[8]
In biological systems, water acts as the solvent, and plasma membranes serve as the semipermeable membranes. Plasma membranes permit the passage of water molecules through protein channels known as aquaporins, as well as small non-polar molecules that diffuse rapidly across the membrane. However, they are impermeable to ions and macromolecules.[9]
Inside the cell, there are macromolecules such as nucleic acids, proteins, glycogen, and supramolecular aggregates, for example, multienzyme complexes, as well as ions, often present at higher concentrations than in the extracellular environment. This difference in solute concentration causes osmotic pressure to drive water from the outside to the inside of the cell.[10]
If this net influx of water is not counterbalanced, the cell swells, and the plasma membrane becomes distended until the cell bursts, a process known as osmotic lysis. Under physiological conditions, this does not occur because, over the course of evolution, several mechanisms have developed to counteract, or in some cases exploit, these osmotic forces.[7] Two of the main strategies are energy-dependent ion pumps, and in plants, bacteria, and fungi, the cell wall.
Energy dependent ion pumps
Ion pumps lower, at the expense of ATP, the intracellular concentration of specific ions relative to their concentration in the extracellular environment, thereby establishing an unequal distribution of ions across the plasma membrane, an ion gradient. In doing so, the cell counterbalances the osmotic forces created by the ions and macromolecules trapped within it.
A classic example of an energy-dependent ion pump is the Na⁺/K⁺-ATPase, which actively reduces the intracellular concentration of Na⁺ compared to the outside.[8]
Cell wall
Plant cells are surrounded by an extracellular matrix known as the cell wall. This structure, being non-expandable and tightly associated with the plasma membrane, enables cells to resist the osmotic forces that would otherwise cause swelling and eventually lysis.[8]
How does it work?
In mature plant cells, vacuoles are the largest organelles, occupying approximately 80% of the total cell volume. These vacuoles accumulate large amounts of solutes, mostly organic and inorganic acids, that osmotically draw water into the cell, causing the vacuoles to swell.
As a result, the tonoplast (the membrane surrounding the vacuole) pushes the plasma membrane outward against the rigid cell wall. The cell wall mechanically resists this force, preventing osmotic lysis.[11]
This internal pressure is known as turgor pressure, which can reach values of up to 2 MPa (approximately 20 atmospheres), about ten times the air pressure in a car tire.[12] Turgor pressure is crucial for the rigidity of non-woody plant tissues and plays a role in:
plant growth;
the wilting of vegetables (due to turgor loss);
plant movements, such as:
circadian movements of leaves;
the snapping shut of Dionaea muscipula (Venus flytrap),
the folding of sensitive plants like Mimosa pudica.[11]
Similarly, in bacteria and fungi, the plasma membrane is surrounded by a cell wall that stably contains internal pressure, thereby preventing osmotic lysis.[12]
Isotonic, hypotonic, and hypertonic solutions
By comparing the osmotic pressure of two solutions separated by a semipermeable membrane, three types of relationships can be defined, as described below.
Isotonic solutions have the same osmotic pressure.
If the solutions have different osmotic pressures, the one with the higher osmotic pressure is termed hypertonic relative to the other.
Conversely, the one with the lower osmotic pressure is termed hypotonic.[1]
In biological systems, the cytosol is typically used as the reference solution. When a cell is placed in a:
Isotonic solution: no net movement of water occurs across the plasma membrane.
Hypertonic solution: water flows out of the cell, leading to cell shrinkage.
Hypotonic solution: water flows into the cell, causing it to swell and potentially burst, this process is known as osmotic lysis.
Effects of osmotic pressure on cells
Solution type
Relative osmotic oressure
Water movement
Effect on the Cell
Isotonic
Equal to cytosol
No net movement
Cell remains stable
Hypertonic
Higher than cytosol
Water flows out of the cell
Cell shrinks (crenation)
Hypotonic
Lower than cytosol
Water flows into the cell
Cell swells and may burst (lysis)
In addition to ion pumps and the cell wall, multicellular organisms have evolved another strategy to counter osmotic forces: surrounding cells with a solution that is isotonic, or nearly isotonic, with respect to the cytosol.[13]
An example is plasma, the liquid component of blood, which, due to the presence of salts and proteins (primarily albumin in humans), has an osmolarity similar to that of the cytosol. This isotonic environment helps prevent excessive water movement into or out of cells.[14]
Osmotic pressure, starch and glycogen
Living organisms store glucose in polymeric forms, glycogen in animals, fungi, and bacteria, and starch in photoautotrophs, rather than as free glucose. This strategy prevents the osmotic pressure exerted by stored carbohydrates from becoming excessively high.
Since osmotic pressure, like the other colligative properties, depends solely on the number of solute particles rather than their mass or size, storing millions of glucose units as a much smaller number of polysaccharide molecules significantly reduces the resulting osmotic pressure.[4]
Here are some illustrative examples
One gram of a polysaccharide (e.g., glycogen or starch) composed of ~1,000 glucose units exerts far less osmotic pressure than just one milligram of free glucose, because the polysaccharide counts as a single solute particle per macromolecule.[15]
In hepatocytes, if all glucose currently stored as glycogen were instead present in its free form, the intracellular glucose concentration would rise to about 0.4 M, compared to the actual polysaccharide concentration of roughly 0.04 μM. This would create a steep osmotic gradient, causing a net inflow of water and potentially leading to osmotic lysis.[7]
Even if cell lysis were avoided, glucose transport into the cell would become energetically unfavorable. In humans, normal blood glucose levels range from 3.33 to 5.56 mmol/L (60–100 mg/dL). If free glucose reached intracellular concentrations of 0.4 M, this would be 72 to 120 times higher than blood glucose levels, requiring significant active transport and energy expenditure to maintain.[16]
Effect of glucose gtorage form on osmotic pressure
Storage form
Molecular units
Approximate concentration
Osmotic effect
Cellular consequences
Free Glucose
~1,000 individual molecules
~0.4 M
Very high osmotic pressure
Water influx → Cell swelling → Risk of lysis
Glycogen/Starch
1 macromolecule (polymer of 1,000 glucose units)
~0.04 μM
Negligible osmotic pressure
Stable osmotic conditions
Osmotic diarrhea
Osmotic diarrhea occurs when diseases lead to the accumulation of non-absorbed, osmotically active solutes in the distal small intestine and colon.[17]
Possible causes include bacterial infections, pancreatic diseases, and celiac disease, an autoimmune enteropathy triggered by gluten intake in genetically predisposed individuals, as well as congenital deficiencies in brush border disaccharidases, such as in lactose intolerance. In these conditions, incomplete carbohydrate digestion may occur due to deficiencies in alpha-amylase and/or one or more disaccharidases.[18] As a result, unabsorbed osmotically active solutes reach the colon, where they can also be fermented by the gut microbiota, part of the human microbiota, producing excess gas (such as hydrogen, carbon dioxide, and methane) and short-chain fatty acids, mainly butyric acid, acetic acid and propionic acid. This leads to a condition known as osmotic-fermentative diarrhea.[19]
Osmotic diarrhea can also result from the use of osmotic laxatives, such as polyethylene glycol (PEG) or magnesium sulfate. Osmotically active solutes from incomplete digestion and osmotic laxatives increase intraluminal osmotic pressure and inhibit the normal absorption of water and electrolytes, leading to looser stools and increased intestinal motility.[20]
Osmotic pressure, galactosemia, and cataracts
Galactose, along with glucose and fructose, is one of the monosaccharides absorbed in the intestine.[21]
Galactose is metabolized primarily in the liver, and to a lesser extent in other organs and tissues. Through the Leloir pathway, it is converted into UDP-glucose and UDP-galactose, which can then be used for both anabolic and catabolic processes.[22]
Mutations in the genes encoding enzymes of the Leloir pathway lead to the accumulation of galactose and cause galactosemia, a genetic metabolic disorder whose only treatment is a galactose-restricted diet.[23] The accumulated galactose is redirected into alternative metabolic pathways, resulting in the production of galactose-related compounds such as galactitol and galactonate.[24]
One of the symptoms of galactosemia is cataracts, and the synthesis and accumulation of galactitol in the lens of the eye appears to be a contributing factor. Why? Galactitol is a poorly metabolized polyol. Due to its low lipophilicity, it does not readily diffuse across cell membranes and tends to accumulate inside the cell. Being osmotically active, its intracellular buildup increases osmotic pressure, leading to a net influx of water. This osmotic effect is believed to be one of the mechanisms through which galactitol contributes to the development of galactosemic cataracts.[25]
References
^ ab Zheng S., Li Y., Shao Y., Li L., Song F. Osmotic pressure and its biological implications. Int J Mol Sci 2024;25(6):3310. doi:10.3390/ijms25063310
^ Bowler M.G. The physics of osmotic pressure. Eur J Phys 2017;38:055102. doi:10.1088/1361-6404/aa7fd3
^ Osmotic pressure, in IUPAC Compendium of Chemical Terminology, 5th ed. International Union of Pure and Applied Chemistry; 2025. Online version 5.0.0, 2025. doi:10.1351/goldbook.O04344
^ ab Hammel H.T. Colligative properties of a solution. Science 1976;192(4241):748-56. doi:10.1126/science.1265478
^ van’t Hoff J.H. The role of osmotic pressure in the analogy between solutions and gases. J Membr Sci 1995;100:39-44. doi:10.1016/0376-7388(94)00232-N
^ Hoff J.H.v.t. Die rolle des osmotischen druckes in der analogie zwischen lösungen und gasen. Z Phys Chem 1887;1U:481-508. doi:10.1515/zpch-1887-0151
^ abcd Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ abc Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002.
^ Agre P., Preston G.M., Smith B.L., Jung J.S., Raina S., Moon C., Guggino W.B., Nielsen S. Aquaporin CHIP: the archetypal molecular water channel. Am J Physiol 1993;265(4 Pt 2):F463-76. doi:10.1152/ajprenal.1993.265.4.F463
^ Phillips R., Kondev J., Theriot J., Garcia H.G. Physical Biology of the Cell. 2nd Edition. Garland Science. 2012. doi:10.1201/9781134111589
^ ab Heldt H-W. Plant biochemistry – 3th Edition. Elsevier Academic Press, 2005.
^ ab Beauzamy L., Nakayama N., and Boudaoud A. Flowers under pressure: ins and outs of turgor regulation in development. Ann Bot 2014;114(7):1517-1533. doi:10.1093/aob/mcu187
^ Alberts B., Heald B., Johnson A.D., Morgan D., Raff M., Roberts K., Walter P. Molecular Biology of the Cell. 7th Edition. Garland Science, 2022.
^ Brinkman J.E., Dorius B., Sharma S. Physiology, Body fluids. [Updated 2023 Jan 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482447/
^ Sadava D.E., Hillis D., Craig Heller H., Berenbaum M.R. Life: the science of biology. 10th Edition. Sinauer Associates. 2014.
^ Nakrani M.N., Wineland R.H., Anjum F. Physiology, Glucose Metabolism. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560599/
^ Bashir A., Sizar O. Laxatives. [Updated 2024 Jan 30]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537246/
^ Wright E.M., Martín M.G., Turk E. Intestinal absorption in health and disease–sugars. Best Pract Res Clin Gastroenterol 2003;17(6):943-56. doi:10.1016/s1521-6918(03)00107-0
Multifunctional enzymes are proteins in which two or more enzymatic activities, catalyzing consecutive steps of a metabolic pathway, are located on the same polypeptide chain. They are thought to have arisen through gene fusion events and, like multienzyme complexes, represent an evolutionary strategy to maximize catalytic efficiency. This configuration provides advantages that would not be possible if these enzymatic activities were present on separate proteins freely dissolved in the cytosol.[1][2]
What advantages do multifunctional enzymes provide?
Living organisms constantly fight against natural processes of decay which, if left unchecked, lead to increasing disorder and ultimately death.
At the molecular level, life is sustained by the extraordinary effectiveness of enzymes in accelerating chemical reactions and minimizing side reactions. The rate of ATP turnover in a mammalian cell gives an idea of how fast cellular metabolism operates: every 1–2 minutes, the entire ATP pool is turned over, hydrolyzed and then regenerated via phosphorylation. This corresponds to a turnover of about 107 ATP molecules per second, and, for the human body, to about 1 gram of ATP every minute.[3] Some enzymes have even achieved catalytic perfection, meaning they are so efficient that nearly every collision with their substrate leads to catalysis.[2]
One of the limiting factors of an enzymatic reaction is the frequency of collisions between enzymes and substrates. The simplest way to increase this frequency would be to increase their concentrations. However, given the vast number of different reactions taking place within the cell, this strategy is not feasible. In other words, there is a physical limit to the concentrations that substrates and enzymes can reach, substrate concentrations which are typically in the micromolar range, and even lower for enzymes.[3] Exceptions include the enzymes of glycolysis in muscle cells and erythrocytes, which can be present at concentrations around 0.1 mM or even higher.[4]
Metabolic channeling
One of the strategies favored by evolution to increase the rate of enzymatic reactions is the selection of molecular structures, such as multifunctional enzymes and multienzyme complexes, that optimize the spatial organization of enzymes within a metabolic pathway. This spatial optimization minimizes the distance that the product of reaction A must travel to reach the active site catalyzing reaction B in the sequence, and so on. This phenomenon is known as substrate channeling, or more broadly, metabolic channeling.[5] In some multifunctional enzymes and multienzyme complexes, channeling is achieved through the presence of intramolecular channels.[6]
Metabolic channeling enhances catalytic efficiency, and therefore the reaction rate, in several ways, outlined below.
It reduces the diffusion of substrates into the bulk solvent, thereby minimizing their dilution. This enables high local substrate concentrations, even when their overall cellular concentrations are low, increasing the frequency of enzyme-substrate collisions.
It reduces the time required for substrates to diffuse from one active site to the next.
It lowers the probability of side reactions.
It decreases the likelihood that unstable intermediates will degrade before reaching the next active site.[2][5]
Multifunctional enzymes also offer advantages in terms of the regulation of their synthesis. Since all enzymatic activities are encoded by a single gene, their expression can be coordinated efficiently.[7]
Finally, like multienzyme complexes, multifunctional enzymes enable coordinated control of catalytic activities. Often, the enzyme that catalyzes the committed step of the pathway is also responsible for the first reaction. This positioning prevents the synthesis of unnecessary products, which would occur if regulation took place downstream, thereby avoiding waste of energy and preventing the depletion of metabolites from other metabolic pathways.[2]
Examples of multifunctional enzymes
Like multienzyme complexes, multifunctional enzymes are also common and play key roles in many metabolic pathways, both anabolic and catabolic.
Below are several noteworthy examples.
Acetyl-CoA carboxylase
Acetyl-CoA carboxylase (ACC; EC 6.4.1.2) is a biotin-dependent carboxylase composed of two catalytic domains, biotin carboxylase (EC 6.3.4.14) and carboxyltransferase, as well as a biotin carboxyl carrier protein (BCCP). ACC catalyzes the synthesis of malonyl-CoA by carboxylating acetyl-CoA, in a reaction that represents the committed step in fatty acid biosynthesis.[7] The reaction occurs in two sequential steps.
In the first step, biotin carboxylase catalyzes the ATP-dependent carboxylation of the nitrogen atom in biotin, which acts as a carrier of carbon dioxide (CO2). The source of CO2 in this reaction is the bicarbonate ion.
In the second step, carboxyltransferase catalyzes the transfer of the carboxyl group from carboxybiotin to acetyl-CoA, forming malonyl-CoA.
Malonyl-CoA then serves as the donor of an activated two-carbon unit to fatty acid synthase (EC 2.3.1.85) during the elongation of fatty acid chains.[8]
In mammals and birds, acetyl-CoA carboxylase is a multifunctional enzyme, with biotin carboxylase, carboxyltransferase, and BCCP activities located on a single polypeptide chain.[9]
In contrast, in bacteria, ACC exists as a multienzyme complex composed of three distinct polypeptide chains: biotin carboxylase, carboxyltransferase, and BCCP.[10]
Both forms, the multifunctional enzyme and the multienzyme complex, are found in higher plants.[8]
Type I fatty acid synthase
Fatty acid synthase (FAS) catalyzes the synthesis of palmitic acid, using malonyl-CoA, the product of the reaction catalyzed by acetyl-CoA carboxylase, as a donor of two-carbon units.
There are two types of FAS.
In fungi and animals, FAS is a multifunctional enzyme known as Type I.
In animals, it exists as a homodimer, with each polypeptide chain containing all seven enzymatic activities, along with an acyl carrier protein (ACP) domain.
In yeast and fungi, FAS is composed of two distinct multifunctional subunits, designated α and β, which assemble into an α6β6 heterododecameric complex.
In most prokaryotes and in plants, FAS is classified as Type II. It is not a multifunctional enzyme, but rather a multienzyme complex composed of separate, individual enzymes and ACP.[11][12][13]
PRA-isomerase:IGP synthase
The biosynthesis of the amino acid tryptophan from chorismate involves several steps, summarized below.
In the first step, glutamine donates a nitrogen atom to the indole ring of chorismate, which is converted into anthranilate, while glutamine is simultaneously converted into glutamate. This reaction is catalyzed by anthranilate synthase (EC 4.1.3.27).
Anthranilate is then phosphoribosylated to form N-(5′-phosphoribosyl)-anthranilate (PRA), in a reaction catalyzed by anthranilate phosphoribosyltransferase (EC 2.4.2.18).
In this step, 5-phosphoribosyl-1-pyrophosphate (PRPP) acts as the donor of a 5-phosphoribosyl group.
In the next step, PRA is isomerized to enol-1-o-carboxyphenylamino-1-deoxyribulose phosphate (CdRP) by PRA isomerase (EC 5.3.1.24).
PRA and CdRP are an example of structural isomer.
CdRP is then converted into indole-3-glycerol phosphate (IGP) in a reaction catalyzed by indole-3-glycerol phosphate synthase (IGP synthase) (EC 4.1.1.48).
Finally, tryptophan synthase (EC 4.2.1.20) catalyzes the last two steps of the pathway: the hydrolysis of IGP to indole, and the condensation of indole with serine to form tryptophan.[11]
In E. coli, PRA isomerase and IGP synthase are located on a single polypeptide chain, making it a bifunctional enzyme.[14] In other microorganisms, such as Bacillus subtilis, Salmonella typhimurium, and Pseudomonas putida, the two catalytic activities are found on separate polypeptide chains.[2] In contrast, tryptophan synthase is a classic example of a multienzyme complex, and one of the best-characterized examples of metabolic channeling.[15][16]
Glutamine-PRPP amidotransferase
Glutamine-PRPP amidotransferase (GPATase; EC 2.4.2.14) catalyzes the first of ten steps in the de novo synthesis of purine nucleotides, specifically, the formation of 5-phosphoribosylamine via transfer of the amide nitrogen from glutamine to PRPP.
In this reaction, glutamine acts as a nitrogen donor.
The reaction proceeds in two steps, which occur at distinct active sites: one located at the N-terminal and the other at the C-terminal region of the enzyme.
In the first step, the N-terminal active site catalyzes the hydrolysis of glutamine, releasing glutamate and ammonia.
In the second step, catalyzed by the C-terminal active site (which has phosphoribosyltransferase activity), the ammonia is attached to the C-1 position of PRPP, forming 5-phosphoribosylamine.
This step also involves inversion of configuration at the ribose C-1 carbon, from α to β, thereby establishing the anomeric form of the future nucleotide.
There are three key regulatory points in the control of purine nucleotide biosynthesis. The reaction catalyzed by GPATase, being the first committed step of the pathway, represents the primary control point.[2]
As in the bacterial carbamoyl phosphate synthetase complex (EC 6.3.4.16), the active sites of this multifunctional enzyme are connected by an intramolecular channel. However, in this case the channel is shorter, approximately 20 Å in length, and is lined with conserved nonpolar (hydrophobic) amino acids.
Due to the lack of hydrogen-bonding groups, the channel does not impede the diffusion of ammonsia between active site.[17][18]
CAD
The de novo synthesis of pyrimidine nucleotides proceeds through a series of enzymatic reactions that, unlike the de novo synthesis of purine nucleotides, begins with the formation of the pyrimidine ring, which is then attached to ribose 5-phosphate. The first three steps of the pathway are catalyzed sequentially by the enzymes carbamoyl phosphate synthetase, aspartate transcarbamoylase (EC 2.1.3.2), and dihydroorotase (EC 3.5.2.3), a sequence that is conserved across all species.
In the first step, carbamoyl phosphate synthetase, which has two enzymatic activities, a glutamine-dependent amidotransferase and a synthase, catalyzes the formation of carbamoyl phosphate from glutamine, bicarbonate ion, and ATP.
In the second step, catalyzed by aspartate transcarbamoylase, carbamoyl phosphate reacts with aspartate to form N-carbamoyl aspartate. This is the committed step of the pathway.
In the final step, dihydroorotase catalyzes the removal of water from N-carbamoyl aspartate, leading to ring closure and the formation of L-dihydroorotate.[8]
In eukaryotes, particularly in mammals, Drosophila, and Dictyostelium (a genus of amoebae), these three enzymatic activities are located on a single polypeptide chain, encoded by a gene derived from a gene fusion event that occurred at least 100 million years ago. The resulting multifunctional enzyme, known by the acronym CAD, functions as a homomultimer composed of three or more subunits.[19]
In contrast, in prokaryotes, the three enzymes exist as separate proteins, and carbamoyl phosphate synthetase is a typical example of a multienzyme complex.[20]
In yeasts, dihydroorotase is located on a distinct polypeptide chain, separate from the other two enzymes.[21]
Interestingly, studies of enzymatic activity have revealed evidence of substrate channeling, particularly between the first two steps. This channeling appears to be more efficient in the yeast protein compared to the mammalian CAD enzyme.[22]
^ abcdef Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
^ ab Alberts B., Johnson A., Lewis J., Morgan D., Raff M., Roberts K., Walter P. Molecular Biology of the Cell. 6th Edition. Garland Science, Taylor & Francis Group, 2015.
^ Maughan D.W., Henkin J.A., Vigoreaux J.O. Concentrations of glycolytic enzymes and other cytosolic proteins in the diffusible fraction of a vertebrate muscle proteome. Mol Cell Proteomics 2005;4(10):1541-9. doi:10.1074/mcp.M500053-MCP200
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ Kondrat S., von Lieres E. Mechanisms and effects of substrate channelling in enzymatic cascades. Methods Mol Biol 2022;2487:27-50. doi:10.1007/978-1-0716-2269-8_3
^ ab Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002.
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ Wang Y., Yu W., Li S., Guo D., He J., Wang Y. Acetyl-CoA carboxylases and diseases. Front Oncol 2022;12:836058. doi:10.3389/fonc.2022.836058
^ Salie M.J., Thelen J.J. Regulation and structure of the heteromeric acetyl-CoA carboxylase. Biochim Biophys Acta 2016;1861(9 Pt B):1207-1213. doi:10.1016/j.bbalip.2016.04.004
^ Priestle J.P., Grütter M.G., White J.L., Vincent M.G., Kania M., Wilson E., Jardetzky T.S., Kirschner K., Jansonius J.N. Three-dimensional structure of the bifunctional enzyme N-(5′-phosphoribosyl)anthranilate isomerase-indole-3-glycerol-phosphate synthase from Escherichia coli. Proc Natl Acad Sci USA 1987;84(16):5690-4. doi:10.1073/pnas.84.16.5690
^ Hyde C.C., Ahmed S.A., Padlan E.A., Miles E.W., and Davies D.R. Three-dimensional structure of the tryptophan synthase multienzyme complex from Salmonella typhimurium. J Biol Chem 1988:263(33);17857-17871. doi:10.1016/S0021-9258(19)77913-7
^ Hyde C.C., Miles E.W. The tryptophan synthase multienzyme complex: exploring structure-function relationships with X-ray crystallography and mutagenesis. Nat Biotechnol 1990:8;27-32. doi:10.1038/nbt0190-27
^ Muchmore C.R.A., Krahn J.M, Smith J.L., Kim J.H., Zalkin H. Crystal structure of glutamine phosphoribosylpyrophosphate amidotransferase from Escherichia coli. Protein Sci 1998:7;39-51. doi:10.1002/pro.5560070104
^ Raushel F.M., Thoden J.B., Holden H.M. The amidotransferase family of enzymes: molecular machines for the production and delivery of ammonia. Biochemistry 1999;38(25):7891-9. doi:10.1021/bi990871p
^ Del Caño-Ochoa F., Moreno-Morcillo M., Ramón-Maiques S. CAD, a multienzymatic protein at the head of de novo pyrimidine biosynthesis. Subcell biochem 2019;93:505-538. doi:10.1007/978-3-030-28151-9_17
^ Washabaugh M.W., Collins K.D. Dihydroorotase from Escherichia coli. Purification and characterization. J Biol Chem 1984;259(5):3293-8.
^ Guan H.H., Huang Y.H., Lin E.S., Chen C.J., Huang C.Y. Structural analysis of Saccharomyces cerevisiae dihydroorotase reveals molecular insights into the tetramerization mechanism. Molecules 2021;26(23):7249. doi:10.3390/molecules26237249
^ Belkaïd M., Penverne B., Hervé G. In situ behavior of the pyrimidine pathway enzymes in Saccharomyces cerevisiae. 3. Catalytic and regulatory properties of carbamylphosphate synthetase: channeling of carbamylphosphate to aspartate transcarbamylase. Arch Biochem Biophys 1988;262(1):171-80. doi:10.1016/0003-9861(88)90179-8
Multienzyme complexes are discrete and stable structures composed of noncovalently associated enzymes that catalyze two or more sequential steps of a metabolic pathway.[1]
They can be considered a step forward in the evolution of catalytic efficiency, as they provide advantages that individual enzymes, even those that have achieved catalytic perfection, would not have on their own.[2]
During evolution, some enzymes have reached a state of virtual catalytic perfection, meaning that nearly every collision with their substrate results in catalysis. Below are several noteworthy examples.
Fumarase (EC 4.2.1.2) catalyzes the seventh reaction of the citric acid cycle, the reversible hydration/dehydration of fumarate’s double bond to form malate.
Acetylcholinesterase (EC 3.1.1.7) catalyzes the hydrolysis of acetylcholine, a neurotransmitter, into choline and acetic acid, which then dissociates to form a hydrogen ion and acetate.
Superoxide dismutase (EC 1.15.1.1) catalyzes the conversion and inactivation of the highly reactive superoxide radical (O2.-) into hydrogen peroxide (H2O2)and water.
Catalase (EC 1.11.1.6) catalyzes the degradation of H2O2 into water and oxygen.[2]
The rate of an enzymatic reaction is partly determined by how often enzymes and substrates collide. A simple way to increase this rate would be to increase enzyme and substrate concentrations. However, within a cell, their concentrations cannot be very high due to the large number of different reactions occurring simultaneously. In fact, most reactants are present in micromolar concentrations (10-6 M), while most enzymes are found at even lower concentrations.[3]
How metabolic channeling optimizes biochemical reactions
To overcome these limitations, evolution has taken different routes to enhance reaction rates. One such route is the optimization of enzyme spatial organization through the formation of multienzyme complexes and multifunctional enzymes. These structures minimize the distance that a reaction product must travel to reach the next active site, which is located close by.[1]
In other words, this leads to substrate (or metabolic) channeling, which can also occur through intramolecular channels connecting active sites.[4]
Metabolic channeling increases not just reaction rate but overall catalytic efficiency in several ways:
minimizing diffusion of substrates and products into bulk solvent, which prevents dilution and maintains high local concentrations, even when overall intracellular concentrations are low;
reducing the time required for substrates to diffuse between successive active sites;
lowering the probability of side reactions;
protecting chemically labile intermediates from degradation by the solvent.[2][4]
Another metabolic advantage, similar to multifunctional enzymes, is that multienzyme complexes allow coordinated regulation of the catalytic activity of their constituent enzymes. Since the enzyme catalyzing the first reaction in a pathway is often the regulatory enzyme, this prevents:
the synthesis of unneeded intermediates that would occur if regulation happened downstream;
the removal of metabolites from other pathways, as well as energy waste.[2]
Examples of multienzyme complexes
As noted above, especially in eukaryotes, multienzyme complexes, like multifunctional enzymes, are common in both anabolic and catabolic pathways, whereas few enzymes diffuse freely in solution. Two examples of multienzyme complexes include the tryptophan synthase complex (EC 4.2.1.20), whose tunnel was the first to be discovered, and bacterial carbamoyl phosphate synthase complex (EC 6.3.4.16).[5][6][7]
2-Ketoacid dehydrogenase family
A classic case of multienzyme organization is the 2-ketoacid (or 2-oxoacid) dehydrogenase family, which includes:
α-ketoglutarate dehydrogenase complex, also called 2-oxoglutarate dehydrogenase (OGDH).[1][8]
α-Ketoglutarate Dehydrogenase Complex
These multienzyme complexes are structurally and functionally similar. PDC, for instance, consists of multiple copies of three enzymes:
pyruvate dehydrogenase (E1; EC 1.2.4.1);
dihydrolipoyl transacetylase (E2; EC 2.3.1.12);
dihydrolipoyl dehydrogenase (E3; EC 1.8.1.4).
The same E1–E2–E3 architecture occurs in all three complexes, both in prokaryotes and eukaryotes. Within a given species, E3 is identical, while E1 and E2 are homologous.[9]
Though substrate-specific, these enzymes share the same cofactors: coenzyme A, NAD+, thiamine pyrophosphate, FAD, and lipoamide. For clarity, their components are labeled:
Note: eukaryotic PDC is the largest known multienzyme complex, larger than a ribosome, and can be visualized by electron microscopy.[10]
PDC links glycolysis to the citric acid cycle, catalyzing the irreversible oxidative decarboxylation of pyruvate to acetyl-CoA, with carbon dioxide (CO2) release and electron transfer to NAD+. Similarly, OGDH and BCKDH catalyze oxidative decarboxylations of α-ketoglutarate (to succinyl-CoA) and branched-chain α-keto acids (from valine, leucine, and isoleucine), involving:
CO2 release;
acyl group transfer to coenzyme A (forming acyl-CoA derivatives);
NAD+ reduction to NADH.
The striking similarities in structure, cofactors, and mechanisms point to a common evolutionary origin.[10]
Tryptophan synthase complex
The tryptophan synthase complex is one of the best-studied examples of substrate channeling.[4] It is present in bacteria and plants, but not in animals.[11] In bacteria, it is composed of two α and two β subunits that associate into αβ dimers, which represent the functional units of the complex. These dimers further assemble into an αββα tetramer.[5][6]
The complex catalyzes the final two steps of tryptophan biosynthesis. In the first step, indole-3-glycerol phosphate undergoes an aldol cleavage, catalyzed by a lyase (EC 4.1.2.8) located on the α subunits, producing indole and glyceraldehyde 3-phosphate. Indole then reaches the active site of the β subunit through an approximately 30 Å-long hydrophobic tunnel that, within each αβ dimer, connects the two active sites. In the second step, in the presence of pyridoxal 5-phosphate, indole condenses with serine to form tryptophan.[4][12]
Acetyl-CoA carboxylase
Acetyl-CoA carboxylase (ACC; EC 6.4.1.2), a member of the biotin-dependent carboxylase family, catalyzes the committed step of fatty acid synthesis: the carboxylation of acetyl-CoA to malonyl-CoA. Malonyl-CoA, in turn, serves as a donor of two-carbon units for the elongation process leading to the synthesis of palmitic acid, catalyzed by fatty acid synthase (EC 2.3.1.85).[13]
In bacteria, ACC is a multienzyme complex composed of two enzymes, biotin carboxylase (EC 6.3.4.14) and carboxytransferase, plus a biotin carboxyl-carrier protein (BCCP).[13]
Conversely, in mammals and birds, it is a multifunctional enzyme, with both enzymatic activities and BCCP present on the same polypeptide chain.[8]
In higher plants, both forms are present.[14]
Carbamoyl phosphate synthetase complex
Another well-characterized example of substrate channeling is the bacterial carbamoyl phosphate synthetase complex, which catalyzes the synthesis of carbamoyl phosphate, required for pyrimidine and arginine synthesis. This multienzyme complex contains an approximately 100 Å-long tunnel that connects its three active sites.
The first active site catalyzes the release of the amide nitrogen from glutamine as an ammonium ion, which enters the tunnel and reaches the second active site, where it is combined with bicarbonate, at the expense of ATP, to yield carbamate. In the last active site, carbamate is phosphorylated to form carbamoyl phosphate.[7][15]
References
^ abc Michal G., Schomburg D. Biochemical pathways. An atlas of biochemistry and molecular biology. 2nd Edition. John Wiley J. & Sons, Inc. 2012.
^ abcde Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
^ Alberts B., Johnson A., Lewis J., Morgan D., Raff M., Roberts K., Walter P. Molecular biology of the cell. 6th Edition. Garland Science, Taylor & Francis Group, 2015.
^ abcd Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ ab Hyde C.C., Ahmed S.A., Padlan E.A., Miles E.W., and Davies D.R. Three-dimensional structure of the tryptophan synthase multienzyme complex from Salmonella typhimurium. J Biol Chem 1988;263(33):17857-17871. doi:10.1016/S0021-9258(19)77913-7
^ ab Hyde C.C., Miles E.W. The tryptophan synthase multienzyme complex: exploring structure-function relationships with X-ray crystallography and mutagenesis. Nat Biotechnol 1990:8;27-32. doi:10.1038/nbt0190-27
^ ab Thoden J.B., Holden H.M., Wesenberg G., Raushel F.M., Rayment I. Structure of carbamoyl phosphate synthetase: a journey of 96 A from substrate to product. Biochemistry 1997;36(21):6305-16. doi:10.1021/bi970503q
^ Zhou Z.H., McCarthy D.B., O’Connor C.M., Reed L.J., and J.K. Stoops. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA 2001;98(26):14802-14807. doi:10.1073/pnas.011597698
^ ab Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ Xie G., Forst C., Bonner C., Jensen R.A. Significance of two distinct types of tryptophan synthase beta chain in Bacteria, Archaea and higher plants. Genome Biol 2002;3(1):RESEARCH0004. doi:10.1186/gb-2001-3-1-research0004
^ Hilario E., Caulkins B.G., Huang Y-M. M., You W., Chang C-E.A., Mueller L.J., Dunn M.F., and Fan L. Visualizing the tunnel in tryptophan synthase with crystallography: insights into a selective filter for accommodating indole and rejecting water. Biochim Biophys Acta 2016;1864(3):268-279. doi:10.1016/j.bbapap.2015.12.006
^ ab Cronan J.E. Jr, Waldrop G.L. Multi-subunit acetyl-CoA carboxylases. Prog Lipid Res 2002;41(5):407-35. doi:10.1016/s0163-7827(02)00007-3
^ Sasaki Y., Nagano Y. Plant acetyl-CoA carboxylase: structure, biosynthesis, regulation, and gene manipulation for plant breeding. Biosci Biotechnol Biochem 2004;68(6):1175-84. doi:10.1271/bbb.68.1175
^ Holden H.M., Thoden J.B., Raushel F.M. Carbamoyl phosphate synthetase: an amazing biochemical odyssey from substrate to product. Cell Mol Life Sci 1999;56(5-6):507-22. doi:10.1007/s000180050448
The pyruvate dehydrogenase complex (PDC) is a mitochondrial multienzyme complex comprising three distinct enzymes:
pyruvate dehydrogenase (E1; EC 1.2.4.1);
dihydrolipoyl transacetylase (E2; EC 2.3.1.12);
dihydrolipoyl dehydrogenase (E3; EC 1.8.1.4).
Subunit stoichiometry and complex size vary among species, with molecular masses ranging from ≈ 4 × 106 to 1 × 107 daltons.[1]
In addition to its core enzymes, the PDC contains several accessory components. These include five essential coenzymes required for catalysis and, in plants, fungi, and in animals such as birds and mammals, two regulatory enzymes: a pyruvate dehydrogenase kinase (EC 2.7.11.2) and a pyruvate dehydrogenase phosphatase (EC 3.1.3.43).
In eukaryotes, the complex also includes an E3-binding protein (E3BP), which contributes to the structural organization of the complex.[2]
The pyruvate dehydrogenase complex catalyzes, through five sequential reactions, the oxidative decarboxylation of pyruvate to acetyl-coenzyme A (acetyl-CoA), producing one molecule of carbon dioxide (CO2) and reducing NAD+ through the transfer of two electrons.[3]
PDC Reaction
The overall reaction is essentially irreversible under physiological conditions (ΔG°′ ≈ −8.0 kcal/mol; −33.4 kJ/mol). During catalysis, the enzymes act in a coordinated manner, with reaction intermediates remaining bound to the complex, enabling efficient substrate channeling and rapid turnover.[4][5]
The Five Reactions Catalyzed by the PDC
Importantly, the PDC catalyzes the same reactions through highly conserved mechanisms in all organisms.[6]
Summary: Key Points
Function: catalyzes the irreversible oxidative decarboxylation of pyruvate to acetyl-CoA.
Structure: mitochondrial multienzyme complex (E1-E2-E3) requiring five cofactors and two regulatory enzymes.
Mechanism and Regulation: utilizes substrate channeling and is tightly controlled by allosteric inhibition and covalent modification.
Metabolic Role: acts as a crucial gatekeeper for carbohydrate metabolism, linking glycolysis to the citric acid cycle.
Five coenzymes are involved in the reactions catalyzed by the pyruvate dehydrogenase complex. They are thiamine pyrophosphate (TPP), flavin adenine dinucleotide (FAD), coenzyme A (CoA), nicotinamide adenine dinucleotide (NAD), and lipoic acid.[1]
Thiamine pyrophosphate
Thiamine pyrophosphate is the active form of thiamine, or vitamin B1. TPP is the coenzyme of pyruvate dehydrogenase, to which it is tightly bound through noncovalent interactions. It is involved in the transfer of hydroxyethyl, or “activated aldehyde,” groups.[2]
Flavin adenine dinucleotide
Flavin adenine dinucleotide is one of the active forms of riboflavin, or vitamin B2; the other is flavin mononucleotide (FMN).
Reduced and Oxidized Forms of Flavin Adenine Dinucleotide
FAD is the coenzyme of dihydrolipoyl dehydrogenase, to which it is tightly bound. Like NAD, it participates in electron transfer, specifically two electrons and two protons (2H+ + 2e−).[5]
Coenzyme A
Coenzyme A consists of a β-mercaptoethylamine group connected to pantothenic acid, or vitamin B5, through an amide linkage, which is in turn bonded to a 3′-phosphoadenosine moiety through a pyrophosphate bridge.
CoA participates in the reaction catalyzed by dihydrolipoyl transacetylase and acts as a carrier of acyl groups.[7]
Coenzyme A and Acetyl-CoA
The β-mercaptoethylamine moiety terminates with a sulfhydryl group (–SH), a reactive thiol crucial to the role played by the coenzyme because acyl groups bonded to it through a thioester linkage have a high standard free energy of hydrolysis. This gives acyl groups a high transfer potential, equal to −7.5 kcal/mol (−31.5 kJ/mol), slightly more exergonic [+0.2 kcal/mol (+1 kJ/mol)] than the hydrolysis of ATP to ADP and Pi.
Therefore, thioesters have a high acyl-group transfer potential and can donate the acyl group to a variety of molecules, that is, the acyl group can be considered an activated group ready for transfer. It can also be stated that formation of the thioester bond conserves part of the free energy released during the oxidation of metabolic fuels.
Note that coenzyme A is also abbreviated as CoA-SH to emphasize the role of the thiol group.[4]
Note: in a thioester bond, a sulfur atom occupies the position where an oxygen atom is found in an ester bond.[8]
Ester and Thioester Bonds
Nicotinamide adenine dinucleotide
Nicotinamide adenine dinucleotide (NAD+) can be synthesized from tryptophan, an essential amino acid, or from niacin, or vitamin B3, or vitamin PP (from “Pellagra-Preventing”), the source of the nicotinamide moiety.
Reduced and Oxidized Forms of Nicotinamide Adenine Dinucleotide
NAD+ participates in the reaction catalyzed by dihydrolipoyl dehydrogenase and, like FAD, carries out electron (hydride) transfer.[4]
Lipoic acid
Unlike the other coenzymes of the pyruvate dehydrogenase complex, lipoic acid does not derive, directly or indirectly, from vitamins and/or essential amino acids, that is, from building blocks that cannot be synthesized de novo and must be supplied through the diet.
It is the coenzyme of dihydrolipoyl transacetylase, to which it is covalently bound through an amide linkage to the ε-amino group of a lysine residue to form lipoyl-lysine, or lipoamide, the so-called lipoyl-lysine arm. It couples electron transfer with acyl-group transfer.[9]
Lipoamide or Lipoyl-lysine
Lipoic acid has two thiol groups that can undergo reversible intramolecular oxidation to form a disulfide bridge (–S–S–), a reaction analogous to that occurring between two cysteine residues (Cys).
Because the disulfide bridge (a cyclic disulfide) can undergo redox reactions, during the reactions catalyzed by the pyruvate dehydrogenase complex it is first reduced to dihydrolipoamide, a dithiol (the reduced form of the prosthetic group), and then reoxidized to the cyclic form.[1]
Note
Many enzymes require small non-protein components, called cofactors, for their catalytic activity. Cofactors may be metal ions or small organic or organometallic molecules and are classified as coenzymes and prosthetic groups.
A prosthetic group is a cofactor that binds tightly to an enzyme by noncovalent or covalent bonds, that is, it is permanently bound to the protein. TPP, FAD, and lipoic acid are considered prosthetic groups in the PDC due to their tight binding.
A coenzyme (e.g. CoA or NAD+) is a cofactor that is not permanently bound to the enzyme.[5]
Cellular location of the pyruvate dehydrogenase complex
In eukaryotes, the pyruvate dehydrogenase complex, like the enzymes of the citric acid cycle and fatty acid oxidation, is located in the mitochondrion, where it is associated with the surface of the inner membrane facing the matrix.
In prokaryotes, it is located in the cytosol.[6]
Functions of the pyruvate dehydrogenase complex
The main functions of the pyruvate dehydrogenase complex are the production of acetyl-CoA and NADH.
The acetyl group linked to coenzyme A, an activated acetate, can be:
oxidized to two carbon dioxide molecules via the reactions of the citric acid cycle, harvesting a portion of the potential energy in the form of ATP or GTP;
used for the synthesis of fatty acids, cholesterol, steroids, isoprenoids, ketone bodies, and acetylcholine.
It is therefore possible to state that, depending on the metabolic conditions and/or the cell type, the pyruvate dehydrogenase complex commits carbon intermediates from amino acid and glucose catabolism to:
the citric acid cycle, and thus to energy production, for example, in skeletal muscle under aerobic conditions, and always in cardiac muscle;
In aerobic organisms, NADH can be oxidized to NAD+ by transferring a hydride ion to Complex I of the mitochondrial electron transport chain, which in turn carries the two electrons to molecular oxygen (O2), allowing the production of 2.5 ATP molecules per pair of electrons.[1]
Note: in anaerobic organisms there are electron acceptors alternative to oxygen, such as sulfate or nitrate.[11]
Conceptually, the pyruvate dehydrogenase complex represents the bridge between glycolysis and the citric acid cycle. However, due to the irreversibility of the overall reaction catalyzed by the multienzyme complex, it acts as a one-way bridge: pyruvate can be decarboxylated and oxidized, and the remaining acetyl unit can be linked to CoA, but the reverse reaction, conversion of acetyl-CoA to pyruvate, is not possible.
The irreversibility of this reaction, together with the absence of alternative pathways, explains why acetyl-CoA, and therefore fatty acids, cannot be used as substrates for gluconeogenesis.[5]
Other sources of acetyl-CoA
Besides pyruvate, the acetyl group of acetyl-CoA can derive from the oxidation of fatty acids and the catabolism of many amino acids. However, regardless of its origin, acetyl-CoA represents an entry compound for new carbon units into the citric acid cycle. It may also be stated that the acetyl group of acetyl-CoA is the form in which most carbon enters the cycle.[10]
Mitochondrial pyruvate transport
In eukaryotes, glycolysis occurs in the cytosol, whereas all subsequent steps of aerobic metabolism, that is, the reactions catalyzed by the pyruvate dehydrogenase complex, the citric acid cycle, the electron transport chain, and oxidative phosphorylation, take place in the mitochondria.[12]
Similarly to most other metabolites and anions, the transport of pyruvate across the outer mitochondrial membrane is probably mediated by a relatively nonspecific, voltage-dependent anion channel. Conversely, its transport across the inner mitochondrial membrane occurs through a specific transporter composed of two proteins named MPC1 and MPC2 (Mitochondrial Pyruvate Carrier), which form a hetero-oligomeric complex in the membrane, catalyzing a pyruvate/H+ symport towards the matrix.[13]
Structure of the pyruvate dehydrogenase complex
Although the pyruvate dehydrogenase complex is composed of multiple copies of three different enzymes and catalyzes the same reactions through similar mechanisms in all organisms in which it is present, it nevertheless displays very different quaternary structures.[6]
Structure of the E. coli complex
The structure of the E. coli multienzyme complex, which has a mass of ≈ 4,600 kDa and a diameter of ≈ 300 Å, was the first to be characterized, thanks to the work of Lester Reed. In this complex, 24 units of dihydrolipoyl transacetylase form a structure with cubic symmetry; the enzymes, associated as trimers, are located at the corners of a cube. Dimers of pyruvate dehydrogenase are associated with the dihydrolipoyl transacetylase core at the center of each edge of the cube, for a total of 24 units. Finally, dimers of dihydrolipoyl dehydrogenase are located at the center of each of the six faces of the cube, for a total of 12 units. Note that the entire complex is composed of 60 units. A similar structure with cubic symmetry is also found in most other Gram-negative bacteria.[14]
Structure of the eukaryotic and Gram-positive bacterial complex
In some Gram-positive bacteria and in eukaryotes, the pyruvate dehydrogenase complex has a dodecahedral form, namely, that of a regular polyhedron with 20 vertices, 12 pentagonal faces, and 30 edges, with icosahedral symmetry (also called I symmetry). For example, the mitochondrial multienzyme complex is the largest known multienzyme complex, with a mass of ≈ 10,000 kDa (10 MDa) and a diameter of ≈ 500 Å, more than five times the size of a ribosome. Notably, it can be visualized by electron microscopy.[15][16]
The complex consists of a dodecahedral core, with a diameter of about 25 nm, formed, as in Gram-negative bacteria, by dihydrolipoyl transacetylase, but composed of 20 trimers of the enzyme (60 units) located at the vertices of the structure. The core is surrounded by 30 units of pyruvate dehydrogenase, one centered on each edge, and 12 units of dihydrolipoyl dehydrogenase, one centered on each face. The entire complex therefore contains 102 units.[2]
Additional subunits
The quaternary structure of the pyruvate dehydrogenase complex is further complicated, as mentioned previously, by the presence of three additional subunits: a pyruvate dehydrogenase kinase, a pyruvate dehydrogenase phosphatase, and the E3-binding protein (E3BP). The kinase and phosphatase are bound to the dihydrolipoyl transacetylase core.[4]
E3BP is bound to each of the 12 pentagonal faces and is therefore present in about 12 copies. It is required for the binding of dihydrolipoyl dehydrogenase to the dihydrolipoyl transacetylase core, as demonstrated by the fact that its partial proteolysis reduces the binding ability of the dehydrogenase.[17]
In the E3-binding protein, one can identify a C-terminal domain, which has no catalytic activity, and a lipoamide-containing domain, similar to that of dihydrolipoyl transacetylase, capable of accepting an acetyl group. However, removal of this domain does not reduce the catalytic activity of the multienzyme complex.[18]
Structure of pyruvate dehydrogenase
Pyruvate dehydrogenase in eukaryotes and some Gram-positive bacteria is composed of two different polypeptide chains, called α and β, which associate to form a 2-fold symmetric α2β2 heterotetramer. Conversely, in E. coli and other Gram-negative bacteria, the two subunits are fused into a single polypeptide chain, and the enzyme is therefore a homodimer.[6][19]
The enzyme contains two active sites.
In the heterotetrameric structure of Bacillus (Geobacillus) stearothermophilus pyruvate dehydrogenase, a Gram-positive bacterium, each thiamine pyrophosphate molecule binds between the N-terminal domains of an α and a β subunit, at the end of a funnel-shaped channel ≈ 21 Å deep that leads to the active site. Its reactive group, the thiazole ring, is positioned closest to the channel entrance. At this entrance, two conserved loops are present, both essential for the catalytic activity of the enzyme and for its regulation.[20]
Active site architecture and structure
X-ray analysis of the B. stearothermophilus enzyme bound to both TPP and the peripheral subunit-binding domain (PSBD) of dihydrolipoyl transacetylase, which interacts with the C-terminal domain of the β subunits, has revealed that, in addition to having a tightly packed heterotetrameric structure, the two active sites differ structurally, particularly in the arrangement of the two conserved loops.[16] Specifically, in one enzyme subunit, in the presence of activated thiamine pyrophosphate, the inner loop is ordered in such a way that it blocks the entrance to the active site, whereas in the other subunit, the corresponding loop is disordered and does not obstruct the entrance.[21]
This observation explains, from a structural standpoint, the differences in substrate-binding rates exhibited by the two active sites. A similar arrangement and asymmetry have been observed in all thiamine pyrophosphate–dependent enzymes whose structures have been determined.
In addition to TPP and a magnesium ion located in each of the two active sites, a third Mg2+ is positioned at the center of the tetramer, within a ≈ 20 Å deep solvent-filled tunnel that connects the two active sites. The tunnel is largely lined by ten conserved amino acid residues from all four subunits, specifically, six glutamate (Glu) and four aspartate (Asp) residues, along with other acidic residues around the TPP aminopyrimidine ring. Notably, there is a complete absence of basic residues to neutralize them. Similar tunnels have been found in all thiamine pyrophosphate-dependent enzymes with known crystal structures, whether dimeric or tetrameric, such as transketolase, an enzyme of the pentose phosphate pathway.[2]
Note: B. stearothermophilus belongs to the phylum Firmicutes and has recently been renamed Geobacillus stearothermophilus.
Role of the acidic tunnel
Mutagenesis experiments on B. stearothermophilus pyruvate dehydrogenase have shown that the tunnel plays a role in the catalytic mechanism.[22][23]
Changing some of the aforementioned acidic residues to neutral amino acids does not alter, compared with the wild-type enzyme:
the efficiency of incorporation of the modified enzyme into the multienzyme complex;
Nevertheless, the rate of decarboxylation is reduced by more than 70% compared with the wild-type enzyme. Likewise, once the multienzyme complex is assembled with the mutant pyruvate dehydrogenase, the overall PDC activity is reduced by more than 85% relative to the wild-type complex.[23]
How does this occur?
Because the distance between the substituted amino acids and the active sites is ≥ 7 Å, that is, the residues are remote from the active sites, they cannot directly influence catalytic activity. Therefore, the catalytic mechanism described below was proposed.[3]
Proton-wire mechanism and communication between the active sites
In the apoenzyme, thiamine pyrophosphate binds rapidly and tightly to the first active site, becomes activated, and the active site closes, thus protecting the zwitterionic thiazolium from the external environment.
Conversely, in the second active site TPP binds but is not activated, and the active site remains in an open conformation.[4]
In the first active site, pyruvate reacts with the thiazolium C-2. Thiamine pyrophosphate in the second active site, acting as a general acid, donates a proton to the first site. The result is decarboxylation in the first active site and activation of the coenzyme in the second site, which then closes.[3]
It should be noted that, whereas activation of the first thiamine pyrophosphate results from its binding to the active site, activation of the second coenzyme, and thus of the second active site, is coupled to the decarboxylation of pyruvate in the first active site. From another point of view: while one active site requires a general acid, the other requires a general base.[2]
Protons are required for catalytic activity, and their transport between the two active sites occurs through the acidic tunnel. They are reversibly shuttled along a chain of donor–acceptor groups provided by glutamate and aspartate residues and by the entrained water, which together act as a proton wire.
Thus, unlike many other enzymes in which communication between active sites occurs through conformational changes and subunit rearrangements, in pyruvate dehydrogenase and other TPP–dependent enzymes, the proton wire is the molecular basis for such communication.[22]
At this point, the holoenzyme has been formed, and the active sites are in dynamic equilibrium, each alternating between the dormant and activated states (flip-flop mechanism). This appears to be the state in which the enzyme exists in vivo at the start of each catalytic cycle.[19]
Implications of proton-wire-mediated asymmetry for catalysis
A consequence of this mechanism is that, as the catalytic cycles proceed, the two active sites remain out of phase with each other; when one active site requires a general acid, the other requires a general base, and vice versa.[22]
Finally, it should be noted that this mechanism allows the loops that close the active sites to switch in a coordinated fashion, thereby:
coordinating substrate uptake and product release;
explaining the asymmetry between the two active sites.[23]
Note: an apoenzyme is an enzyme lacking its cofactors. Conversely, a holoenzyme is the apoenzyme together with its cofactors. The apoenzyme is catalytically inactive, whereas the holoenzyme is catalytically active.[5]
Structure of dihydrolipoyl transacetylase
Three functionally distinct domains can be identified in the structure of dihydrolipoyl transacetylase: an N-terminal lipoyl domain, a peripheral subunit-binding domain, and a C-terminal catalytic (or acyltransferase) domain. These domains are connected by 20- to 40-residue linkers that are rich in alanine and proline, hydrophobic residues interspersed with charged ones. These linkers are highly flexible and largely extended, which allows the three domains to remain separated from each other.[25][26]
E2 Domains
Note: flexible linkers are also present in E3BP.
The lipoyl domain
The N-terminal lipoyl domain is composed of ≈ 80 amino acid residues and is so called because it binds lipoic acid. The number of these domains varies by species, ranging from one to three. For example, there is one domain in Enterococcus faecalis, two in mammals, and three in Azotobacter vinelandii and E. coli.[27][28]
The linkage between the ɛ-amino group of a lysine residue and lipoic acid results in a flexible arm, the lipoyl-lysine, which has a maximum extended length of ≈ 14 Å. When considering the length of the lipoyl-lysine arm and the highly flexible 20- to 40-residue linker (which can span greater than 140 Å distance between active sites), the resulting flexible tether is capable of swinging the lipoyl group between the active sites of pyruvate dehydrogenase and dihydrolipoyl dehydrogenase, as well as interacting with neighboring dihydrolipoyl transacetylases within the core.[2][29]
Notably, the number of these tethers is 3 × 24 = 72 in E. coli, whereas in mammals it is 2 × 60 = 120, based on the number of N-terminal domains and the number of dihydrolipoyl transacetylase units.
A single pyruvate dehydrogenase can therefore acetylate numerous dihydrolipoyl transacetylases, and one dihydrolipoyl dehydrogenase can reoxidize many dihydrolipoamide groups.
Moreover, the following also occur:
interchange of acetyl groups between the lipoyl groups of the dihydrolipoyl transacetylase core;
exchange of both acetyl groups and disulfides between the tethered arms.[25]
The peripheral subunit-binding domain
PSBD is composed of ≈ 35 amino acid residues arranged into a globular structure that binds both pyruvate dehydrogenase and dihydrolipoyl dehydrogenase; that is, it helps hold the multienzyme complex together.[30]
The C-terminal catalytic domain
The C-terminal catalytic domain, which contains the active site, consists of ≈ 250 amino acid residues arranged into a hollow, cage-like structure containing channels large enough to allow substrates and products to diffuse in and out. For example, CoA and lipoamide, the two substrates of dihydrolipoyl transacetylase, bind in their extended conformations at opposite ends of a channel located at the interface between each pair of subunits in each trimer.[26][31]
Structure of dihydrolipoyl dehydrogenase
The structure of dihydrolipoyl dehydrogenase has been deduced from studies of the enzyme in several microorganisms. It has a homodimeric structure, with each ≈ 470-residue polypeptide chain folded into four domains, from the N-terminal to the C-terminal end: a FAD-binding domain, a NAD+-binding domain, a central domain, and an interface domain. All domains participate in the formation of the active site.[32]
FAD is almost completely buried inside the protein because, unlike a thiol or NADH, it is easily oxidized and must therefore be protected from the surrounding solution. In fact, in the absence of the nicotinamide coenzyme, the phenolic side chain of a tyrosine residue (e.g., Tyr181 in the Gram-negative bacterium Pseudomonas putida) covers the NAD+-binding pocket so as to protect FADH2 from exposure to the solvent.[2]
Conversely, when NAD+ occupies the active site, the phenolic side chain of the this tyrosine residue becomes interposed between the nicotinamide ring and the flavin ring.[33]
In the oxidized form of the enzym, a redox-active disulfide bridge is present. It forms between two cysteine residues located in a highly conserved segment of the polypeptide chain, e.g., Cys43 and Cys48 in P. putida, and is positioned on the side of the flavin ring opposite the nicotinamide ring. This bridge connects consecutive turns of a distorted α-helix; without this distortion, the Cα atoms of the two cysteine residues would be too far apart to allow disulfide bond formation.[34]
Dihydrolipoyl dehydrogenase therefore has two electron acceptors: FAD and the redox-active disulfide bridge.
Note: the heterocyclic rings of NAD and FAD are parallel and in contact through van der Waals interactions; the sulfur atom of Cys48 is also in van der Waals contact with the flavin ring on the side opposite the NAD ring.[2]
Reaction of pyruvate dehydrogenase
In the reaction sequence catalyzed by the components of the pyruvate dehydrogenase complex, pyruvate dehydrogenase catalyzes the first two steps:
the decarboxylation of pyruvate to form CO2 and the hydroxyethyl-TPP intermediate;
the reductive acetylation of the lipoyl group of dihydrolipoyl transacetylase.
The first reaction is essentially identical to the reaction catalyzed by pyruvate decarboxylase (EC 4.1.1.1), which carries out a non-oxidative decarboxylation during glucose fermentation to ethanol. What differs is the fate of the hydroxyethyl group bound to thiamine pyrophosphate: in the reaction catalyzed by pyruvate dehydrogenase, this group is transferred to the next enzyme in the sequence, dihydrolipoyl transacetylase, whereas in the reaction catalyzed by pyruvate decarboxylase it is converted into acetaldehyde.[4]
Catalytic mechanism: the first stage
In thiamine pyrophosphate-dependent enzymes, the thiazolium ring is the active center, but only in its dipolar carbanion, or ylid, form, that is, a zwitterion (German for “hybrid ion”) with a positive charge on N-3 and a negative charge on C-2. Conversely, the positively charged thiazolium ring (positively charged nitrogen and no charge on C-2) can be considered the “dormant” or inactive form.[35]
The reaction begins with the nucleophilic attack by the C-2 carbanion on the carbonyl carbon of pyruvate, which has the oxidation state of an aldehyde, leading to the formation of a covalent bond between the coenzyme and pyruvate.
Then, cleavage of the C-1–C-2 bond of pyruvate occurs. This results in the release of the carboxyl group, namely C-1 as CO2, while the remaining carbon atoms, C-2 and C-3, remain bound to thiamine pyrophosphate as a hydroxyethyl group. Cleavage of the C-1–C-2 bond, and thus pyruvate decarboxylation, is favored because the negative charge on carbon C-2, which would otherwise be unstable, is stabilized by the positively charged N-3 of the thiazolium ring, an iminium nitrogen (C=N+). This electrophilic, electron-deficient structure acts as an electron sink, in which the carbanion electrons can be delocalized by resonance.[36]
At this point, the resonance-stabilized intermediate can be protonated to form hydroxyethyl–TPP.[5]
Note: this first reaction catalyzed by pyruvate dehydrogenase is the step at which the pyruvate dehydrogenase complex exerts its substrate specificity; furthermore, it is the slowest of the five reactions and thus rate-limiting for the overall process.[1]
Catalytic Mechanism of Pyruvate Dehydrogenase
Catalytic mechanism: the second stage
Pyruvate dehydrogenase then catalyzes the oxidation of the hydroxyethyl group to an acetyl group and its transfer to the lipoyl-lysyl arm of dihydrolipoyl transacetylase. The reaction begins with the formation of a carbanion on the hydroxyl-bearing carbon of hydroxyethyl–TPP, via removal of its proton by an enzyme base.[5]
The carbanion carries out a nucleophilic attack on the lipoamide disulfide, forming a high-energy acetyl-thioester bond with one of the two –SH groups. During this reaction, oxidation of the hydroxyethyl group to an acetyl group occurs simultaneously with reduction of the lipoamide disulfide bond: the two electrons removed from the hydroxyethyl group are used to reduce the disulfide. This reaction is therefore a reductive acetylation accompanied by regeneration of the active form of pyruvate dehydrogenase, namely the enzyme with thiazolium C-2 in its deprotonated, ylid (dipolar carbanion) form.[36]
Note that the energy derived from the oxidation of the hydroxyethyl group to an acetyl group drives the formation of the thioester bond between the acetyl group and lipoamide.[4]
Note: as previously mentioned, the lipoyl-lysyl arm, arranged in an extended conformation and able to access the active site of pyruvate dehydrogenase where TPP is bound, enables the transfer of the acetyl group, generated by oxidation of hydroxyethyl–TPP, to the lipoamide moiety. The swinging arm then delivers the acetyl group to the active site of dihydrolipoyl transacetylase for transfer to coenzyme A. It can subsequently move to the active site of dihydrolipoyl dehydrogenase.[2]
A deeper look at thiamine pyrophosphate
Thiamine pyrophosphate consists of three chemical moieties on which its chemistry and enzymology depend: a thiazolium ring, a 4-aminopyrimidine ring, and the diphosphate side chain.[37]
The diphosphate side chain anchors the cofactor to the enzyme through electrostatic interactions between its negatively charged phosphoryl groups and the positively charged Ca2+ and Mg2+ ions, which are themselves bound to highly conserved sequences, Gly-Asp-Gly-(GDG) and Gly-Asp-Gly-X26-Asn-Asn (GDG-X26-NN), respectively.[38]
The thiazolium ring plays the central role in catalysis because of its ability to form the C-2 carbanion, which acts as a nucleophilic center on the C-2 atom.[39]
Note: as mentioned previously, once bound to the enzyme, thiamine pyrophosphate positions itself in the active site so that the thiazolium ring lies near the entrance of the channel leading to the active site.[5]
The aminopyrimidine ring has a dual function:
it anchors the coenzyme, holding it in place;
it has a specific catalytic role, participating in acid–base catalysis, as demonstrated by studies with TPP analogs in which the three nitrogen atoms of the ring were substituted in turn. These studies showed that the N-1′ atom and the N-4′ amino group are essential, whereas the N-3′ atom is required to a lesser extent.[4]
How the dipolar carbanion of thiamine pyrophosphate is formed
Three tautomeric forms of the aminopyrimidine ring can be identified in the enzyme-bound coenzyme (before substrate entry):
the canonical 4′-aminopyrimidine tautomer;
the N-1 protonated form (the 4-aminopyrimidinium ion);
Evidence indicates that the 1′,4′-imino tautomer is the form that undergoes deprotonation before the substrate enters the active site. The C-2 proton of the thiazolium ring is “much more acidic than most =C–H groups in other molecules.” Its unusually high acidity, i.e., the ease with which the C-2 proton dissociates, is due to the presence of the quaternary nitrogen in the thiazolium ring, a positively charged atom capable of stabilizing the resulting carbanion by electrostatic attraction.[41]
In the deprotonation reaction, the amino group of the aminopyrimidine ring appears to play an essential role: it acts as a base and is ideally positioned to accept the proton. However, in the 4′-aminopyrimidine tautomer, one of its protons sterically clashes with the C-2 proton, and its pKa is too low to perform the deprotonation efficiently.[37]
A mechanism was therefore proposed in which the side chain of a conserved glutamate residue, for example, βGlu59 in Bacillus stearothermophilus or Glu51 in Saccharomyces uvarum (brewer’s yeast) pyruvate decarboxylase, donates a proton to the aminopyrimidine ring. This converts it to the 1′,4′-iminopyrimidine tautomer, which then accepts the C-2 proton and reverts to the canonical 4′-aminopyrimidine form, enabling formation of the carbanion.[2]
Note: formation of the C-2 carbanion occurs through an intramolecular proton transfer.
Deprotonation of thiamine pyrophosphate
Loss of the C-2 proton of the thiazolium ring converts it from a positively charged ring into a dipolar ion (zwitterion). This change in charge state induces a conformational change in one of the two conserved loops located at the entrance of the active site channel, specifically the inner loop, which in turn causes the channel to close to the surrounding aqueous environment. In this closed conformation, the thiazolium carbanion is protected from electrophiles.[42]
To summarize: deprotonation of thiamine pyrophosphate leads to closure of the active site and protection of the newly formed dipolar carbanion. Thus, TPP-dependent enzymes are active only in the closed conformation.
Conversely, in the other active site (since these enzymes typically have two active sites per dimer), thiamine pyrophosphate is not in the ylid form, the channel remains open, and that site is inactive.[5]
Reaction of dihydrolipoyl transacetylase
In the reaction sequence catalyzed by the components of the pyruvate dehydrogenase complex, dihydrolipoyl transacetylase catalyzes the third step: the transfer of the acetyl group from acetyl-dihydrolipoamide to CoA to form acetyl-CoA and dihydrolipoamide, the fully reduced dithiol form of lipoamide.
It should be noted that the acetyl group, initially linked by a thioester bond to one of the –SH groups of lipoamide, is subsequently transferred to the –SH group of coenzyme A, also via a thioester bond, hence the term transthioesterification.[4]
Catalytic mechanism
During the reaction, the sulfhydryl group of coenzyme A performs a nucleophilic attack on the carbonyl carbon of the acetyl group of acetyl-dihydrolipoamide (bound to dihydrolipoyl transacetylase). This forms a transient tetrahedral intermediate, which then decomposes to produce dihydrolipoamide–dihydrolipoyl transacetylase and acetyl-CoA.
Catalytic Mechanism of Dihydrolipoyl Transacetylase
As mentioned earlier, the mobility of the lipoyl-lysyl arm plays a central role in this mechanism.[43][44]
Reaction of dihydrolipoyl dehydrogenase
In the reaction sequence catalyzed by the pyruvate dehydrogenase complex, dihydrolipoyl dehydrogenase catalyzes the fourth and fifth steps. The enzyme mediates the electron transfers required to regenerate the disulfide bridge of the lipoyl group of dihydrolipoyl transacetylase, that is, to restore the oxidized form of the prosthetic group, thus completing the catalytic cycle of the transacetylase.[5]
The reaction follows a ping-pong catalytic mechanism: it proceeds through two successive half-reactions in which each of the two substrates, NAD+ and dihydrolipoamide, reacts in the absence of the other. During the first half-reaction, release of the first product and formation of an enzyme intermediate occur before the second substrate binds, and the enzyme undergoes a structural change. During the second half-reaction, release of the second product occurs, and the enzyme returns to its initial state, again through a conformational change.[2]
According to the ping-pong kinetic mechanism:
in the first half-reaction, dihydrolipoamide is oxidized to lipoamide;
in the second half-reaction, NAD+ is reduced to NADH.[4][45]
Catalytic mechanism: first half-reaction
Below, the reaction mechanism of P. putida dihydrolipoyl dehydrogenase is described.
In the first half-reaction, oxidized dihydrolipoyl dehydrogenase (E), i.e., the enzyme containing the Cys43–Cys48 disulfide, binds dihydrolipoamide (LH2) to form the enzyme–dihydrolipoamide complex (E–LH2). Subsequently, a sulfur atom of dihydrolipoamide performs a nucleophilic attack on the sulfur of Cys43, forming the lipoamide–Cys43 disulfide bridge (E–S–S–L), while the sulfur of Cys48 is released as a thiolate ion (S48−).[34]
The proton on the second thiol group of lipoamide is then abstracted by histidine (His) 451, which acts as a general acid–base catalyst, leading to the formation of a second thiolate ion, this time on the lipoamide (E–S–S–L–S⁻). This thiolate, through a nucleophilic attack, displaces the sulfur of Cys43, S43, aided by general acid catalysis from His451, which donates a proton to S43. The catalytic action of His451 is essential, as demonstrated by mutagenesis studies in which its substitution with a glutamine residue causes the enzyme to retain ≈ 0.4% of the wild-type catalytic activity.[18]
Then, the thiolate anion S48− interacts, through non-covalent contacts, with the flavin ring near the 4a position; that is, an electron pair of S48−, acting as electron donor, is partially transferred to the oxidized flavin ring, which acts as the electron acceptor. The resulting structure is called a charge-transfer complex.
Meanwhile, the phenolic side chain of Tyr181 continues to hinder access to the flavin ring, thereby protecting it from oxidation by O2.[2]
Dihydrolipoamide Oxidation via E3
To summarize, what occurs is an interchange reaction of disulfide bridges leading to the formation of the oxidized form of lipoamide, the first product, which is released, and the reduced form of dihydrolipoyl dehydrogenase.
Catalytic mechanism: second half-reaction
The second half-reaction involves the reduction of NAD+ to NADH + H+ by electron transfer from the reactive disulfide of the enzyme via FAD.
Oxidation of Reduced E3
It begins with the entry of NAD+ into the active site and its binding to form the E–H2–NAD+ complex. It should be noted that the entry of the coenzyme causes the phenolic side chain of Tyr181 to be pushed aside by the nicotinamide ring.
Following the collapse of the charge-transfer complex, a covalent bond is formed between flavin atom C-4a and S48, accompanied by the extraction of a proton from S43 by flavin atom N-5, yielding the corresponding thiolate anion (S43−).
S43− then carries out a nucleophilic attack on S48, leading to the formation of the redox-active disulfide bridge between Cys43 and Cys48, followed by the cleavage of the covalent bond between S48 and flavin atom C-4a, producing the reduced FADH− anion with a negative charge on atom N-1.
It should be noted that dihydrolipoyl dehydrogenase is now in the oxidized form (E).[34]
FADH− has a transient existence because it instantly transfers a hydride ion to nicotinamide atom C-4, which is juxtaposed to flavin atom N-5. This results in the formation of FAD and of the second product of the reaction, NADH, which is released.[1]
Functional implications and comparison with glutathione reductase
To summarize, what occurs is that the electrons removed from the hydroxyethyl group, which derives from pyruvate, pass via FAD to NAD+. The catalytic cycle of dihydrolipoyl dehydrogenase is therefore completed, with the enzyme and its coenzymes in their oxidized forms. At this point, the catalytic cycle of the entire pyruvate dehydrogenase complex is also completed, and the complex is ready for a new reaction cycle.[1]
Note: unlike the thiazolium ring of thiamine pyrophosphate, FAD does not act as an electron trap or electron sink, but rather as an electron conduit between the reduced redox-active disulfide and NAD+.
The catalytic mechanism of dihydrolipoyl dehydrogenase has been determined by analogy with that of glutathione reductase (EC 1.8.1.7), which is 33% identical and whose structure is more extensively characterized.[46] It should, however, be noted that although the two enzymes catalyze similar reactions, these generally occur in opposite directions:
dihydrolipoyl dehydrogenase uses NAD+ to oxidize two –SH groups to a disulfide (–S–S–);
glutathione reductase uses NADPH to reduce a –S–S– to two thiol groups.
Nevertheless, their active sites are closely superimposable.[2]
Regulation of the pyruvate dehydrogenase complex
In mammals, the regulation of the activity of the pyruvate dehydrogenase complex is essential in both the fed and fasted states. In fact, this multienzyme complex plays a central role in metabolism because, by catalyzing the irreversible oxidative decarboxylation of pyruvate, it represents the entry point of carbon flux into the citric acid cycle or lipid and synthesis (fed state). This flux derives from all carbohydrate sources and ≈ 50% of the carbon skeletons of glucogenic amino acids, which, taken together, account for ≈ 60% of daily caloric intake.[47]
The importance of regulating the conversion of pyruvate into acetyl-CoA is also underscored by the fact that mammals, although able to produce glucose from pyruvate, cannot synthesize it from acetyl-CoA because of the irreversibility of the pyruvate dehydrogenase reaction and the absence of alternative pathways. Thus, inhibition of the activity of the complex allows glucose, and amino acids that can be converted into pyruvate, such as alanine, to be spared when other fuels, for example acetyl-CoA from fatty acid oxidation, are available.[48]
This explains why the activity of the complex is tightly regulated by:
feedback inhibition;
nucleotides;
covalent modifications, namely phosphorylation and dephosphorylation of specific target proteins.[5]
Regulation by feedback inhibition and the energy status of the cell
The activity of the dephosphorylated form of the pyruvate dehydrogenase complex is regulated by feedback inhibition.
Acetyl-CoA and NADH allosterically inhibit the enzymes that catalyze their formation, dihydrolipoyl transacetylase and dihydrolipoyl dehydrogenase, respectively.[49]
In addition, CoA and acetyl-CoA, as well as NAD+ and NADH, compete for binding sites on E2 and E3, respectively, which catalyze reversible reactions. This means that, when the ratios [acetyl-CoA]/[CoA] and [NADH]/[NAD+] are high, the transacetylation and dehydrogenation reactions proceed in reverse. Consequently, dihydrolipoyl transacetylase cannot accept the hydroxyethyl group from TPP because it is maintained in the acetylated form. This causes thiamine pyrophosphate to remain bound to pyruvate dehydrogenase in its hydroxyethyl form, which, in turn, decreases the rate of pyruvate decarboxylation. Hence, high [acetyl-CoA]/[CoA] and [NADH]/[NAD+] ratios indirectly influence pyruvate dehydrogenase activity.[1]
PDC Activity: Regulation by Feedback Inhibition
Links with fatty acid oxidation
Acetyl-CoA and NADH are also produced by fatty acid oxidation, which, like the reactions of the pyruvate dehydrogenase complex, takes place within the mitochondrion. This means that the cell, by regulating the activity of the multienzyme complex, preserves carbohydrate stores when fatty acids are available for energy. This reciprocal relationship between the oxidation of glucose and fatty acids, characterized by the inhibition of glucose utilization when fatty acid availability is high, is known as the Randle cycle (or glucose–fatty acid cycle). For example, during the fasted state, the liver, skeletal muscle, and many other organs and tissues rely primarily on fatty acid oxidation for energy. Conversely, the activity of the multienzyme complex is increased in the fed state, when many different types of cells and tissues mainly use glucose as a fuel.
More generally, when the production of NADH and/or acetyl-CoA exceeds the capacity of the cell to use them for ATP production, the activity of the pyruvate dehydrogenase complex is inhibited. The same is true when there is no need for additional ATP to be produced. In fact, the activity of the multienzyme complex is also sensitive to the energy charge of the cell. Through allosteric mechanisms, high ATP levels inhibit the activity of the pyruvate dehydrogenase component of the complex, whereas high ADP levels, which signal that the energy charge of the cell may be falling, activate it, thus committing the carbon skeletons of carbohydrates and some amino acids to energy production.
Note: in skeletal muscle, the activity of the pyruvate dehydrogenase complex increases with increased aerobic activity, resulting in a greater dependence on glucose as a fuel source.[4]
Regulation by phosphorylation/dephosphorylation
Unlike prokaryotes, in mammals the activity of the pyruvate dehydrogenase complex is also regulated by covalent modifications, namely, the phosphorylation and dephosphorylation of three specific serine residues on the α-subunit of pyruvate dehydrogenase, the enzyme that catalyzes the first and irreversible step of the overall reaction sequence.
Note: since mammalian pyruvate dehydrogenase is a heterotetramer, there are six potential phosphorylation sites.[1]
PDC Activity: Regulation by Covalent Modifications
Phosphorylation, which inactivates pyruvate dehydrogenase and thereby blocks the overall reaction sequence, is catalyzed by a Mg2+-dependent pyruvate dehydrogenase kinase. Two of the aforementioned serine residues are located on the more C-terminal loop at the entrance of the substrate channel leading to the respective active site, and phosphorylation of only one of them is sufficient to inactivate pyruvate dehydrogenase, thus demonstrating the out-of-phase coupling between its active sites.[50][51]
Conversely, in the dephosphorylated state the complex is active. Dephosphorylation is catalyzed by a specific protein phosphatase, a Ca2+-activated pyruvate dehydrogenase phosphatase.[52]
The activities of pyruvate dehydrogenase kinase and pyruvate dehydrogenase phosphatase are, in turn, subject to allosteric regulation by several modulators.[53]
Regulation of pyruvate dehydrogenase kinase
The activity of pyruvate dehydrogenase kinase depends on the ratios of [NADH]/[NAD+], [acetyl-CoA]/[CoA], and [ATP]/[ADP], as well as on the pyruvate concentration in the mitochondrial matrix.[54]
High ratios of [NADH]/[NAD+] and [acetyl-CoA]/[CoA], such as those occurring during the oxidation of fatty acids and ketone bodies, activate the kinase. As a result, pyruvate dehydrogenase is phosphorylated, and the pyruvate dehydrogenase complex is inhibited. This allows tissues, such as cardiac muscle, to preserve glucose when fatty acids and/or ketone bodies are being used for energy, because acetyl-CoA synthesis from pyruvate, and therefore from carbohydrates (and some amino acids), is turned off. Conversely, when the concentrations of NAD+ and coenzyme A are high, the activity of the kinase is inhibited and the multienzyme complex is active. Therefore, acetyl-CoA and NADH, two of the three end products of the reactions catalyzed by the pyruvate dehydrogenase complex, control their own synthesis allosterically, both directly and indirectly, by regulating the activity of pyruvate dehydrogenase kinase.
High ratio of [ATP]/[ADP] activates the kinase and thereby inhibits the pyruvate dehydrogenase complex. Note: unlike many other kinases, such as those involved in regulating glycogen metabolism, pyruvate dehydrogenase kinase is not regulated by cAMP levels, but rather by molecules that signal changes in the energy status of the cell and the availability of biosynthetic intermediates, ATP and NADH, and acetyl-CoA, respectively.
Pyruvate allosterically inhibits pyruvate dehydrogenase kinase. When its levels are high, it binds to the kinase and inactivates it; pyruvate dehydrogenase is not phosphorylated, and the pyruvate dehydrogenase complex remains active.
Pyruvate dehydrogenase kinase is also activated by interaction with dihydrolipoyl transacetylase in its acetylated form, i.e., when acetyl-dihydrolipoamide is present.
Other activators of the kinase are potassium and magnesium ions.[2][53]
Regulation of pyruvate dehydrogenase phosphatase
The activity of pyruvate dehydrogenase phosphatase depends on the ratios of [NADH]/[NAD+] and [acetyl-CoA]/[CoA], as well as on the concentration of Ca2+ in the mitochondrial matrix.
Low ratios of [NADH]/[NAD+] and [acetyl-CoA]/[CoA] activate the phosphatase; pyruvate dehydrogenase is dephosphorylated, and the pyruvate dehydrogenase complex is activated. Conversely, when these ratios are high, phosphatase activity decreases, kinase activity increases, and the multienzyme complex is inhibited.
Calcium ion activates pyruvate dehydrogenase phosphatase.
Ca2+ is an important second messenger that signals that the cell requires more energy. Therefore, when its levels are high, as in cardiac muscle cells after epinephrine stimulation or in skeletal muscle cells during muscular contraction, the phosphatase is active, the complex is dephosphorylated, and thus active.
Insulin also contributes to the regulation of the pyruvate dehydrogenase complex by activating pyruvate dehydrogenase phosphatase. In response to increases in blood glucose, the hormone stimulates glycogen synthesis and the synthesis of acetyl-CoA, a precursor for lipid biosynthesis.[2][53]
Fasting and subsequent refeeding also affect the activity of the multienzyme complex.
In tissues such as skeletal muscle, cardiac muscle, and kidney, fasting significantly decreases the activity of the complex, whereas refeeding reverses the inhibition produced by fasting. In the brain, however, these variations are not observed, because the activity of the pyruvate dehydrogenase complex is essential for ATP production.[1][55]
References
^ abcdefghij Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ abcdefghijklmno Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
^ abc Patel M.S., Nemeria N.S., Furey W., Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem 2014;289(24):16615-16623. doi:10.1074/jbc.R114.563148
^ abcdefghijk Berg J.M., Tymoczko J.L., Gatto G.J. Jr., Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
^ abcd Bothe S.N., Zdanowicz R. Structural diversity of pyruvate dehydrogenase complexes. FEBS Lett 2025. doi:10.1002/1873-3468.70140
^ Czumaj A., Szrok-Jurga S., Hebanowska A., Turyn J., Swierczynski J., Sledzinski T., Stelmanska E. The pathophysiological role of CoA. Int J Mol Sci 2020;21(23):9057. doi:10.3390/ijms21239057
^ Solomons T.W.G., Fryhle C.B., Snyder S.A. Solomons’ organic chemistry. 12th Edition. John Wiley & Sons Incorporated, 2017.
^ Solmonson A., DeBerardinis R.J. Lipoic acid metabolism and mitochondrial redox regulation. J Biol Chem 2018;293(20):7522-7530. doi:10.1074/jbc.TM117.000259
^ ab Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 4th Edition. St. Louis: Elsevier, 2018.
^ Lu Z., Imlay J.A. When anaerobes encounter oxygen: mechanisms of oxygen toxicity, tolerance and defence. Nat Rev Microbiol 2021;19(12):774-785. doi:10.1038/s41579-021-00583-y
^ Chaudhry R., Varacallo M.A. Biochemistry, Glycolysis. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482303/
^ Zangari J., Petrelli F., Maillot B., Martinou J.C. The multifaceted pyruvate metabolism: role of the mitochondrial pyruvate carrier. Biomolecules 2020;10(7):1068. doi:10.3390/biom10071068
^ Reed L.J., Pettit F.H., Eley M.H., Hamilton L., Collins J.H., Oliver R.M. Reconstitution of the Escherichia coli pyruvate dehydrogenase complex. Proc Natl Acad Sci USA. 1975;72(8):3068-72. doi:10.1073/pnas.72.8.3068
^ Stoops J.K., Cheng R.H., Yazdi M.A., et al. On the unique structural organization of the Saccharomyces cerevisiae pyruvate dehydrogenase complex. J Biol Chem 1997;272(9):5757-64. doi:10.1074/jbc.272.9.5757
^ ab Zhou Z.H., McCarthy D.B., O’Connor C.M., Reed L.J., Stoops J.K. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA 2001;98(26):14802-7. doi:10.1073/pnas.011597698
^ Forsberg B.O., Aibara S., Howard R.J., Mortezaei N., Lindahl E. Arrangement and symmetry of the fungal E3BP-containing core of the pyruvate dehydrogenase complex. Nat Commun 2020;11(1):4667. doi:10.1038/s41467-020-18401-z
^ ab Ciszak E.M., Makal A., Hong Y.S., Vettaikkorumakankauv A.K., Korotchkina L.G., Patel M.S. How dihydrolipoamide dehydrogenase-binding protein binds dihydrolipoamide dehydrogenase in the human pyruvate dehydrogenase complex. J Biol Chem 2006;281(1):648-55. doi:10.1074/jbc.M507850200
^ ab Wang C., Ma C., Xu Y., et al. Dynamics of the mammalian pyruvate dehydrogenase complex revealed by in-situ structural analysis. Nat Commun 2025;16(1):917. doi:10.1038/s41467-025-56171-8
^ Milne J.L., Wu X., Borgnia M.J., Lengyel J.S., Brooks B.R., Shi D., Perham R.N., Subramaniam S. Molecular structure of a 9-MDa icosahedral pyruvate dehydrogenase subcomplex containing the E2 and E3 enzymes using cryoelectron microscopy. J Biol Chem 2006;281(7):4364-70. doi:10.1074/jbc.M504363200
^ Kale S., Arjunan P., Furey W., Jordan F. A dynamic loop at the active center of the Escherichia coli pyruvate dehydrogenase complex E1 component modulates substrate utilization and chemical communication with the E2 component. J Biol Chem 2007;282(38):28106-16. doi:10.1074/jbc.M704326200
^ abc Frank R.A.W., Titman C.M., Pratap J.V., Luisi B.F., Perham R.N. A molecular switch and proton wire synchronize the active sites in thiamine enzymes. Science 2004;306(5697):872-876. doi:10.1126/science.1101030
^ abc Nemeria N.S., Arjunan P., Chandrasekhar K., Mossad M., Tittmann K., Furey W., Jordan F. Communication between thiamin cofactors in the Escherichia coli pyruvate dehydrogenase complex E1 component active centers: evidence for a “direct pathway” between the 4′-aminopyrimidine N1′ atoms. J Biol Chem 2010;285(15):11197-209. doi:10.1074/jbc.M109.069179
^ Arjunan P., Nemeria N., Brunskill A., Chandrasekhar K., Sax M., Yan Y., Jordan F., Guest J.R., Furey W. Structure of the pyruvate dehydrogenase multienzyme complex E1 component from Escherichia coli at 1.85 A resolution. Biochemistry 2002;41(16):5213-21. doi:10.1021/bi0118557
^ ab Perham R.N. Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu Rev Biochem 2000;69:961-1004. doi:10.1146/annurev.biochem.69.1.961
^ ab Wang J., Nemeria N.S., Chandrasekhar K., et al. Structure and function of the catalytic domain of the dihydrolipoyl acetyltransferase component in Escherichia coli pyruvate dehydrogenase complex. J Biol Chem 2014;289(22):15215-30. doi:10.1074/jbc.M113.544080
^ Allen A.G., Perham R.N. Two lipoyl domains in the dihydrolipoamide acetyltransferase chain of the pyruvate dehydrogenase multienzyme complex of Streptococcus faecalis. FEBS Lett 1991;287(1-2):206-10. doi:10.1016/0014-5793(91)80052-5
^ Hanemaaijer R., de Kok A., Jolles J., Veeger C. The domain structure of the dihydrolipoyl transacetylase component of the pyruvate dehydrogenase complex from Azotobacter vinelandii. Eur J Biochem 1987;169(2):245-52. doi:10.1111/j.1432-1033.1987.tb13604.x
^ Hale G., Perham R.N. Amino acid sequence around lipoic acid residues in the pyruvate dehydrogenase multienzyme complex of Escherichia coli. Biochem J 1980;187(3):905-8. doi:10.1042/bj1870905
^ Hipps D.S., Packman L.C., Allen M.D., Fuller C., Sakaguchi K., Appella E., Perham R.N. The peripheral subunit-binding domain of the dihydrolipoyl acetyltransferase component of the pyruvate dehydrogenase complex of Bacillus stearothermophilus: preparation and characterization of its binding to the dihydrolipoyl dehydrogenase component. Biochem J 1994;297(Pt 1):137-43. doi:10.1042/bj2970137
^ Schulze E., Westphal A.H., Obmolova G., Mattevi A., Hol W.G., de Kok A. The catalytic domain of the dihydrolipoyl transacetylase component of the pyruvate dehydrogenase complex from Azotobacter vinelandii and Escherichia coli. Expression, purification, properties and preliminary X-ray analysis. Eur J Biochem 1991;201(3):561-8. doi:10.1111/j.1432-1033.1991.tb16315.x
^ Brautigam C.A., Chuang J.L., Tomchick D.R., Machius M., Chuang D.T. Crystal structure of human dihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causing mutations. J Mol Biol 2005;350(3):543-52. doi:10.1016/j.jmb.2005.05.014
^ abc Mattevi A., Obmolova G., Sokatch J.R., Betzel C., Hol W.G. The refined crystal structure of Pseudomonas putida lipoamide dehydrogenase complexed with NAD+ at 2.45 A resolution. Proteins 1992;13(4):336-51. doi:10.1002/prot.340130406
^ Kern D., Kern G., Neef H., Tittmann K., Killenberg-Jabs M., Wikner C., Schneider G., Hübner G. How thiamine diphosphate is activated in enzymes. Science 1997;275(5296):67-70. doi:10.1126/science.275.5296.67
^ ab Jordan F. Current mechanistic understanding of thiamin diphosphate-dependent enzymatic reactions. Nat Prod Rep 2003;20(2):184-201. doi:10.1039/b111348h
^ ab Nemeria N.S., Chakraborty S., Balakrishnan A., Jordan F. Reaction mechanisms of thiamin diphosphate enzymes: defining states of ionization and tautomerization of the cofactor at individual steps. FEBS J 2009;276(9):2432-46. doi:10.1111/j.1742-4658.2009.06964.x
^ Hawkins C.F., Borges A., Perham R.N. A common structural motif in thiamin pyrophosphate-binding enzymes. FEBS Lett 1989;255(1):77-82. doi:10.1016/0014-5793(89)81064-6
^ Breslow R. Rapid deuterium exchange in thiazolium salts. J Am Chem Soc 1957;79:1762-1763. doi:10.1021/ja01564a064
^ Baykal A.T., Kakalis L., Jordan F. Electronic and nuclear magnetic resonance spectroscopic features of the 1′,4′-iminopyrimidine tautomeric form of thiamin diphosphate, a novel intermediate on enzymes requiring this coenzyme. Biochemistry 2006;45(24):7522-8. doi:10.1021/bi060395k
^ Meyer D., Neumann P., Koers E., Sjuts H., Lüdtke S., Sheldrick G.M., Ficner R., Tittmann K. Unexpected tautomeric equilibria of the carbanion-enamine intermediate in pyruvate oxidase highlight unrecognized chemical versatility of thiamin. Proc Natl Acad Sci USA 2012;109(27):10867-72. doi:10.1073/pnas.1201280109
^ Shaanan B., Chipman D.M. Reaction mechanisms of thiamin diphosphate enzymes: new insights into the role of a conserved glutamate residue. FEBS J 2009 May;276(9):2447-53. doi:10.1111/j.1742-4658.2009.06965.x
^ Yang Y.S., Frey P.A. Dihydrolipoyl transacetylase of Escherichia coli. Formation of 8-S-acetyldihydrolipoamide. Biochemistry 1986;25(25):8173-8. doi:10.1021/bi00373a008
^ Škerlová J., Berndtsson J., Nolte H., Ott M., Stenmark P. Structure of the native pyruvate dehydrogenase complex reveals the mechanism of substrate insertion. Nat Commun 2021;12(1):5277. doi:10.1038/s41467-021-25570-y
^ Massey V., Gibson Q.H., Veeger C. Intermediates in the catalytic action of lipoyl dehydrogenase (diaphorase). Biochem J 1960;77(2):341-51. doi:10.1042/bj0770341
^ Rice D.W., Schulz G.E., Guest J.R. Structural relationship between glutathione reductase and lipoamide dehydrogenase. J Mol Biol 1984;174(3):483-96. doi:10.1016/0022-2836(84)90332-2
^ Denton R.M., Randle P.J., Bridges B.J., et al. Regulation of mammalian pyruvate dehydrogenase. Mol Cell Biochem 1975;9(1):27-53. doi:10.1007/BF01731731
^ Gray L.R., Tompkins S.C., Taylor E.R. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-2604. doi:10.1007/s00018-013-1539-2
^ Harris R.A., Bowker-Kinley M.M., Huang B., Wu P. Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul 2002;42:249-59. doi:10.1016/s0065-2571(01)00061-9
^ Kolobova E., Tuganova A., Boulatnikov I., Popov K.M. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J 2001;358(Pt 1):69-77. doi:10.1042/0264-6021:3580069
^ Hiromasa Y., Fujisawa T., Aso Y., Roche T.E. Organization of the cores of the mammalian pyruvate dehydrogenase complex formed by E2 and E2 plus the E3-binding protein and their capacities to bind the E1 and E3 components. J Biol Chem 2004;279(8):6921-33. doi:10.1074/jbc.M308172200
^ Roche T.E., Baker J.C., Yan X., et al. Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog Nucleic Acid Res Mol Biol 2001;70:33-75. doi:10.1016/s0079-6603(01)70013-x
^ abc Pettit F.H., Pelley J.W., Reed L.J. Regulation of pyruvate dehydrogenase kinase and phosphatase by acetyl-CoA/CoA and NADH/NAD ratios. Biochem Biophys Res Commun 1975;65(2):575-82. doi:10.1016/s0006-291x(75)80185-9
^ Patel M.S., Korotchkina L.G. Regulation of the pyruvate dehydrogenase complex. Biochem Soc T 2006;34(2):217-222. doi:10.1042/bst0340217
^ Small L., Brandon A.E., Quek L.E., Krycer J.R., James D.E., Turner N., Cooney G.J. Acute activation of pyruvate dehydrogenase increases glucose oxidation in muscle without changing glucose uptake. Am J Physiol Endocrinol Metab 2018;315(2):E258-E266. doi:10.1152/ajpendo.00386.2017
The pentose phosphate pathway, also known as the phosphogluconate pathway, is a metabolic route common to all living organisms that oxidizes glucose and serves as an alternative to glycolysis. It branches off after the formation of glucose 6-phosphate (G6P) and is also referred to as the hexose monophosphate shunt, where shunt denotes a metabolic diversion or bypass.
Its primary function is the production, in varying proportions, of NADPH, a reduced coenzyme, and ribose 5-phosphate, a five-carbon phosphorylated sugar (i.e., a pentose phosphate), from which the pathway derives its name.[1]
Conceptually, the pathway consists of two phases: an oxidative phase, in which NADPH is generated, and a non-oxidative phase, which produces ribose 5-phosphate and other phosphorylated carbohydrates.[2]
It is estimated that over 10% of cellular glucose is metabolized via this pathway, which notably oxidizes glucose without directly producing or consuming ATP.[3]
Summary: Key Points
Definition: a cytosolic pathway for the oxidative metabolism of glucose, alternative to glycolysis, whose main function is the production, in variable proportions, of NADPH and ribose 5-phosphate.
Fate of NADPH: produced in the oxidative phase, it provides the reducing power required for reductive biosyntheses and antioxidant defense mechanisms.
Fate of ribose 5-phosphate: produced in the non-oxidative phase, it is used for the synthesis of nucleotides and various coenzymes.
Key enzymes: glucose 6-phosphate dehydrogenase, regulated by the NADP+/NADPH ratio, and transketolase, a thiamine pyrophosphate–dependent enzyme.
Metabolic flexibility: the pathway adapts to cellular needs, operating in different modes to favor the production of NADPH, ribose 5-phosphate, or the recovery of carbon in the form of glycolytic intermediates.
The first evidence of the pentose phosphate pathway emerged in the 1930s through the work of Otto Warburg, who received the Nobel Prize in Physiology or Medicine in 1931 for his work on cellular metabolism and respiratory enzymes.[4]
Warburg and his collaborator Erwin Christian discovered NADP during studies on the oxidation of glucose 6-phosphate to 6-phosphogluconate.
Further evidence came from observations that glucose metabolism continued in tissues even in the presence of glycolysis inhibitors such as fluoride and iodoacetate ions, which inhibit enolase (EC 4.2.1.11) and glyceraldehyde 3-phosphate dehydrogenase (EC 1.2.1.12), respectively.[5]
However, the pathway was fully elucidated only in the 1950s, thanks to the work of several researchers, notably Efraim Racker, Bernard Horecker, Frank Dickens, and Fritz Lipmann, who received the Nobel Prize in Physiology or Medicine in 1953 for his discovery of coenzyme A, not specifically for the pentose phosphate pathway, although his work was highly relevant in the broader context of intermediary metabolism.[6][7]
Main function
The primary function of the pentose phosphate pathway is the production of NADPH and ribose 5-phosphate.[8]
NADPH is needed for reductive biosynthesis, such as the synthesis of fatty acids, cholesterol, steroid hormones and two non-essential amino acids, proline and tyrosine, from glutamate and phenylalanine, respectively, as well as for the reduction of oxidized glutathione. In these reactions, the reduced coenzyme acts as an electron donor, more precisely as a hydride ion donor (:H−), a species composed of one proton and two electrons.[1][9]
Reduced and Oxidized Forms of Nicotinamide Adenine Dinucleotide Phosphate
Note: In vertebrates, approximately half of the NADPH required for the reductive steps of fatty acid synthesis is produced via the pentose phosphate pathway. The remainder is generated by the reaction catalyzed by the malic enzyme (EC 1.1.1.40).[10]
Malate + NADP+ ⇌ Pyruvate + NADPH + CO2
Ribose 5-phosphate is used in the synthesis of nucleotides and nucleic acids (DNA and RNA), as well as ATP, and coenzymes such as coenzyme A, NAD, NADP, and FAD. It also plays a role in the biosynthesis of the essential amino acids tryptophan and histidine.[8] This five-carbon phosphorylated sugar is not used directly; instead, it is converted to 5-phosphoribosyl 1-pyrophosphate (PRPP) in a reaction catalyzed by ribose phosphate pyrophosphokinase, also known as PRPP synthase (EC 2.7.6.1).[10]
Ribose 5-phosphate + ATP → 5-Phosphoribosyl 1-pyrophosphate + AMP
Additional functions
In addition to producing NADPH and ribose 5-phosphate, the pentose phosphate pathway also serves other functions, both anabolic and catabolic.
In yeasts and many bacteria, it is involved in the catabolism of the five-carbon sugars ribose, xylose, and arabinose.[9]
In humans, it also contributes to the catabolism of the aforementioned pentoses, as well as of less common sugars containing three, four, or seven carbon atoms, derived from the diet or from endogenous sources. These include:
pentoses released during the breakdown of structural carbohydrates;
ribose 5-phosphate generated from nucleotide catabolism.[11][12]
In photosynthetic organisms, it contributes to carbon dioxide (CO2) fixation during the Calvin cycle.[9]
In addition to ribose 5-phosphate, the pathway also provides several intermediates for other biosynthetic processes, including:
erythrose 4-phosphate, used in the synthesis of the aromatic amino acids phenylalanine, tryptophan, and tyrosine;
ribulose 5-phosphate, a precursor in riboflavin (vitamin B2) biosynthesis;
sedoheptulose 7-phosphate, which, in Gram-negative bacteria, is involved in the synthesis of heptose units found in the lipopolysaccharide layer of the outer membrane.[1][13]
Sites of the pentose phosphate pathway
In animal cells and bacteria, the hexose monophosphate shunt, like glycolysis, fatty acid synthesis, and most reactions of gluconeogenesis, occurs in the cytosol. Notably, glycolysis, gluconeogenesis, and the pentose phosphate pathway are interconnected through several shared enzymes and intermediates.[12]
In plant cells, the pentose phosphate pathway takes place in the plastids, and its intermediates can reach the cytosol through membrane pores in these organelles.[10]
In humans, the expression of the enzymes involved in the pathway varies significantly between tissues. Relatively high levels are found in the liver, adrenal cortex, testes, ovaries, thyroid, mammary glands during lactation, and in red blood cells.[2] In all these tissues, a constant supply of NADPH is required to sustain reductive biosynthesis and/or to counteract reactive oxygen species (ROS), which can damage sensitive cellular structures such as DNA, membrane lipids, and proteins. This protection is largely achieved through the reduction of oxidized glutathione (GSSG) to reduced glutathione (GSH), the major intracellular antioxidant in erythrocytes and most other cells. This reaction is catalyzed by glutathione reductase (EC 1.8.1.7).[14]
High levels of phosphogluconate pathway enzymes are also found in rapidly dividing cells, such as enterocytes, skin cells, bone marrow cells, embryonic cells, and, under pathological conditions, cancer cells. These cells require a continuous supply of ribose 5-phosphate for nucleic acid synthesis.
Conversely, these enzymes are present at very low levels in skeletal muscle, where the pentose phosphate pathway is virtually absent. In this tissue, glucose 6-phosphate is primarily utilized for energy production through glycolysis and the citric acid cycle.[2]
Oxidative phase of the pentose phosphate pathway
The oxidative phase of the pentose phosphate pathway consists of two irreversible oxidation reactions, the first and third steps, and one hydrolysis.
Summary of the Reactions of the Pentose Phosphate Pathway
During this phase, glucose 6-phosphate is converted into ribulose 5-phosphate, a five-carbon phosphorylated sugar that serves as the starting substrate for the reactions of the non-oxidative phase.
This conversion is accompanied by the formation of two molecules of NADPH and the release of carbon 1 of glucose as CO2.[15]
The overall equation is:
Oxidation of glucose 6-phosphate to 6-phosphoglucono-δ-lactone
In the first step of the oxidative phase, glucose 6-phosphate dehydrogenase (G6PD; EC 1.1.1.49) catalyzes the oxidation of glucose 6-phosphate to 6-phosphoglucono-δ-lactone, an intramolecular ester.
This occurs via the transfer of a hydride ion (H−) from carbon 1 of glucose 6-phosphate to NADP+, which acts as the oxidizing agent.[12]
Note: this reaction produces the first molecule of NADPH in the pentose phosphate pathway.
The reaction catalyzed by G6PD is unique to the pathway. As in most metabolic pathways, the first reaction that is unique, known as the committed step, is essentially irreversible.
In the liver, it has a ΔG of −17.6 kJ/mol (−4.21 kcal/mol), and it is highly allosterically regulated. Indeed, glucose 6-phosphate dehydrogenase serves as the major control point for the flux of metabolites through the pentose phosphate pathway.[5]
G6PD expression and immune function
In humans, the highest levels of G6PD are found in neutrophils and macrophages, two types of phagocytic cells.
During inflammation, these cells use NADPH to generate superoxide radicals (O2•−) from molecular oxygen, in a reaction catalyzed by NADPH oxidase (EC 1.6.3.1).
2 O2 + NADPH → 2 O2•− + NADP+ + H+
Superoxide radicals are used to generate, for antimicrobial defense, both other reactive oxygen species (ROS) and reactive nitrogen species (RNS), such as:[2]
hydrogen peroxide (H2O2), via the reaction catalyzed by superoxide dismutase (SOD; EC 1.15.1.1)
2 O2•− + 2 H+ → H2O2 + O2
peroxynitrite (ONOO−), via the reaction with nitric oxide (•NO)
O2•− + •NO → ONOO−
hydroperoxide radical (HOO•)
O2•− + H+ → HOO•
Catalytic mechanism of G6PD
The catalytic mechanism of glucose 6-phosphate dehydrogenase has been studied in great detail in the microorganism Leuconostoc mesenteroides, whose enzyme displays the unusual ability to use NAD+ and/or NADP+ as coenzyme.
Catalytic Mechanism of Glucose 6-Phosphate Dehydrogenase
The enzyme does not require metal ions for its activity. Instead, an amino acid residue in the active site acts as a general base, capable of abstracting a proton from the hydroxyl group attached to carbon 1 of glucose 6-phosphate.[16]
In the bacterial enzyme, this function is performed by the Nɛ2 atom of the imidazole ring in a histidine side chain. This nitrogen atom contains a lone pair of electrons that enables it to act as a nucleophile.
Its action facilitates the oxidation of glucose 6-phosphate, a cyclic hemiacetal with C-1 in the aldehyde oxidation state, into a cyclic ester, specifically a lactone.
Simultaneously, the hydride ion is transferred from C-1 of glucose 6-phosphate to the C-4 position of the nicotinamide ring of NADP+ to form NADPH.
Since this histidine residue is conserved in many of the glucose 6-phosphate dehydrogenase sequences identified so far, it is likely that this catalytic mechanism is common to G6PD enzymes across a wide range of organisms.[17]
Regulation of G6PD
Glucose 6-phosphate dehydrogenase is the primary control point in the pentose phosphate pathway and the main regulator of NADPH production.[18]
In humans, G6PD exists in two forms:
an inactive monomeric form;
an active form, which exists in a dimer–tetramer equilibrium.[14]
One of the main modulators of its activity is the cytosolic NADP+/NADPH ratio.
NADPH acts as a competitive inhibitor, while NADP+ is both a substrate and a structural activator, stabilizing the enzyme’s active conformation by binding to a regulatory site near the dimer interface, helping to maintain the dimeric (active) structure.
Regulation of G6PD Activity
Under typical metabolic conditions, the NADP+/NADPH ratio is low.
As a result, NADP+ availability is limited, less coenzyme binds to the enzyme, and G6PD activity is reduced, regardless of expression levels. In these conditions, the oxidative phase of the pathway is virtually inactive.
Conversely, in cells where NADPH-consuming reactions are active (e.g., fatty acid synthesis, antioxidant defense), the cytosolic concentration of NADPH decreases, while NADP+ increases.
This change promotes the activation of G6PD, triggering the oxidative phase of the hexose monophosphate pathway.
Therefore, the fate of glucose 6-phosphate is influenced by the cell’s current demand for NADPH.[19][20]
A second regulatory mechanism involves the accumulation of acyl-CoAs, intermediates in fatty acid biosynthesis.[8] These molecules bind to the dimeric form of G6PD and induce its dissociation into monomers, resulting in the loss of catalytic activity.[14]
Insulin also plays a role by upregulating the expression of the genes encoding glucose 6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase.
Thus, in the well-fed state, insulin enhances the carbon flux through the pentose phosphate pathway, increasing NADPH production
Note: insulin also promotes fatty acid synthesis, which depends on a continuous supply of NADPH.[15]
Hydrolysis of 6-phosphoglucono-δ-lactone to 6-phosphogluconate
In the second step of the oxidative phase, 6-phosphoglucono-δ-lactone is hydrolyzed to form 6-phosphogluconate, a linear molecule.
Although 6-phosphoglucono-δ-lactone is hydrolytically unstable and can undergo spontaneous (nonenzymatic) ring-opening at a significant rate, this hydrolysis reaction is greatly accelerated in the cell by the enzyme 6-phosphogluconolactonase (EC 3.1.1.31).[21]
The reaction is catalyzed by 6-phosphogluconate dehydrogenase (EC 1.1.1.44), an enzyme that requires Mg2+ ions for activity.[5]
6-Phosphogluconate + NADP+ → Ribulose 5-phosphate + NADPH + CO2
Catalytic mechanism of 6-phosphogluconate dehydrogenase
The catalytic mechanism of 6-phosphogluconate dehydrogenase closely resembles that of isocitrate dehydrogenase (EC 1.1.1.41), a key enzyme of the citric acid cycle.[5]
It proceeds via acid-base catalysis through a three-step mechanism, involving two strictly conserved amino acid residues, a lysine (Lys) and a glutamate (Glu).[22]
In humans:
Lys185, which acts as both a general base and a general acid;
Catalytic Mechanism of 6-Phosphogluconate Dehydrogenase
Step 1: oxidation
In the first step, 6-phosphogluconate is oxidized to a β-keto acid, specifically 3-keto-6-phosphogluconate.
The ε-amino group of Lys185 acts as a general base, abstracting a proton from the hydroxyl group at C-3 of 6-phosphogluconate.
This promotes the transfer of a hydride ion (H−) from C-3 of the substrate to C-4 of the nicotinamide ring in NADP+, yielding NADPH.
The 3-keto intermediate and NADPH are released from the active site.
Step 2: decarboxylation
The 3-keto-6-phosphogluconate formed is highly unstable and undergoes a spontaneous decarboxylation, resulting in:
the formation of CO2 (from C-1 of the original glucose 6-phosphate);
the intermediate cis-1,2-enediol of ribulose 5-phosphate, a high-energy tautomer.
In this step Lys185 acts as a general acid, donating a proton to the C-3 carbonyl oxygen, facilitating the decarboxylation.
Step 3: Keto–enol tautomerization
In the final step, the cis-1,2-enediol intermediate undergoes a stereospecific keto–enol tautomerization to form ribulose 5-phosphate.
Glu192 donates a proton to the C-1 of the enediol.
Simultaneously, the ε-amino group of Lys185 acts as a base, abstracting a proton from the hydroxyl group on C-2.
The result is the formation of ribulose 5-phosphate, completing the oxidative phase of the pentose phosphate pathway.[22][23]
Non-oxidative phase of the pentose phosphate pathway
In the non-oxidative phase of the pentose phosphate pathway, several phosphorylated carbohydrates are produced, and their fate depends on the cell’s relative needs for NADPH, ribose 5-phosphate, and ATP.
This phase consists of five freely reversible steps, involving a series of interconversions of phosphorylated sugars.
It begins with two initial reactions: an isomerization and an epimerization.
These transform ribulose 5-phosphate into:
Epimerases (EC 5.1), a subclass of isomerases, catalyze configurational changes at a single asymmetric carbon atom, typically via a deprotonation/protonation mechanism.
In contrast, isomerases mediate the rearrangement of chemical groups between different carbon atoms.[24]
Note: enzymatic isomerizations and epimerizations play a key role in carbohydrate metabolism, enabling the interconversion of sugars with distinct functional properties.[25]
Isomerization of ribulose 5-phosphate to ribose 5-phosphate
In the first step of the non-oxidative phase, ribulose 5-phosphate (a ketose) is converted into its corresponding aldose, ribose 5-phosphate, via an isomerization reaction.
The reaction is catalyzed by phosphopentose isomerase, also known as ribose 5-phosphate isomerase (EC 5.3.1.6).
Ribulose 5-phosphate ⇄ Ribose 5-phosphate
This is an example of functional group isomerism, where a ketone is converted into an aldehyde.[1]
Catalytic mechanism of phosphopentose isomerase
The catalytic mechanism of phosphopentose isomerase resembles that of phosphohexose isomerase (EC 5.3.1.9), a glycolytic enzyme. Both involve the formation of a high-energy intermediate, cis-1,2-enediol, through a proton-transfer mechanism that is common in aldose–ketose isomerizations.[26]
In Escherichia coli, the mechanism (described here in the direction of ribulose 5-phosphate formation from ribose 5-phosphate, as occurs in the Calvin cycle of photosynthesis) proceeds as follows.
Catalytic Mechanism of Phosphopentose Isomerase
Step 1: ring opening
The furanose ring of ribose 5-phosphate is opened by interaction with an aspartic acid residue (Asp81), which accepts a proton from the hydroxyl group bound to C-1.
It is likely that a water molecule donates the proton required for this step.
Note: in aqueous solution, spontaneous ring opening of ribose is rare, less than 0.5% occurs naturally.
Step 2: formation of the enediol intermediate
A glutamic acid residue (Glu103) acts as a general base, abstracting a proton from the C-2 carbon. Simultaneously, Asp81 donates a proton. This concerted action leads to the formation of the cis-1,2-enediol intermediate.
Step 3: protonation and product formation
The now protonated Glu103 acts as a general acid, donating a proton to C-1 of the enediol intermediate. At the same time, Asp81 accepts a proton from the hydroxyl group on C-2. These proton transfers lead to the formation of ribulose 5-phosphate.
During ribose 5-phosphate synthesis from ribulose 5-phosphate, as occurs in the pentose phosphate pathway, phosphopentose isomerase functions in reverse, following the same general principles of acid-base catalysis.[27]
Epimerization of ribulose 5-phosphate to xylulose 5-phosphate
The other metabolic fate of ribulose 5-phosphate in the pentose phosphate pathway is its epimerization to xylulose 5-phosphate, a ketose like ribulose 5-phosphate.
This reaction is catalyzed by phosphopentose epimerase (EC 5.1.3.1).[5]
Ribulose 5-Phosphate ⇄ Xylulose 5-Phosphate
Note: xylulose 5-phosphate also acts as a regulatory molecule. It inhibits gluconeogenesis and stimulates glycolysis by modulating the intracellular concentration of fructose 2,6-bisphosphate in the liver.[28]
Catalytic mechanism of phosphopentose epimerase
Like the reactions catalyzed by 6-phosphogluconate dehydrogenase and ribose 5-phosphate isomerase, this reaction also proceeds through the formation of an enediol intermediate.
However, in this case, the double bond is formed between C-2 and C-3, rather than between C-1 and C-2.[5]
During the reaction, an amino acid residue in the enzyme’s active site acts as a general base and abstracts a proton from C-3, leading to the formation of the cis-2,3-enediol intermediate.
Next, an acidic amino acid residue donates a proton back to C-3, but from the opposite face, resulting in inversion at C-3 and formation of xylulose 5-phosphate.[29]
Catalytic Mechanism of Phosphopentose Epimerase
Up to this point, the hexose monophosphate shunt has generated the following, for each molecule of glucose 6-phosphate metabolized:
a pool of three pentose 5-phosphates, ribulose 5-phosphate, ribose 5-phosphate, and xylulose 5-phosphate, which coexist at equilibrium;
two molecules of NADPH.
In the next three steps (steps 6 through 8), the enzymes transketolase (EC 2.2.1.1) and transaldolase (EC 2.2.1.2), key enzymes of the pentose phosphate pathway, catalyze a series of carbon skeleton rearrangements.
These reactions lead to the formation of three-, four-, six-, and seven-carbon sugar phosphates, which can be redirected into other metabolic pathways, depending on the cell’s metabolic needs.[12]
By analyzing the flow of metabolites across different pathways, it becomes clear that the concerted action of transketolase and transaldolase allows the non-oxidative phase of the pentose phosphate pathway to interact with:
glycolysis;
gluconeogenesis;
pathways for the synthesis of various vitamins, coenzymes, and nucleic acid precursors.[1]
Transketolase
Transketolase is the rate-limiting enzyme of the non-oxidative phase of the pentose phosphate pathway and is the first enzyme to act downstream of ribose 5-phosphate isomerase and phosphopentose epimerase.[30]
Discovered independently in 1953 by Horecker and Racker, and named by Racker, transketolase catalyzes the transfer of a two-carbon unit from a ketose donor to an aldose acceptor. This occurs in both the sixth and eighth steps of the pathway.
The ketose donors are typically:
Taking the forward reactions as an example, in the sixth step, the ketose donor xylulose 5-phosphate transfers a two-carbon unit to the aldose acceptor ribose 5-phosphate. This reaction generates glyceraldehyde 3-phosphate, which corresponds to the three-carbon remainder of xylulose 5-phosphate, and sedoheptulose 7-phosphate, a seven-carbon sugar that participates in the subsequent step of the pathway.[31]
In the eighth step, xylulose 5-phosphate once again acts as the donor, but this time the acceptor is erythrose 4-phosphate. The products of this reaction are another molecule of glyceraldehyde 3-phosphate and fructose 6-phosphate.
It is worth noting that three out of the four products generated by transketolase, namely, two molecules of glyceraldehyde 3-phosphate and one of fructose 6-phosphate, are also key intermediates of glycolysis.[30]
Beyond xylulose 5-phosphate, sedoheptulose 7-phosphate, and fructose 6-phosphate, transketolase is also capable of using other 2-keto sugars and a variety of aldose phosphates as substrates.[32]
Transketolase requires thiamine pyrophosphate (TPP) as a cofactor. TPP, the biologically active form of thiamine (vitamin B1), is tightly bound to the enzyme and participates in the transfer of activated aldehyde intermediates by stabilizing the two-carbon carbanions formed during the reaction.[8]
Catalytic mechanism of transketolase
The carbon atom located between the sulfur and nitrogen atoms of the thiazolium ring of thiamine pyrophosphate, namely, the C-2 atom, is significantly more acidic than most =CH groups in other molecules. This is due to the adjacent positively charged nitrogen, which electrostatically stabilizes the carbanion formed upon proton dissociation. As a result, the C-2 proton is readily abstracted, generating a carbanion. This proton abstraction is catalyzed by transketolase.
The resulting carbanion attacks the carbonyl carbon of the donor ketose substrate. In step 6, this is xylulose 5-phosphate (or sedoheptulose 7-phosphate in the reverse reaction); in step 8, it is again xylulose 5-phosphate (or fructose 6-phosphate in reverse). Taking the forward direction of step 6 as an example, the covalent adduct formed between thiamine pyrophosphate and xylulose 5-phosphate undergoes cleavage at the C-2–C-3 bond. This reaction produces glyceraldehyde 3-phosphate, which is released, and a two-carbon hydroxyethyl group, which remains covalently bound to the C-2 atom of the thiazolium ring.
Catalytic Mechanism of Transketolase
The negative charge on this hydroxyethyl intermediate is stabilized by the thiazolium ring of thiamine pyrophosphate. The positively charged nitrogen in the ring acts as an electron sink, delocalizing the carbanion’s negative charge via resonance. In this way, the thiazolium ring provides an electron-deficient (electrophilic) environment that stabilizes the intermediate.
Subsequently, the hydroxyethyl-TPP group condenses with ribose 5-phosphate, the aldehyde acceptor substrate, via nucleophilic attack on the aldehyde carbon. This forms a new covalent adduct with thiamine pyrophosphate.
Finally, cleavage of the adduct releases sedoheptulose 7-phosphate and regenerates the TPP carbanion.[8][12][30]
Transaldolase
Transaldolase was discovered in 1953 by Horecker and Smyrniotis in brewer’s yeast, identified as Saccharomyces cerevisiae. In the seventh step of the pentose phosphate pathway, this enzyme catalyzes the transfer of a three-carbon unit from the donor substrate sedoheptulose 7-phosphate to the acceptor substrate glyceraldehyde 3-phosphate. The reaction yields fructose 6-phosphate and erythrose 4-phosphate.
As in the reactions catalyzed by transketolase, the donor of the carbon unit is a ketose, while the acceptor is an aldose. In the reverse direction, the donor becomes fructose 6-phosphate and the acceptor erythrose 4-phosphate.[9]
Catalytic mechanism of transaldolase
Unlike transketolase, transaldolase does not require a cofactor for its activity.
The reaction proceeds in two steps: an aldol cleavage followed by an aldol condensation. Below, the catalytic mechanism of E. coli transaldolase B is described, taking as an example the forward reaction that produces erythrose 4-phosphate and fructose 6-phosphate.
Step 1: aldol cleavage and intermediate formation
In the first step, the ε-amino group of a lysine residue (Lys132) in the active site performs a nucleophilic attack on the carbonyl carbon (C-2) of sedoheptulose 7-phosphate. This occurs after a proton transfer to a glutamic acid residue (Glu96), mediated by a water molecule. The result is the formation of a carbinolamine intermediate.
Catalytic Mechanism of Transaldolase
Subsequently, the elimination of a water molecule leads to the formation of an enzyme-bound imine, also known as a Schiff base. This step also involves the transfer of a proton from Glu96 to the catalytic water molecule.
Note: this enzyme–substrate covalent intermediate is similar to that formed during the reaction catalyzed by aldolase (EC 4.1.2.13) in glycolysis.
Then, an aspartic acid residue (Asp17) acts as a base and abstracts a proton from the hydroxyl group at C-4. This facilitates the cleavage of the C–C bond between C-3 and C-4, releasing erythrose 4-phosphate, an aldose. The remaining three-carbon carbanion stays covalently bound to the enzyme and is stabilized by resonance. Just like in the transketolase mechanism, the positive charge on the Schiff base nitrogen acts as an electron sink, stabilizing the negative charge on the carbanion.[5][8][12][33]
Step 2: aldol condensation and product release
After the release of erythrose 4-phosphate, the acceptor substrate glyceraldehyde 3-phosphate enters the active site.
The previously formed carbanion performs a nucleophilic attack on the carbonyl carbon of glyceraldehyde 3-phosphate. This aldol condensation results in the formation of a new carbon–carbon bond and a covalently bound ketose intermediate.
The final step involves the hydrolysis of the Schiff base. This reaction restores the free ε-amino group of Lys132 and releases fructose 6-phosphate, a ketose and the second product of the reaction. The enzyme is now regenerated and ready for a new catalytic cycle.
As previously mentioned, in the eighth step of the pentose phosphate pathway, transketolase catalyzes the synthesis of fructose 6-phosphate and glyceraldehyde 3-phosphate from erythrose 4-phosphate and xylulose 5-phosphate, linking the activities of transaldolase and transketolase in the non-oxidative phase of the pathway.[33]
The cell’s need for NADPH, ribose 5-phosphate, and ATP
From a molecular point of view, the fate of glucose 6-phosphate depends largely on the relative activities of the enzymes that metabolize it through glycolysis and the pentose phosphate pathway, particularly phosphofructokinase-1 (PFK-1; EC 2.7.1.11) and glucose 6-phosphate dehydrogenase, both of which are highly regulated.[1]
PFK-1 is inhibited when ATP and/or citrate concentrations increase, that is, when the cell’s energy charge is high. Conversely, it is activated when AMP and/or fructose 2,6-bisphosphate concentrations increase, indicating a low energy charge. As a result, when the energy charge is high, the carbon flux, specifically the flux of glucose 6-phosphate, through glycolysis decreases.[34][35]
G6PD, in contrast, is inhibited by NADPH and by acyl-CoAs, which are intermediates in fatty acid biosynthesis. Thus, when cytosolic NADPH levels rise, the pentose phosphate pathway is downregulated. However, when NADPH levels fall, the inhibition is relieved, the pathway is reactivated, and both NADPH and ribose 5-phosphate are synthesized.[8][19]
Therefore, depending on the cell’s current demand for ATP, NADPH, and ribose 5-phosphate, reactions from glycolysis and the pentose phosphate pathway can be strategically combined to prioritize the synthesis of specific metabolites. This metabolic flexibility is also made possible by the fact that the non-oxidative phase of the pentose phosphate pathway is primarily controlled by substrate availability.
The four main scenarios are outlined below.[12]
When the need for NADPH is much greater than that for ribose 5-phosphate or ATP
When the cellular requirement for NADPH far exceeds that for ribose 5-phosphate or ATP, namely, when no additional ATP is needed and the energy charge is high, glucose 6-phosphate enters the pentose phosphate pathway and is fully oxidized to CO2. These metabolic conditions are typically found in adipose tissue during fatty acid synthesis.
Through a combination of reactions from the non-oxidative phase and selected gluconeogenic reactions, namely, those catalyzed by triose phosphate isomerase (EC 5.3.1.1), aldolase (EC 4.1.2.13), phosphohexose isomerase (EC 5.3.1.9), and fructose 1,6-bisphosphatase (EC 3.1.3.11), six molecules of ribulose 5-phosphate can be converted into five molecules of glucose 6-phosphate. In this way, the non-oxidative phase supports the continuation of the oxidative phase by replenishing the starting substrate.
The process can be divided into three groups of reactions.
Oxidative phase reactions
These reactions are catalyzed by the enzymes of the oxidative phase, producing two molecules of NADPH and one molecule of ribulose 5-phosphate per glucose 6-phosphate.
Non-oxidative phase reactions
These include the enzymes phosphopentose epimerase, ribose 5-phosphate isomerase, transketolase, and transaldolase, which convert ribulose 5-phosphate into fructose 6-phosphate and glyceraldehyde 3-phosphate.
Gluconeogenic reactions
The fructose 6-phosphate and glyceraldehyde 3-phosphate produced in the non-oxidative phase are recycled into glucose 6-phosphate via gluconeogenic reactions. These reactions allow the cycle to restart.
The net result of the last two groups of reactions is the conversion of six molecules of ribulose 5-phosphate into five molecules of glucose 6-phosphate.
6 Ribulose 5-phosphate+ H2O → 5 Glucose 6-phosphate + Pi
Finally, by summing all three groups of reactions, the oxidative phase, non-oxidative phase, and gluconeogenic steps, the overall reaction is:
Glucose 6-phosphate + 12 NADP+ + 7 H2O → 6 CO2 + 12 NADPH + 12 H+ + Pi
Thus, one molecule of glucose 6-phosphate, through six complete cycles of the pentose phosphate pathway (coupled with specific gluconeogenic reactions), is oxidized to six molecules of CO2. In the process, twelve molecules of NADPH are generated, with no net production of ribose 5-phosphate.[5][8][12]
When the need for NADPH and ATP is much greater than that for ribose 5-phosphate
When the cell requires significantly more NADPH than ribose 5-phosphate, and the energy charge is low, that is, there is a need for ATP, the ribulose 5-phosphate formed in the oxidative phase of the pentose phosphate pathway is converted into fructose 6-phosphate and glyceraldehyde 3-phosphate through the reactions of the non-oxidative phase. These two intermediates then enter glycolysis, where they are oxidized to pyruvate (the conjugate base of pyruvic acid), with the concomitant production of ATP.
The net reaction is as follows:
If the cell requires additional ATP, the pyruvate produced can be further oxidized via the citric acid cycle. Conversely, if there is no need for further ATP production, the carbon skeleton of pyruvate can be diverted into various biosynthetic pathways.
Note: as in the previous scenario, there is no net production of ribose 5-phosphate.[5][8][12]
When the need for ribose 5-phosphate is much greater than that for NADPH
When significantly more ribose 5-phosphate than NADPH is required, as in rapidly dividing cells with high rates of nucleotide synthesis, precursors of DNA, the oxidative phase of the pentose phosphate pathway is bypassed, and NADPH is not produced. Instead, the rapid utilization of ribose 5-phosphate causes its intracellular levels to drop, thereby stimulating its synthesis. This is possible because the reactions of the non-oxidative phase are readily reversible.
In this metabolic context, most of the glucose 6-phosphate is first converted into fructose 6-phosphate and glyceraldehyde 3-phosphate via glycolysis. These intermediates are then used by transaldolase and transketolase to generate ribose 5-phosphate and xylulose 5-phosphate. The latter is further converted into ribose 5-phosphate through the actions of phosphopentose epimerase and ribose 5-phosphate isomerase.
The overall reaction is:
Under these metabolic conditions, there is a functional interplay between glycolysis and the non-oxidative phase of the pentose phosphate pathway, with the latter running in the direction of ribose 5-phosphate synthesis.
Note: no metabolites return to glycolysis in this case.[5][8][12]
When the needs for ribose 5-phosphate and NADPH are balanced
If the metabolic needs of the cell are satisfied by one molecule of ribose 5-phosphate and two molecules of NADPH per molecule of glucose 6-phosphate metabolized, the predominant reactions are those of the oxidative phase and the one catalyzed by ribose 5-phosphate isomerase.
The net reaction is:
Under these metabolic conditions as well, no metabolites return to glycolysis.[5][8][12]
Metabolic scenarios compared
Scenario
Predominant pathway
Main output
Notes
High NADPH demand, low ribose 5-P or ATP need
Full oxidative phase + gluconeogenic recycling
↑ NADPH (12 per G6P), CO2 (6), no net ribose 5-P
Supports fatty acid synthesis (e.g., in adipose tissue)
High NADPH and ATP demand, low ribose 5-P need
Oxidative phase + glycolysis
NADPH (6), ATP (8), pyruvate (5), NADH (5), CO2 (3)
Supports both biosynthesis and energy production
High ribose 5-P demand, low NADPH need
Non-oxidative phase from glycolytic intermediates
Ribose 5-P (6 per 6 G6P), consumes ATP
Found in rapidly dividing cells (e.g., cancer, bone marrow)
Balanced need for ribose 5-P and NADPH
Oxidative phase + ribose 5-P isomerase
Ribose 5-P (1), NADPH (2), CO2; no return to glycolysis
Typical of balanced anabolic demand
Refences
^ abcdef Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ abcd Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009.
^ Gebril H.M., Avula B., Wang Y.H., Khan I.A., Jekabsons M.B. (13)C metabolic flux analysis in neurons utilizing a model that accounts for hexose phosphate recycling within the pentose phosphate pathway. Neurochem Int 2016;93:26-39. doi:10.1016/j.neuint.2015.12.008
^ abcdefghijk Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
^ abcd Samland A.K., Sprenger G.A. Transaldolase: from biochemistry to human disease. Int J Biochem Cell Biol 2009;41(7):1482-1494. doi:10.1016/j.biocel.2009.02.001
^ abc Michal G., Schomburg D. Biochemical pathways. An atlas of biochemistry and molecular biology. 2nd Edition. John Wiley & Sons, 2012.
^ Stincone A., Prigione A., Cramer T., Wamelink M.M., Campbell K., Cheung E., Olin-Sandoval V., Grüning N.M., Krüger A., Tauqeer Alam M., Keller M.A., Breitenbach M., Brindle K.M., Rabinowitz J.D., Ralser M. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc 2015;90(3):927-63. doi:10.1111/brv.12140
^ abcdefghijk Berg J.M., Tymoczko J.L., Gregory J.G. Jr., Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
^ Sprenger G.A. Genetics of pentose-phosphate pathway enzymes of Escherichia coli K-12. Arch Microbiol 1995;164(5):324-30. doi:10.1007/BF02529978
^ abc Au S.W.N., Gover S., Lam V.M.S., Adams M.J. Human glucose-6-phosphate dehydrogenase: the crystal structure reveals a structural NADP+ molecule and provides insights into enzyme deficiency. Structure 2000;8(3):293-303. doi:10.1016/S0969-2126(00)00104-0
^ Levy H.R. Glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides. Biochem Soc Trans. 1989 Apr;17(2):313-5. doi:10.1042/bst0170313
^ Cosgrove M.S., Naylor C., Paludan S., Adams M.J., Richard Levy H. On the mechanism of the reaction catalyzed by glucose 6-phosphate dehydrogenase. Biochemistry 1998;37(9);2759-2767. doi:10.1021/bi972069y
^ Stanton R.C. Glucose 6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012;64(5):362-9. doi:10.1002/iub.1017
^ abc Patra K.C., Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci 2014;39(8):347-354. doi:10.1016/j.tibs.2014.06.005
^ García-Domínguez E., Carretero A., Viña-Almunia A., Domenech-Fernandez J., Olaso-Gonzalez G., Viña J., Gomez-Cabrera M.C. Glucose 6-P dehydrogenase – An antioxidant enzyme with regulatory functions in skeletal muscle during exercise. Cells 2022;11(19):3041. doi:10.3390/cells11193041
^ Phégnon L., Pérochon J., Uttenweiler-Joseph S., Cahoreau E., Millard P., Létisse F. 6-Phosphogluconolactonase is critical for the efficient functioning of the pentose phosphate pathway. FEBS J 2024;291(20):4459-4472. doi:10.1111/febs.17221
^ ab Ruda G.F., Campbell G., Alibu V.P., Barrett M.P., Brenk R., Gilbert I.H. Virtual fragment screening for novel inhibitors of 6-phosphogluconate dehydrogenase. Bioorg Med Chem 2010;18(14):5056-62. doi:10.1016/j.bmc.2010.05.077
^ ab Hanau S., Montin K., Cervellati C., Magnani M., Dallocchio F. 6-Phosphogluconate dehydrogenase mechanism: evidence for allosteric modulation by substrate. J Biol Chem 2010;285(28):21366-21371. doi:10.1074/jbc.M110.105601
^ Allard S.T., Giraud M.F., Naismith J.H. Epimerases: structure, function and mechanism. Cell Mol Life Sci 2001;58(11):1650-65. doi:10.1007/PL00000803
^ Samuel J., Tanner M.E. Mechanistic aspects of enzymatic carbohydrate epimerization. Nat Prod Rep 2002;19(3):261-77. doi:10.1039/b100492l
^ Bartkevihi L., Caruso Í.P., Martins B., Pires, J.R.M., Oliveira D.M.P., Anobom C.D., Almeida, F.C.L. Insights into the substrate uptake mechanism of Mycobacterium tuberculosis ribose 5-phosphate isomerase and perspectives on drug development. Biophysica 2023;3:139-157. doi:10.3390/biophysica3010010
^ Zhang R., Andersson C.E., Savchenko A., Skarina T., Evdokimova E., Beasley S., Arrowsmith C.H., Edwards A.M., Joachimiak A., Mowbray S.L. Structure of Escherichia coli ribose-5-phosphate isomerase: a ubiquitous enzyme of the pentose phosphate pathway and the Calvin cycle. Structure 2003;11(1):31-42. doi:10.1016/S0969-2126(02)00933-4
^ Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003;100:5107-5112. doi:10.1073/pnas.0730817100
^ Jelakovic S., Kopriva S., Süss K-H, Schulz G.E. Structure and catalytic mechanism of the cytosolic D-ribulose-5-phosphate 3-epimerase from rice. J Mol Biol 2003;326:127-135. doi:10.1016/S0022-2836(02)01374-8
^ abc Kochetov G.A., Solovjeva O.N. Structure and functioning mechanism of transketolase. Biochim Biophys Acta 2014;1844(9):1608-1618. doi:10.1016/j.bbapap.2014.06.003
^ Solovjeva O.N. Regulation of enzymes with identical subunits on the example of transketolase. Open J Anal Bioanal Chem 2022;6(1):004-012. doi:10.17352/ojabc.000024
^ Sharkey T.D. Pentose phosphate pathway reactions in photosynthesizing cells. Cells 2021;10(6):1547. doi:10.3390/cells10061547
^ ab Lehwess-Litzmann A., Neumann P., Parthier C., Lüdtke S., Golbik R., Ficner R., Tittmann K. Twisted Schiff base intermediates and substrate locale revise transaldolase mechanism. Nat Chem Biol 2011;7(10):678-84. doi:10.1038/nchembio.633
^ Van Schaftingen E., Hers H-G. Inhibition of fructose-1,6-bisphosphatase by fructose-2,6-bisphosphate. Proc Natl Acad Sci USA 1981;78(5):2861-2863. doi:10.1073/pnas.78.5.2861
^ Van Schaftingen E., Jett M-F., Hue L., Hers H-G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA 1981;78(6):3483-3486. doi:10.1073/pnas.78.6.3483
Glycolysis, also known as the glycolytic pathway, from the Greek words glykys (“sweet”) and lysis (“dissolution” or “breakdown”), is the sequence of enzymatic reactions that occur in the cytosol and, even in the absence of oxygen, convert glucose, a six-carbon sugar, into two molecules of pyruvate, a three-carbon compound. This process is accompanied by the production of two molecules of ATP and NADH.[1]
Glycolysis, which evolved before a significant accumulation of oxygen in the atmosphere, is the metabolic pathway with the highest carbon flux in most living cells and is present in nearly all organisms.[1]
Because it does not require oxygen, glycolysis played a crucial role during the first two billion years of life’s evolution, representing one of the earliest biological mechanisms for energy extraction from organic molecules. In addition, it supplies key intermediates for aerobic catabolism and numerous biosynthetic pathways.[2]
Note: glycolysis is also known as the Embden–Meyerhof–Parnas pathway, named after Gustav Embden, Otto Meyerhof, and Jakub Karol Parnas, the researchers who elucidated the pathway in muscle tissue.[3]
Summary: Key Points
Definition: a sequence of 10 cytosolic enzymatic reactions which, even in the absence of oxygen, convert glucose into pyruvate, with the simultaneous production of 2 molecules of ATP and 2 of NADH.
Fates of pyruvate: in the presence of oxygen, pyruvate can enter the Krebs cycle, while in the absence of oxygen it can be converted via lactic or alcoholic fermentation into lactate and ethanol, respectively.
Regulation: mainly mediated by three key enzymes (hexokinase, phosphofructokinase-1, and pyruvate kinase) through allosteric mechanisms, covalent modifications, and hormonal signals.
The development of biochemistry progressed in parallel with the study of glucose metabolism, particularly glycolysis, the first metabolic pathway to be fully elucidated.[4][5]
Although the pathway was not completely mapped until the 1940s, a key discovery was made in 1897, by chance. A year earlier, German chemist M. Hahn, while attempting to obtain and preserve cell-free protein extracts of yeast, had encountered difficulties with their stability. His colleague Hans Buchner, recalling a method used for preserving jams, suggested adding sucrose to the extract.[6]
Eduard Buchner, Hans’ brother, applied this idea and observed that the solution produced bubbles. He concluded that fermentation was occurring, an entirely unexpected finding. According to Pasteur’s assertion in 1860, fermentation was thought to be inseparably linked to living cells. Eduard’s experiments, however, demonstrated that it could also occur in their absence. These results disproved the vitalist dogma and marked a turning point in the emergence of modern biochemistry.[7]
For this work, Eduard Buchner was awarded the Nobel Prize in Chemistry in 1907, becoming the first of several scientists later honored for discoveries related to glycolysis. Subsequent studies with muscle extracts revealed that many of the reactions involved in lactic fermentation were identical to those in alcoholic fermentation, highlighting the fundamental unity of biochemistry.[8]
As noted, glycolysis was eventually fully elucidated during the first half of the 20th century, thanks to the contributions of Gerty and Carl Cori, Carl Neuberg, Arthur Harden, William Young, Jacob Parnas, Otto Warburg, Hans von Euler-Chelpin, Gustav Embden, and Otto Meyerhof. Warburg and von Euler-Chelpin clarified the pathway in yeast, while Embden and Meyerhof did so in muscle tissue during the 1930s.[3]
Importance of glycolysis
Glycolysis is essential for most living cells, both as a source of energy and as a provider of precursors for many other metabolic pathways. The rate of carbon flow through glycolysis, that is, the amount of glucose converted to pyruvate per unit of time, is regulated to satisfy these two fundamental cellular demands.
Although relatively inefficient energetically, glycolysis can proceed in the absence of oxygen, a condition under which life first evolved and one still encountered by many modern cells, both eukaryotic and prokaryotic. Several examples illustrate this adaptability.[9]
Anaerobic glycolysis
In animals, skeletal muscle exhibits activity-dependent anaerobiosis; it can function without oxygen for short periods. During intense exercise, when ATP demand exceeds oxygen delivery, muscles continue to operate anaerobically, though only temporarily.
The cornea of the eye provides another example, because it is poorly vascularized.[9]
Many microorganisms also thrive in low-oxygen environments such as deep water, soil, or skin pores. Some, like Clostridium perfringens, Clostridium tetani, and Clostridium botulinum, which cause gangrene, tetanus, and botulism, respectively, are obligate anaerobes that cannot survive in the presence of oxygen.[10]
Glycolysis in glucose-dependent cells
Glycolysis also sustains cells and tissues where glucose is the sole source of energy, including:
red blood cells, which lack mitochondria,
sperm cells;
the brain (which can also utilize ketone bodies during glucose scarcity);
A similar pattern occurs in plants. Many aquatic plants and starch-storing tissues (e.g., potato tubers) rely on glucose as their main energy source.[12]
Note: some organisms, known as facultative anaerobes, can survive both in the presence and absence of oxygen, switching between aerobic and anaerobic metabolism. For instance, Mytilus species exhibit habitat-dependent anaerobiosis, comparable to that of muscle tissue.[13]
Aerobic glycolysis
Under aerobic conditions, in cells containing mitochondria, glycolysis represents the first stage of the metabolic pathway that leads to the complete oxidation of glucose to carbon dioxide (CO2) and water (H2O), producing ATP.[14]
Sources of building blocks
Several glycolytic intermediates, for example, glucose 6-phosphate (G6P), fructose 6-phosphate (F6P), and dihydroxyacetone phosphate (DHAP), serve as building blocks for other metabolic pathways. These include glycogen synthesis, as well as the biosynthesis of fatty acids, triglycerides, nucleotides, certain amino acids, and 2,3-bisphosphoglycerate (2,3-BPG).[15]
Glycolysis as a Source of Building Blocks
Sequence of reactions
The ten steps that constitute glycolysis can be divided into two phases.
The first, known as the preparatory phase, consists of five steps and begins with the conversion of glucose to fructose 1,6-bisphosphate (F1,6BP) through three enzymatic reactions: phosphorylation at C-6, isomerization, and a second phosphorylation at C-1, with the consumption of two ATP molecules.
Fructose 1,6-bisphosphate is then cleaved into two phosphorylated three-carbon compounds: glyceraldehyde 3-phosphate (G3P) and dihydroxyacetone phosphate. Finally, DHAP is isomerized into a second molecule of glyceraldehyde 3-phosphate. Thus, in the preparatory phase, one glucose molecule is split into two molecules of glyceraldehyde 3-phosphate, with a net consumption of two ATP molecules.
The Glycolytic Pathway
The second phase, the payoff phase, consists of the remaining five steps. In this phase, the two molecules of glyceraldehyde 3-phosphate are converted into two molecules of pyruvate, with the simultaneous production of four ATP molecules. Part of the energy stored in the chemical bonds of glucose is extracted and conserved in the form of ATP. In addition, reducing equivalents are captured and stored in the form of NADH, the reduced form of the coenzyme NAD+ (nicotinamide adenine dinucleotide). The metabolic fate of NADH depends on the cell type and whether conditions are aerobic or anaerobic.
Note: the glucose metabolized via the glycolytic pathway may originate either from extracellular glucose entering the cell through specific membrane transporters (derived from the bloodstream) or from intracellular glucose 6-phosphate produced by glycogenolysis.[11]
Reaction 1
In the first step of glycolysis, glucose is phosphorylated to glucose 6-phosphate at the expense of one ATP molecule.
Glucose + ATP → G6P + ADP
In most cells, this reaction is catalyzed by hexokinase (EC 2.7.1.1), an enzyme present in all organisms. In humans, four isoforms of hexokinase are expressed.[16] Both hexokinase and pyruvate kinase (EC 2.7.1.40), two of the key kinases of glycolysis, require a divalent metal ion such as magnesium (Mg2+) or manganese (Mn2+) for activity. Mg2+ binds to ATP, forming the MgATP2− complex, which is the true substrate of the enzyme rather than free ATP. The nucleophilic attack by the hydroxyl group (–OH) of glucose on the terminal (γ) phosphorus atom of ATP is facilitated by Mg2+, which stabilizes the negative charges on the phosphoryl groups of the nucleotide triphosphate.
The reaction involves the formation of a phosphoester bond between the phosphoryl group and the hydroxyl group at the C-6 position of glucose. This step is thermodynamically unfavorable and requires an input of energy, provided by ATP.[3]
Energetics and irreversibility
In the absence of ATP, the direct phosphorylation of glucose at C-6 by inorganic phosphate (Pi) is endergonic, with a standard Gibbs free energy change (ΔG°′) of +13.8 kJ/mol (+3.3 kcal/mol).
ATP hydrolysis to ADP and Pi has a ΔG°’ of –30.5 kJ/mol (–7.3 kcal/mol), an exergonic process.
The net ΔG°’ of the coupled reaction is –16.7 kJ/mol (–4.0 kcal/mol).
Under physiological conditions, the reaction is even more favorable, with a ΔG of ≈ –33.5 kJ/mol (–8.0 kcal/mol). Consequently, this step is considered essentially irreversible.[17]
Note: phosphorylation is a fundamental biochemical reaction catalyzed by kinases, a subclass of transferases. Kinases catalyze the transfer of the terminal phosphoryl group from a nucleoside triphosphate (usually ATP) to an acceptor nucleophile, forming a phosphoester bond. Specifically, hexokinase catalyzes the transfer of the γ-phosphoryl group of ATP to a variety of hexoses, including glucose, fructose, and mannose.[5]
Reaction 1 – Phosphorylation of Glucose
Property
Details
Reaction
Glucose + ATP → G6P + ADP
Enzyme
Hexokinase. In humans: four isoforms.
Cofactors
Mg2+ (as MgATP2−) or Mn2+
ΔG°′
–16.7 kJ/mol (net) (–4.0 kcal/mol)
ΔG
≈ –33.5 kJ/mol (–8.0 kcal/mol)
Reversibility
Irreversible under physiological conditions
Reaction 2
In the second step of glycolysis, G6P, an aldose, is isomerized to fructose 6-phosphate, a ketose.
G6P ⇄ F6P
This reaction is catalyzed by phosphoglucose isomerase (also known as phosphohexose isomerase or glucose phosphate isomerase; EC 5.3.1.9). The enzyme requires Mg2+ for activity. Glucose 6-phosphate and fructose 6-phosphate represent a case of functional group isomerism.
The ΔG°′ of the reaction is +1.7 kJ/mol (+0.4 kcal/mol), whereas the ΔG is ≈ –2.5 kJ/mol (–0.6 kcal/mol). These small values indicate that the reaction operates near equilibrium and is therefore readily reversible.
The reaction involves a shift of the carbonyl group from C-1 in the open-chain form of glucose 6-phosphate to C-2 in the open-chain form of fructose 6-phosphate.[3]
Mechanism and importance
The enzyme first opens the ring of glucose 6-phosphate, since both hexoses are mainly present in cyclic form in aqueous solution.
It then catalyzes the isomerization of the open-chain intermediate, shifting the carbonyl group from C-1 to C-2.
Finally, it promotes ring closure, forming fructose 6-phosphate, which adopts a five-membered furanose structure.[5]
Phosphoglucose Isomerase Reaction
This isomerization is crucial because it prepares the substrate for subsequent reactions.
In the third step, phosphorylation occurs at the C-1 position, which requires a hydroxyl group rather than a carbonyl group.
In the fourth step, cleavage of the bond between C-3 and C-4 is facilitated by the presence of a carbonyl group at C-2, generated during this isomerization.[11]
Reaction 2 – Isomerization of glucose 6-phosphate
Property
Details
Reaction
G6P ⇄ F6P
Enzyme
Phosphoglucose isomerase
Cofactors
Mg2+
ΔG°′
+1.7 kJ/mol (+0.4 kcal/mol)
ΔG
≈ –2.5 kJ/mol (–0.6 kcal/mol)
Reversibility
Reversible (near equilibrium)
Reaction 3
In the third step of glycolysis, F6P undergoes a second phosphorylation. Phosphofructokinase-1 (PFK-1; EC 2.7.1.11) catalyzes the phosphorylation of fructose 6-phosphate at the C-1 position to yield fructose 1,6-bisphosphate, at the expense of one ATP molecule.
F6P + ATP → F1,6BP + ADP
PFK-1 is named to distinguish it from phosphofructokinase-2 (PFK-2; EC 2.7.1.105), which catalyzes the formation of fructose 2,6-bisphosphate (F2,6BP) from fructose 6-phosphate.[1]
Like the hexokinase-catalyzed reaction, this ATP-dependent phosphorylation step is essentially irreversible. Its irreversibility results from coupling to ATP hydrolysis, which provides the necessary free energy. Phosphorylation of fructose 6-phosphate by inorganic phosphate alone is endergonic, with a ΔG°′ of +16.3 kJ/mol (+3.9 kcal/mol). When coupled to ATP hydrolysis, however, the overall reaction becomes exergonic, with a ΔG°′ of –14.2 kJ/mol (–3.4 kcal/mol) and a cellular ΔG of ≈ –22.2 kJ/mol (–5.3 kcal/mol).[3]
While hexokinase functions primarily to trap glucose inside the cell, PFK-1 commits glucose-derived carbon to glycolysis rather than directing it toward glycogen synthesis or other biosynthetic pathways. Unlike glucose 6-phosphate, fructose 1,6-bisphosphate cannot readily enter alternative metabolic routes, making this the first committed step of glycolysis.
Committed steps are typically catalyzed by allosterically regulated enzymes, preventing the unnecessary accumulation of intermediates and end products. PFK-1 exemplifies this regulation, as it is subject to activation and inhibition by metabolites that reflect both the cell’s energy status and hormonal signals.[5]
Reaction 3 – Phosphorylation of fructose 6-phosphate
Property
Details
Reaction
F6P + ATP → F1,6BP + ADP
Enzyme
Phosphofructokinase-1 (PFK-1)
Cofactors
Mg2+ (as MgATP2–)
ΔG°′
–14.2 kJ/mol (–3.4 kcal/mol)
ΔG
≈ –22.2 kJ/mol (–5.3 kcal/mol)
Reversibility
Irreversible (first committed step of glycolysis)
PPi dependent phosphofructokinase
In some protists and bacteria, and possibly in all plants, a variant phosphofructokinase uses pyrophosphate (PPi) instead of ATP as the phosphoryl donor. This alternative reaction has a ΔG°′ of –12.1 kJ/mol (–2.9 kcal/mol).[18]
F6P + PPi → F1,6BP + Pi
Reaction 4
In the fourth step of glycolysis, fructose 1,6-bisphosphate aldolase (commonly known as aldolase; EC 4.1.2.13) catalyzes the reversible cleavage of F1,6BP into two triose phosphates: glyceraldehyde 3-phosphate (an aldose) and dihydroxyacetone phosphate (a ketose). The enzyme cleaves the carbon–carbon bond between C-3 and C-4 of the fructose backbone.[1]
F1,6-BP ⇄ DHAP + G3P
From this step onward, all glycolytic intermediates are three-carbon compounds, whereas the preceding intermediates were six-carbon sugars.[11]
Note on nomenclature
The name “aldolase” derives from the reverse reaction, i.e., the condensation of an aldehyde and a ketone to form a β-hydroxyketone (an aldol), in a process known as aldol condensation.
Energetics and reversibility
The ΔG°′ of the cleavage reaction is +23.8 kJ/mol (+5.7 kcal/mol), with an equilibrium constant (Keq) of approximately 10−4 M. These values indicate that under standard conditions the reaction would not favor the forward (cleavage) direction. However, in the cellular context, the products are rapidly consumed in subsequent reactions, maintaining their concentrations at low levels. As a result, the actual free energy change is about –1.3 kJ/mol (–0.3 kcal/mol), showing that the reaction is near equilibrium and easily reversible.[5]
Reaction 4 – Cleavage of fructose 1,6-bisphosphate
Property
Details
Reaction
F1,6 ⇄ DHAP + G3P
Enzyme
Aldolase (Fructose 1,6-bisphosphate aldolase)
ΔG°′
+23.8 kJ/mol (+5.7 kcal/mol)
ΔG
≈ –1.3 kJ/mol (–0.3 kcal/mol)
Reversibility
Reaction near equilibrium; readily reversible
Note
The enzyme name originates from the reverse aldol condensation reaction
Reaction 5
Of the two products generated in the previous step, glyceraldehyde 3-phosphate directly enters the second phase of glycolysis. In contrast, DHAP is not part of the main glycolytic route and must first be isomerized into glyceraldehyde 3-phosphate. This reversible reaction is catalyzed by triose phosphate isomerase (EC 5.3.1.1).
DHAP ⇄ G3P
Triose phosphate isomerase catalyzes a keto–enol tautomerization involving the transfer of a hydrogen atom from C-1 to C-2 of DHAP. As a consequence of this isomerization, the carbon atoms C-1, C-2, and C-3 of glucose become chemically indistinguishable from C-6, C-5, and C-4, respectively. The net result of reactions 4 and 5 is therefore the production of two molecules of glyceraldehyde 3-phosphate: one formed directly in reaction 4, and the other via isomerization.[3]
The ΔG°′ of this step is +7.5 kJ/mol (+1.8 kcal/mol), while the actual ΔG under cellular conditions is about +2.5 kJ/mol (+0.6 kcal/mol). At equilibrium, ≈ 96% of the triose phosphate pool exists as DHAP. Nonetheless, the reaction proceeds efficiently toward G3P because this intermediate is rapidly consumed in the following glycolytic step, driving the reaction forward.[1]
One notable feature of triose phosphate isomerase is its extraordinary catalytic efficiency. The enzyme is considered kinetically “perfect” because it accelerates the reaction rate by a factor of 1010 compared to a simple catalyst (e.g., acetate ion). Its catalytic efficiency, expressed as the Kcat/KM ratio, is about 2 x 108 M–1 s–1, approaching the diffusion-controlled limit. Thus, the rate-limiting factor is not the chemistry of the reaction itself but the rate at which substrate and enzyme encounter each other.[19]
Energetics and metabolic relevance
From a thermodynamic perspective, reactions 4 and 5 together are unfavorable, with a combined ΔG°′ of +31.3 kJ/mol (+7.5 kcal/mol). However, when the hydrolysis of two ATP molecules in earlier steps is taken into account, the overall ΔG°′ for the preparatory phase (steps 1–5) is only +2.1 kJ/mol (+0.5 kcal/mol). The corresponding Keq is ≈ 0.43. Importantly, under physiological conditions, the actual ΔG is strongly negative (≈ –56.8 kJ/mol; –13.6 kcal/mol), ensuring the pathway proceeds forward.[17]
Beyond glycolysis, DHAP can also serve as a precursor in lipid metabolism. It can be reduced to glycerol 3-phosphate by cytosolic glycerol 3-phosphate dehydrogenase (EC 1.1.1.8)
DHAP + NADH + H+ ⇄ Glycerol 3-phosphate + NAD+
This reaction links carbohydrate metabolism to lipid biosynthesis, as glycerol 3-phosphate is an essential precursor for triacylglycerols and phospholipids. In adipose tissue and the small intestine, this pathway represents a major source of glycerol 3-phosphate.[11]
Reaction 5 – Isomerization of DHAP to G3P
Property
Details
Reaction
DHAP ⇄ G3P
Enzyme
Triose phosphate isomerase
ΔG°′
+7.5 kJ/mol (+1.8 kcal/mol)
ΔG
≈ +2.5 kJ/mol (+0.6 kcal/mol), but pulled forward by rapid G3P utilization
Equilibrium
≈ 96% DHAP at equilibrium
Catalytic efficiency
Kinetically “perfect”: Kcat/KM ≈ 2 x 108 M–1s–1
Additional role
DHAP can be reduced to glycerol 3-phosphate, linking glycolysis to lipid metabolism
Reaction 6
In the sixth step of glycolysis, and the first of the payoff phase, glyceraldehyde 3-phosphate dehydrogenase (EC 1.2.1.12) catalyzes the oxidation of G3P to 1,3-bisphosphoglycerate (1,3-BPG), coupled with the reduction of NAD+ to NADH.[3]
G3P + NAD+ + Pi ⇄ 1,3-BPG + NADH + H+
This is the first of two glycolytic reactions that conserve the chemical energy required for the subsequent synthesis of ATP.
Mechanism and energetics
The reaction consists of two tightly coupled processes.
Oxidation: the aldehyde group of G3P is oxidized to a carboxylic acid, with NAD+ acting as the oxidizing agent. This process is strongly exergonic (ΔG°′ = –43.1 kJ/mol, −10.3 kcal/mol).
Phosphorylation: a high-energy acyl phosphate bond is formed between the new carboxyl group and inorganic phosphate, yielding 1,3-BPG. This step is highly endergonic (ΔG°′ = +49.3 kJ/mol, +11.8 kcal/mol).[11]
Although each process alone would be thermodynamically unbalanced, their tight coupling allows the energy released from oxidation to drive acyl phosphate formation. The overall reaction has a ΔG°′ of +6.3 kJ/mol (+1.5 kcal/mol) and a cellular ΔG of ≈ +2.5 kJ/mol (+0.6 kcal/mol), both slightly endergonic.[17]
Thus, free energy is conserved in the form of the high-energy acyl phosphate bond of 1,3-BPG, rather than being lost as heat.[20]
Reaction 6 – Oxidation and Phosphorylation of G3P
Property
Details
Reaction
G3P + NAD+ + Pi ⇄ 1,3-BPG + NADH + H+
Enzyme
Glyceraldehyde 3-phosphate dehydrogenase
Cofactor
NAD+ (reduced to NADH)
ΔG°′
+6.3 kJ/mol (+1.5 kcal/mol)
ΔG
≈ +2.5 kJ/mol (+0.6 kcal/mol)
Importance
First energy-conserving step; formation of high-energy acyl phosphate bond
Note on NAD+ reduction
The reversible reduction of NAD⁺ involves the transfer of two hydrogen atoms from the substrate, the aldehyde group of glyceraldehyde 3-phosphate undergoing oxidation: one as a hydride ion (2 e− + H+) to the nicotinamide ring, and the other as a free proton released into solution.[5]
Half-reactions:
NAD+ + 2 e– + 2 H+ → NADH + H+
NADP+ + 2 e– + 2 H+ → NADPH + H+
Reaction 7
In the seventh step of glycolysis, phosphoglycerate kinase (EC 2.7.2.3) catalyzes the transfer of a high-energy phosphoryl group from the acyl phosphate of 1,3-BPG to ADP, forming ATP and 3-phosphoglycerate (3-PG).[1]
1,3-BPG + ADP ⇄ 3-PG + ATP
The reaction has a ΔG°′ of −18.5 kJ/mol (−4.4 kcal/mol), making it exergonic, although under cellular conditions the free energy change is ≈ +1.3 kJ/mol (+0.3 kcal/mol).[17]
Energetics and reversibility
The high phosphoryl-transfer potential of the acyl phosphate group is harnessed to phosphorylate ADP in a process known as substrate-level phosphorylation. In other words, part of the energy released during the oxidation of glyceraldehyde 3-phosphate in the previous step is conserved in the synthesis of ATP.[5]
This is the first step in glycolysis where the chemical energy of glucose is captured as ATP. Since aldolase and triose phosphate isomerase (steps 4 and 5) yield two molecules of glyceraldehyde 3-phosphate per glucose, this step generates two ATP molecules, thereby offsetting the ATP investment made earlier (steps 1 and 3).
The enzyme name refers to the reverse reaction, i.e., the phosphorylation of 3-phosphoglycerate to form 1,3-bisphosphoglycerate using ATP. Like all enzymes, phosphoglycerate kinase can catalyze the reaction in both directions. The reverse reaction occurs in gluconeogenesis and in photosynthetic CO2 fixation.[21]
Reaction 7 – Substrate-level Phosphorylation by Phosphoglycerate Kinase
Property
Details
Reaction
1,3-BPG + ADP ⇄ 3-PG + ATP
Enzyme
Phosphoglycerate kinase
ΔG°′
−18.5 kJ/mol (−4.4 kcal/mol)
ΔG
≈ +1.3 kJ/mol (+0.3 kcal/mol)
Process
Substrate-level phosphorylation; ATP generation from the high-energy acyl phosphate of 1,3-BPG
ATP Yield
2 ATP per glucose (since 2 × G3P enter the payoff phase)
Importance
First ATP-generating step in glycolysis; compensates the ATP invested in the preparatory phase
Reversibility
Reversible reaction; reverse direction occurs in gluconeogenesis and photosynthesis
Coupling with reaction 6
Reactions 6 and 7 together constitute an energy-coupling system, with 1,3-BPG as the common intermediate.
While the synthesis of 1,3-BPG is endergonic (ΔG°′ = +6.3 kJ/mol; +1.5 kcal/mol), the subsequent phosphoglycerate kinase reaction is strongly exergonic (ΔG°′ = −18.5 kJ/mol, −4.4 kcal/mol). The net ΔG°′ is −12.2 kJ/mol (−2.9 kcal/mol), sufficient to drive not only the preceding dehydrogenase step, but also the aldolase and triose phosphate isomerase reactions.
G3P + ADP + Pi + NAD+ ⇄ 3-PG + ATP + NADH + H+
The phosphoryl group of 3-phosphoglycerate has a relatively low transfer potential (ΔG°′ ≈ −12.5 kJ/mol; −3 kcal/mol), insufficient to directly drive ATP synthesis. Therefore, in the next steps of glycolysis, 3-phosphoglycerate is converted into phosphoenolpyruvate (PEP), a compound with much higher phosphoryl-transfer potential.[3]
Reaction 8
In the eighth step of glycolysis, 3-PG is converted into 2-phosphoglycerate (2-PG) in a reversible reaction catalyzed by phosphoglycerate mutase (EC 5.4.2.1). This reaction has a very small free energy change, with ΔG°′ = +4.4 kJ/mol (+1.1 kcal/mol) and ΔG ≈ +0.8 kJ/mol (+0.2 kcal/mol).[17]
Phosphoglycerate mutase belongs to the class of mutases, enzymes that catalyze intramolecular group transfers. In this case, it catalyzes the transfer of a phosphoryl group from the C-3 to the C-2 position of 3-PG. Mutases are a subclass of isomerases.[22]
3-PG ⇄ 2-PG
Mechanism and significance
The mechanism of this reaction varies among organisms. For example, in yeast and rabbit muscle, the reaction proceeds in two steps and involves the formation of phosphoenzyme intermediates.
In the first step, a phosphoryl group bound to a histidine residue in the enzyme’s active site is transferred to the hydroxyl group at C-2 of 3-PG, generating 2,3-bisphosphoglycerate.
In the second step, the enzyme acts as a phosphatase, converting 2,3-BPG into 2-phosphoglycerate, while the phosphoryl group at C-3 is transferred back to the histidine residue, regenerating the phosphorylated enzyme (enzyme–His–phosphate).
Schematically:
It is important to note that the phosphoryl group in 2-phosphoglycerate is not the same as the one originally present in 3-phosphoglycerate.[5]
Reaction 8 – Conversion of 3-phosphoglycerate to 2-phosphoglycerate
Property
Details
Reaction
3-PG ⇄ 2-PG
Enzyme
Phosphoglycerate mutase
Cofactor
Requires 2,3-bisphosphoglycerate for activation
ΔG°′
+4.4 kJ/mol (+1.1 kcal/mol)
ΔG
≈ +0.8 kJ/mol (+0.2 kcal/mol)
Mechanism
Proceeds via a phosphoenzyme intermediate and transient formation of 2,3-bisphosphoglycerate
Importance
Rearranges the phosphoryl group to prepare for the high-energy intermediate phosphoenolpyruvate
2,3-BPG and the Rapoport–Luebering shunt
Approximately once every 100 catalytic cycles, 2,3-BPG dissociates from the enzyme, leaving it unphosphorylated and inactive. The enzyme can be reactivated by binding a new molecule of 2,3-BPG, which must therefore be present in the cytosol to maintain optimal activity. Small but sufficient amounts of 2,3-BPG are found in most cells, with the notable exception of red blood cells, where it is synthesized through the Rapoport–Luebering shunt. In erythrocytes, 2,3-BPG also acts as an allosteric inhibitor of hemoglobin, lowering its affinity for oxygen.[23][24]
Note: 3-phosphoglycerate also serves as a precursor for the biosynthesis of serine, from which glycine and cysteine are derived. The first step in serine biosynthesis is catalyzed by phosphoglycerate dehydrogenase (EC 1.1.1.95), which oxidizes 3-phosphoglycerate to 3-phosphohydroxypyruvate while reducing NAD+ to NADH. This step is rate-limiting, as serine acts as a feedback inhibitor of the enzyme.[25]
Reaction 9
In the ninth step of the glycolytic pathway, 2-PG is dehydrated to form phosphoenolpyruvate, an enol, in a reversible reaction catalyzed by enolase (EC 4.2.1.11).
2-PG ⇄ PEP + H2O
This reaction requires Mg2+, which stabilizes the enolic intermediate formed during the process.[11]
The ΔG°’ of the reaction is +7.5 kJ/mol (+1.8 kcal/mol), while ΔG = –3.3 kJ/mol (–0.8 kcal/mol), indicating that the reaction proceeds readily in vivo.[17]
Phosphoryl transfer potential and tautomerization
Like 1,3-bisphosphoglycerate, phosphoenolpyruvate possesses a phosphoryl group with a very high transfer potential, sufficient to allow ATP synthesis.
But why is the free energy of hydrolysis of PEP so high?
Although 2-PG and PEP contain nearly the same total amount of metabolic energy relative to complete oxidation to CO2, H2O and Pi, the dehydration of 2-PG redistributes this energy, leading to very different standard free energies of hydrolysis:
–17.6 kJ/mol (–4.2 kcal/mol) for 2-phosphoglycerate (a phosphoric ester);
–61.9 kJ/mol (–14.8 kcal/mol) for phosphoenolpyruvate (an enol phosphate).
This difference arises because the phosphoryl group of PEP locks the molecule in its unstable enol form. In the final step of glycolysis, when PEP donates its phosphoryl group to ADP, ATP and the enol form of pyruvate are generated. The enol form of pyruvate is unstable and rapidly tautomerizes nonenzymatically into the more stable keto form, which predominates at physiological pH (≈7).
Thus, the extraordinarily high phosphoryl-transfer potential of PEP is due primarily to the subsequent enol–keto tautomerization of pyruvate, which provides a strong thermodynamic driving force.[5][26]
Reaction 9 – Dehydration of 2-phosphoglycerate to phosphoenolpyruvate
PEP stores energy in an unstable enol form; tautomerization of pyruvate (enol → keto) drives high phosphoryl-transfer potential
Reaction 10
In the final step of glycolysis, pyruvate kinase catalyzes the transfer of a phosphoryl group from PEP to ADP, producing pyruvate and ATP. This is the second example of substrate-level phosphorylation in the pathway.
PEP + ADP → Pyruvate + ATP
Pyruvate kinase is a tetrameric enzyme and, like PFK-1, it is highly regulated. It contains binding sites for several allosteric effectors. In vertebrates, at least three isozymes exist: the M isozyme (predominant in muscle and brain) and the L isozyme (predominant in liver) are the best characterized. Although they share many properties, they differ in their regulation by hormones such as glucagon, epinephrine, and insulin.
Enzyme activity is also stimulated by potassium ions (K⁺) and other monovalent cations.[1][11][27]
The reaction is essentially irreversible, with a ΔG°′ of –31.4 kJ/mol (–7.5 kcal/mol) and a cellular ΔG of ≈ –16.7 kJ/mol (–4.0 kcal/mol). This strong thermodynamic favorability is largely due to the spontaneous tautomerization of pyruvate from its unstable enol form to its stable keto form.[5]
Spontaneous Tautomerization of Pyruvate
Energetics and reversibility
Of the –61.9 kJ/mol (–14.8 kcal/mol) released from the hydrolysis of the phosphoryl group of PEP, nearly half is conserved in the formation of the phosphoanhydride bond between ADP and Pi, whose ΔG°’ is –30.5 kJ/mol (–7.3 kcal/mol). The remaining energy (–31.4 kJ/mol or –7.5 kcal/mol) serves as the driving force that pushes the reaction toward ATP production.[17]
While the reaction catalyzed by phosphoglycerate kinase (step 7) restores the ATP “debt” from the preparatory phase, the pyruvate kinase reaction yields a net gain of two ATP molecules per glucose.
Reaction 10 – Conversion of PEP to pyruvate
Property
Details
Reaction
PEP + ADP → Pyruvate + ATP
Enzyme
Pyruvate kinase
Cofactors / Activators
K+ and other monovalent cations; allosteric regulation (isozyme-specific)
ΔG°′
–31.4 kJ/mol (–7.5 kcal/mol)
ΔG
≈ –16.7 kJ/mol (–4.0 kcal/mol)
Isozymes
M isozyme (muscle, brain); L isozyme (liver); regulated differently by hormones
Importance
Second substrate-level phosphorylation; net gain of 2 ATP per glucose; irreversibility ensured by pyruvate tautomerization
Fate of NADH and pyruvate
Glycolysis yields 2 NADH, 2 ATP, and 2 pyruvate molecules per molecule of glucose.[1]
For glycolysis to continue, NADH must be reoxidized to NAD+. This coenzyme, derived from vitamin B3 (niacin), is present in the cytosol at a concentration of ≤ 10−5M, far lower than the amount of glucose metabolized within a few minutes, and therefore must be continuously regenerated. The regeneration of NAD+ from NADH constitutes the final step of the glycolytic pathway and can proceed through either aerobic or anaerobic routes, both of which involve pyruvate. These pathways are essential for maintaining the cell’s redox balance.[28]
Pyruvate itself is a versatile metabolite that can participate in several metabolic fates, both anabolic and catabolic, depending on the cell type, the energy status of the cell, and the availability of oxygen.[29]
Three Catabolic Pathways of Pyruvate from Glycolysis
With the exception of certain bacterial variations, exploited, for example, in the food industry for the production of specific fermented products such as cheese, pyruvate typically follows one of three catabolic routes:
reduction to lactate, via lactic acid fermentation;
reduction to ethanol, via alcoholic fermentation;
aerobic oxidation.
These pathways allow glycolysis to proceed under both anaerobic and aerobic conditions. Thus, the catabolic fate of glucose carbon skeletons depends on cell type, energetic state, and oxygen availability.[11]
Lactic acid fermentation
In animals, aside from a few exceptions, and in many microorganisms, when oxygen availability is insufficient to meet the cell’s energy demands, or when the cell lacks mitochondria, the pyruvate generated by glycolysis is reduced to lactate in the cytosol. This reaction is catalyzed by lactate dehydrogenase (EC 1.1.1.27).[11]
Pyruvate + NADH + H+ ⇄ Lactate + NAD+
Here, pyruvate acts as the electron acceptor and is reduced to lactate, while NAD+ is regenerated. The equilibrium strongly favors lactate formation, as indicated by the ΔG°′ of –25.1 kJ/mol (–6.0 kcal/mol).[30]
The overall equation for the conversion of glucose to lactate, known as lactic acid fermentation, is:
Glucose + 2 Pi + 2 ADP → 2 Lactate + 2 ATP + 2 H2O
Discovered by Louis Pasteur, who famously described it as “la vie sans l’air”, fermentation is a metabolic process that:
extracts energy from glucose, conserving it as ATP;
Although NAD+ and NADH do not appear in the overall reaction, both are essential intermediates. Thus, there is no net oxidation or reduction: the hydrogen-to-carbon ratio remains unchanged in the conversion of glucose (C6H12O6) to lactate (C3H6O3).[1]
However, from an energetic standpoint, fermentation captures only a small fraction of the total chemical energy stored in glucose.
Fate of lactic acid
In humans, much of the lactate produced is directed into the Cori cycle, where it is transported to the liver and used for glucose synthesis via gluconeogenesis. This process shifts part of the metabolic load from extrahepatic tissues, such as skeletal muscle during intense exercise (e.g., a 200-meter sprint, when glycolysis can increase up to 2,000-fold almost instantaneously), to the liver.[32]
Unlike skeletal muscle, which releases lactate into venous blood, cardiac muscle can take up and utilize lactate an energy substrate. This is due to its strictly aerobic metabolism and to the properties of its tissue-specific isozyme of lactate dehydrogenase, LDH-1. Consequently, a portion of the lactate released by active skeletal muscle is directly consumed by the heart.[33]
Note: in microorganisms, lactic acid fermentation produces lactate that contributes to the flavor and aroma of foods such as sauerkraut (fermented cabbage) and soured milk.[34]
Alcoholic fermentation
In microorganisms such as brewer’s and baker’s yeast, in certain plant tissues, and in some invertebrates and protists, pyruvate may be converted to ethanol under hypoxic or anaerobic conditions, with the concomitant release of CO2. This occurs in two enzymatic steps.[32]
In the first step, pyruvate undergoes non-oxidative decarboxylation to form acetaldehyde. This essentially irreversible reaction is catalyzed by pyruvate decarboxylase (EC 4.1.1.1), which requires Mg2+ and thiamine pyrophosphate, a coenzyme derived from thiamine (vitamin B1). Pyruvate decarboxylase is absent in vertebrates and organisms that carry out lactic acid fermentation.
In the second step, acetaldehyde is reduced to ethanol in a reaction catalyzed by alcohol dehydrogenase (EC 1.1.1.1). This enzyme contains a bound zinc ion at its active site. NADH provides the reducing equivalents and is oxidized to NAD⁺. At physiological pH, the equilibrium strongly favors ethanol formation.
The overall process, known as alcoholic fermentation, is summarized by the equation:
Glucose + 2 Pi + 2 ADP → 2 Ethanol + 2 CO2 + 2 ATP + 2 H2O
As in lactic acid fermentation, no net oxidation–reduction occurs.[35]
Alcoholic fermentation is the biochemical basis for the production of beer and wine. The CO2 released by brewer’s yeast generates the bubbles characteristic of beer, champagne, and sparkling wines, while the CO2 produced by baker’s yeast causes dough to rise.[36]
Fate of pyruvate and NADH under aerobic conditions
In cells that contain mitochondria and under aerobic conditions, the predominant situation in both multicellular organisms and many unicellular species, the oxidation of NADH and the catabolism of pyruvate proceed through distinct but interconnected pathways.
Within the mitochondrial matrix, pyruvate is first converted to acetyl-CoA by the pyruvate dehydrogenase complex, a large multienzyme complex. During this oxidative decarboxylation, pyruvate loses, as part of a carboxyl group, one carbon atom in the form of CO2, and the remaining two-carbon fragment is transferred to coenzyme A, forming acetyl-CoA.[37][38]
Pyruvate + NAD+ + CoA → Acetyl-CoA + CO2 + NADH + H+
The acetyl group of acetyl-CoA is then completely oxidized to CO2 via the citric acid cycle, with the concurrent production of NADH and FADH2. The pyruvate dehydrogenase complex thus constitutes a crucial metabolic link between glycolysis, which occurs in the cytosol, and the citric acid cycle, which takes place in the mitochondrial matrix.[32]
Electrons generated during glycolysis are transferred into the mitochondria indirectly: cytosolic NADH reduces specific shuttle intermediates, which are then reoxidized inside the mitochondrial matrix. In this way, NADH is effectively reoxidized to NAD+ in the cytosol, while the reduced intermediates deliver electrons to Complex I of the electron transport chain. From there, electrons are passed through the chain to oxygen, producing H2O. This electron transfer provides the free energy required for ATP synthesis via oxidative phosphorylation.
Electrons carried by NADH formed during the pyruvate dehydrogenase reaction and the citric acid cycle, as well as those derived from FADH2, follow a similar path toward oxidative phosphorylation.[39]
Note: FADH₂ does not transfer its reducing equivalents to Complex I but instead donates them to Complex II.[40]
Anabolic fates of pyruvate
Under anabolic conditions, the carbon skeleton of pyruvate can follow fates other than complete oxidation to CO2 or reduction to lactate. After its conversion to acetyl-CoA, for example, pyruvate may serve as a precursor for fatty acid biosynthesis or for the synthesis of the amino acid alanine.[41][42]
Glycolysis and ATP production
In the glycolytic pathway, one molecule of glucose is degraded into two molecules of pyruvate.
During the preparatory phase, two molecules of ATP are consumed per molecule of glucose in the reactions catalyzed by hexokinase and PFK-1.
During the payoff phase, four molecules of ATP are produced via substrate-level phosphorylation in the reactions catalyzed by phosphoglycerate kinase and pyruvate kinase.
Thus, the net gain is two ATP molecules per glucose molecule. In addition, the reaction catalyzed by glyceraldehyde 3-phosphate dehydrogenase produces two molecules of NADH per glucose.
The overall ΔG°′ of glycolysis is –85 kJ/mol (–20.3 kcal/mol). This value results from the difference between:
the ΔG°′ of glucose conversion into two molecules of pyruvate (–146 kJ/mol; –34.9 kcal/mol), and
the ΔG°′ required for ATP formation from ADP and Pi: 2 × 30.5 kJ/mol = 61 kJ/mol (2 × 7.3 kcal/mol = 14.6 kcal/mol).
The two reactions can be written as:
Glucose + 2 NAD+ → 2 Pyruvate + 2 NADH + 2 H+
2 ADP + 2 Pi → 2 ATP + 2 H2O
Summing these gives the overall glycolytic equation:
By canceling common terms on both sides, this reduces to the standard overall equation shown above.[17]
Free energy changes of glycolytic reactions
The direction and regulation of glycolysis depend on the free energy changes of its individual steps. The table below summarizes the standard and cellular ΔG values for each reaction, showing that only a few steps are highly exergonic and essentially irreversible under physiological conditions.[17]
A graphical version is shown below for quick reference.
Free energy changes of glycolytic reactions
Step
Reaction
ΔG°′ (kJ/mol, kcal/mol)
ΔG (kJ/mol, kcal/mol)
1
Glu + ATP → G6P + ADP
–16.7 (–4.0)
–33.5 (–8.0)
2
G6P ⇄ F6P
+1.7 (+0.4)
–2.5 (–0.6)
3
F6P + ATP → F1,6BP + ADP
–14.2 (–3.4)
–22.2 (–5.3)
4
F1,6P ⇄ DHAP + G3P
+23.8 (+5.7)
–1.3 (–0.3)
5
DHAP ⇄ G3P
+7.5 (+1.8)
+2.5 (+0.6)
6
G3P + NAD+ + Pi ⇄ 1,3-BPG + NADH + H+
+6.3 (+1.5)
+2.5 (+0.6)
7
1,3-BPG + ADP ⇄ 3-PG + ATP
–18.5 (–4.4)
+1.3 (+0.3)
8
3-PG ⇄ 2-PG
+4.4 (+1.1)
+0.8 (+0.2)
9
2-PG ⇄ PEP + H2O
+7.5 (+1.8)
–3.3 (–0.8)
10
PEP + ADP → Pyr + ATP
–31.4 (–7.5)
–16.7 (–4.0)
ΔG°′ values from: Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
ΔG values from: Berg J.M., Tymoczko J.L., Gregory J.G. Jr, Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
Energy Changes of Glycolytic Reactions
ATP production under anaerobic conditions
Under anaerobic conditions, there is no additional ATP production beyond glycolysis, regardless of the metabolic fate of pyruvate, whether it is converted to lactate, ethanol, or other molecule.[43]
Therefore, glycolysis extracts only a small fraction of the chemical energy contained in a glucose molecule, which has a total energy yield of approximately 2,840 kJ/mol (679 kcal/mol) when fully oxidized to CO2 and H2O. In contrast, the conversion of glucose to two molecules of pyruvate releases only 146 kJ/mol, corresponding to ≈ 5% [(146/2,840) × 100] of the total available chemical energy. As a result, pyruvate retains most of the energy of the original hexose. Likewise, the four electrons carried by the NADH molecules produced during step 6 of glycolysis cannot contribute to ATP production under anaerobic conditions.[17]
In lactic acid fermentation, the ΔG°′ for the conversion of one molecule of glucose to two molecules of lactate is –183.6 kJ/mol (–43.9 kcal/mol). Of this free energy, ≈ 33.2% [(61/183.6) × 100] is stored in the two molecules of ATP formed. This is lower than the ≈ 41.8% efficiency of energy storage observed during glycolysis when glucose is converted to pyruvate.[44]
It should be noted that under physiological conditions, the free energy required to synthesize ATP from ADP and Pi is significantly higher than under standard conditions. Consequently, only about 50% of the energy released during glycolysis is actually stored in ATP in vivo.[1]
ATP production under aerobic conditions
Under aerobic conditions, cells with mitochondria can extract far more chemical energy from glucose and conserve it as ATP than under anaerobic conditions.[14]
The two molecules of NADH produced during glycolysis carry four reducing equivalents. When these electrons are transferred through the mitochondrial electron transport chain via oxidative phosphorylation, they yield approximately 2 to 3 ATP per electron pair. Consequently, the conversion of one molecule of glucose into two molecules of pyruvate can generate a total of 6 to 8 ATP: 2 ATP via substrate-level phosphorylation during glycolysis and 4 to 6 ATP through oxidative phosphorylation.[40]
Note: the exact number of ATP molecules generated from cytosolic NADH depends on the shuttle system used to transfer reducing equivalents into the mitochondria.
Furthermore, if we consider the complete aerobic oxidation of glucose through the coordinated action of glycolysis, the pyruvate dehydrogenase complex, the citric acid cycle, the electron transport chain, and oxidative phosphorylation, the ATP yield is much higher. According to Lehninger’s Principles of Biochemistry, the total ATP yield per glucose molecule ranges from 30 to 32. More recent estimates suggest a net production of approximately 29.85 ATP per glucose molecule, or 29.38 ATP if the ATP derived from GTP in the citric acid cycle is exported to the cytosol. In either case, aerobic metabolism yields roughly 15 times more ATP than anaerobic metabolism.[1]
Feeder pathways of glycolysis
Carbohydrates other than glucose, both simple and complex, can also be catabolized via glycolysis after their enzymatic conversion into one of the glycolytic intermediates.
Among the most important are:
glycogen and starch, two major storage polysaccharides;
monosaccharides including glucose, galactose, fructose, and the less common mannose.[45]
In the intestine, starch and disaccharides are hydrolyzed by digestive enzymes during carbohydrate digestion. The resulting monosaccharides, released directly or derived from the diet, are absorbed and transported into the venous circulation. From there, they reach the liver through the portal vein, which serves as the primary site for their metabolic processing.[46]
Glycogen and starch
For details on the phosphorolytic degradation of starch and endogenous glycogen, refer to the dedicated sections.
Fructose
Under physiological conditions, the liver removes much of the ingested fructose from the bloodstream before it can reach extrahepatic tissues.[47]
The hepatic pathway for converting fructose into glycolytic intermediates consists of several steps.[32]
In the first step, fructose is phosphorylated to fructose 1-phosphate (F1P) at the expense of one ATP molecule. This reaction, catalyzed by fructokinase (EC 2.7.1.4), requires Mg2+.
Fructose + ATP → F1P + ADP
As with glucose, phosphorylation traps fructose inside the cell.
In the second step, fructose 1-phosphate aldolase catalyzes an aldol cleavage of fructose 1-phosphate, yielding dihydroxyacetone phosphate and glyceraldehyde.
F1P → DHAP + Glyceraldehyde
Dihydroxyacetone phosphate is a direct glycolytic intermediate and, once converted to glyceraldehyde 3-phosphate, can continue through the pathway.
By contrast, glyceraldehyde is not a glycolytic intermediate and must be phosphorylated to glyceraldehyde 3-phosphate at the expense of another ATP molecule. This reaction, catalyzed by triose kinase (EC 2.7.1.28), also requires Mg2+.
Glyceraldehyde + ATP → G3P + ADP
Thus, in hepatocytes, one molecule of fructose is converted into two molecules of glyceraldehyde 3-phosphate, at the cost of two ATP molecules, just as with glucose.[48]
Fructose + 2 ATP → 2 G3P + 2 ADP
Fructose and hexokinase
In addition to the hepatic fructokinase pathway, extrahepatic tissues can also metabolize fructose through the action of hexokinase, albeit with a markedly lower affinity for fructose than for glucose. In these tissues, such as skeletal muscle, kidney, and adipose tissue, fructokinase is absent. Instead, fructose enters glycolysis as fructose 6-phosphate, following its phosphorylation at C-6 catalyzed by hexokinase.
Fructose + ATP → F6P + ADP
However, the enzyme’s affinity for fructose is about 20 times lower than for glucose. Consequently, in hepatocytes, where glucose is abundant, or in skeletal muscle under anaerobic conditions, when glucose is the preferred fuel, only small amounts of fructose 6-phosphate are produced.[11][47]
In contrast, in adipose tissue, fructose levels can exceed those of glucose, and its phosphorylation by hexokinase is not significantly inhibited. Thus, in this tissue, the formation of fructose 6-phosphate represents the primary entry point of fructose into glycolysis.[49]
Metabolic implications
It is important to note that in the liver, fructose, being phosphorylated at C-1, enters glycolysis at the level of triose phosphates, downstream of the reaction catalyzed by PFK-1, the key regulatory enzyme of glycolytic flux.
Conversely, when fructose is phosphorylated at C-6, it enters glycolysis upstream of PFK-1.[9]
Sorbitol
Fructose represents the entry point into glycolysis for sorbitol, a sugar naturally present in many fruits and vegetables, and also widely used as a sweetener and stabilizer.
In the liver, sorbitol dehydrogenase (EC 1.1.1.14) catalyzes the oxidation of sorbitol to fructose in a reaction that requires zinc ions and NAD+ as a cofactor.[32] This enzyme also participates in the polyol pathway, which interconverts glucose, sorbitol, and fructose and plays an important role in tissues such as the liver, seminal vesicles, and retina.[48]
Sorbitol + NAD+ → Fructose + NADH + H+
Galactose
Galactose, primarily derived from the intestinal digestion of lactose, is metabolized in the liver via the Leloir pathway, which converts it into glucose 1-phosphate.[50]
The metabolic fate of glucose 1-phosphate depends on the energy status of the cell.
Under conditions favoring glucose storage, glucose 1-phosphate is directed toward glycogen synthesis.
Under conditions favoring glucose utilization as a fuel, glucose 1-phosphate is isomerized to glucose 6-phosphate in the reversible reaction catalyzed by phosphoglucomutase (EC 5.4.2.2):
G1P ⇄ G6P
Glucose 6-phosphate can then either enter glycolysis for energy production or be dephosphorylated by glucose 6-phosphatase (EC 3.1.3.9) to free glucose, which can be released into the bloodstream.[51][52]
Mannose
Mannose is found in various dietary polysaccharides, glycolipids, and glycoproteins. In the intestine, it is released from these macromolecules, absorbed, and then transported to the liver. There, it is phosphorylated at C-6 to form mannose 6-phosphate (M6P) in a reaction catalyzed by hexokinase.
Mannose + ATP → M6P + ADP
The resulting sugar phosphate is subsequently isomerized to fructose 6-phosphate by mannose 6-phosphate isomerase (EC 5.3.1.8).[53]
M6P ⇄ F6P
Regulation of glycolysis
The flux of carbon through the glycolytic pathway is tightly regulated in response to metabolic conditions, both intracellular and extracellular, in order to satisfy two major needs:
the production of ATP;
the supply of precursors for biosynthetic reactions.
In the liver, glycolysis and gluconeogenesis are reciprocally regulated to avoid a wasteful expenditure of energy: when one pathway is active, the other is inhibited. Evolutionarily, this control was achieved by selecting distinct enzymes to catalyze the essentially irreversible reactions of the two pathways. These enzymes are regulated independently, thereby avoiding the risk of a futile cycle (also known as a substrate cycle) that would result if both reactions occurred simultaneously at high rates. Such precise regulation would not be possible if a single enzyme catalyzed both directions of the reaction.[7]
The control of glycolysis primarily involves three enzymes: hexokinase, phosphofructokinase-1, and pyruvate kinase. Their activities are regulated through:
allosteric modifications, which occur on a millisecond timescale and are rapidly reversible;
covalent modifications (phosphorylation and dephosphorylation), which take place on a timescale of seconds;
changes in enzyme concentration, due to altered rates of synthesis and/or degradation, which occur over hours.
Note: the main regulatory enzymes of gluconeogenesis are pyruvate carboxylase (EC 6.4.1.1) and fructose 1,6-bisphosphatase (FBPasi-1; EC 3.1.3.11).[1][11]
Hexokinase
In humans, hexokinase exists as four tissue-specific isozymes, hexokinase I, II, III, and IV, each of which is encoded by a distinct gene.[16]
Hexokinase I is the predominant isozyme in the brain. In skeletal muscle, both hexokinase I and II are expressed, contributing approximately 70–75% and 25–30% of total hexokinase activity, respectively.[9]
Hexokinase IV, also known as glucokinase (EC 2.7.1.2), is primarily expressed in hepatocytes and pancreatic β-cells, where it is the predominant isozyme. In the liver, glucokinase participates, together with glucose 6-phosphatase, in the substrate cycle between glucose and glucose 6-phosphate. Glucokinase differs from the other hexokinase isozymes in both its kinetic and regulatory properties.[54]
Note: isozymes (or isoenzymes) are distinct proteins that catalyze the same reaction but generally differ in kinetic and regulatory properties, subcellular localization, or cofactor requirements. They may coexist within the same organism, the same tissue, or even the same cell.[55]
Kinetic properties of hexokinase isozymes
The kinetic properties of hexokinase I, II, and III are broadly similar.
Hexokinase I and II have Km values for glucose of 0.03 mM and 0.1 mM, respectively. These low Km values allow the enzymes to function efficiently at normal blood glucose concentrations (4–5 mM).
By contrast, glucokinase has a high Km for glucose (≈ 10 mM), meaning it is active primarily when blood glucose levels are elevated, such as after a carbohydrate-rich meal.[9]
Regulation of the activity of hexokinases I–III
Hexokinases I–III are allosterically inhibited by glucose 6-phosphate, the product of their reaction.[16] This feedback mechanism prevents the accumulation of glucose 6-phosphate in the cytosol when glucose is not required for:
At the same time, this regulation allows glucose to remain in the bloodstream, available to other tissues. For example, inhibition of phosphofructokinase-1 leads to the accumulation of fructose 6-phosphate, which, via the phosphoglucose isomerase reaction, increases glucose 6-phosphate levels. Consequently, inhibition of PFK-1 indirectly inhibits the activity of hexokinases I–III.[56]
Coordination of hexokinase and GLUT4 in skeletal muscle
In skeletal muscle, the activities of hexokinase I and II are tightly coordinated with those of GLUT4, a glucose transporter with a Km of ≈ 5 mM. Translocation of GLUT4 to the plasma membrane is stimulated by both insulin and physical activity. The combined action of GLUT4-mediated glucose uptake and cytosolic hexokinase activity maintains a balance between glucose entry and its phosphorylation.[9]
Since blood glucose concentrations typically range between 4 and 5 mM, GLUT4-mediated uptake increases intracellular glucose to levels sufficient to saturate, or nearly saturate, hexokinase, allowing the enzyme to operate at or near its Vmax.[57]
Hepatic glucokinase
Glucokinase differs in three key respects from hexokinases I–III and is particularly well-suited to the liver’s role in regulating blood glucose levels.[54][58]
Kinetic and functional properties
As previously mentioned, glucokinase has a Km for glucose of about 10 mM, much higher than that of hexokinases I–III and also higher than fasting blood glucose levels (4–5 mM). In the liver, where glucokinase is the predominant hexokinase isoenzyme, its role is to provide glucose 6-phosphate for the synthesis of glycogen and fatty acids. The activity of glucokinase is linked to that of GLUT2, the main glucose transporter in hepatocytes, which also has a high Km for glucose (approximately 10 mM).[16]
Hence, GLUT2 is highly active when blood glucose levels are elevated, rapidly equilibrating glucose concentrations between the cytosol of hepatocytes and the blood. Under such conditions, glucokinase is active and converts glucose into G6P. Due to its high Km, glucokinase activity continues to rise even as intracellular glucose concentrations reach or exceed 10 mM. Therefore, the rate of glucose uptake and phosphorylation is directly determined by blood glucose levels.[59]
Conversely, when glucose availability is low, its cytosolic concentration in hepatocytes is also low, much lower than the Km of glucokinase, which ensures that glucose produced through gluconeogenesis and/or glycogenolysis is not phosphorylated and can exit the cell.[60] A similar situation also occurs in pancreatic β-cells, where the GLUT2/glucokinase system aligns intracellular glucose 6-phosphate levels with blood glucose levels, allowing the cells to detect and respond to hyperglycemia.[61]
Regulation by glucokinase regulatory protein
Unlike hexokinases I–III, glucokinase is not inhibited by glucose 6-phosphate, that is, it is not subject to product inhibition, and continues to catalyze glucose phosphorylation even when glucose 6-phosphate accumulates. Glucokinase is inhibited by reversible binding to the glucokinase regulatory protein (GKRP), a liver-specific regulator. This inhibition involves the sequestration of glucokinase in the nucleus, separating it from other glycolytic enzymes.[60][62]
Regulation of Hepatic Glucokinase Activity
The binding between glucokinase and GKRP is strengthened by fructose 6-phosphate, whereas it is weakened by glucose and fructose 1-phosphate.
In the absence of glucose, glucokinase adopts its “super-open” conformation, which is capable of binding to GKRP. Rising cytosolic glucose concentrations trigger a concentration-dependent shift to the “closed” conformation, its active form, which cannot bind GKRP. As a result, glucokinase becomes active and is no longer inhibited.
Note that fructose 1-phosphate is present in the hepatocyte only when fructose is metabolized. Therefore, fructose relieves glucokinase inhibition by disrupting its interaction with GKRP.[63]
Physiological significance
After a carbohydrate-rich meal, blood glucose levels rise. Glucose enters hepatocytes via GLUT2 and then diffuses into the nucleus through nuclear pores. There, glucose promotes the transition of glucokinase to its closed, active conformation, which is no longer accessible to GKRP. This allows glucokinase to diffuse into the cytosol, where it phosphorylates glucose.
Conversely, when glucose concentration declines, such as during fasting when blood glucose levels may fall below 4 mM, glucose levels in hepatocytes also drop. Under these conditions, fructose 6-phosphate binds to GKRP, enhancing its affinity for glucokinase and resulting in strong inhibition of the enzyme. This mechanism ensures that, at low blood glucose levels, the liver does not compete with other organs, primarily the brain, for glucose.
Within the cell, fructose 6-phosphate is in equilibrium with glucose 6-phosphate due to the action of phosphoglucose isomerase. By binding to GKRP, fructose 6-phosphate contributes to the suppression of glucokinase activity, thereby preventing the accumulation of glycolytic intermediates.
To sum up, when blood glucose levels are within the normal range, glucose is phosphorylated primarily by hexokinases I–III. When blood glucose levels are high, glucokinase contributes to glucose phosphorylation as well.[9][64]
Regulation of phosphofructokinase-1 activity
Phosphofructokinase-1 is the key control point for carbon flow through glycolysis.[65]
In addition to substrate binding sites, the enzyme has several binding sites for allosteric effectors.
ATP, citrate, and hydrogen ions act as allosteric inhibitors of the enzyme, whereas AMP, Pi, and fructose 2,6-bisphosphate function as allosteric activators.[66][67][68]
Regulation of PFK 1 and Fructose 1,6-Bisphosphatase
It is worth noting that ATP, an end product of glycolysis, is also a substrate of phosphofructokinase-1. The enzyme possesses two binding sites for the nucleotide: a low-affinity regulatory site and a high-affinity substrate site.[9]
What allosteric effectors signal
ATP, AMP, and Pi signal the energy status of the cell.
PFK-1 activity increases when the energy charge of the cell is low, that is, when ATP is needed, and decreases when the energy charge is high, that is, when ATP levels are elevated.[69]
How this happens
When ATP is produced faster than it is consumed, its intracellular concentration becomes elevated. Under these conditions, ATP binds to its allosteric site and inhibits PFK-1 by reducing the enzyme’s affinity for fructose 6-phosphate.
From a kinetic perspective, increased ATP concentration alters the relationship between enzyme activity and substrate concentration, changing the hyperbolic velocity curve of fructose 6-phosphate into a sigmoidal one and thereby increasing the Km for the substrate.
However, under most physiological conditions, intracellular ATP concentration does not vary significantly. For example, during vigorous exercise, ATP concentration in muscle may decrease by about 10% compared to resting levels, whereas the glycolytic rate changes much more than would be expected from such a small reduction.
When ATP consumption exceeds its production, concentrations of ADP and AMP rise, particularly AMP, due to the reaction catalyzed by adenylate kinase (EC 2.7.4.3), which forms ATP and AMP from two ADP molecules.
ADP + ADP ⇄ ATP + AMP
The equilibrium constant of the reaction is:
Keq = [ATP][AMP]/[ADP]2 = 0.44
Under physiological conditions, the concentrations of ADP and AMP are approximately 10% and often less than 1% of the ATP concentration, respectively. Therefore, considering that the total adenylate pool remains constant over the short term, even a small decrease in ATP concentration leads, due to adenylate kinase activity, to a much larger relative increase in AMP concentration. In turn, AMP acts by reversing the inhibition caused by ATP.
Thus, the activity of phosphofructokinase-1 depends on the cellular energy status:
when ATP is plentiful, enzyme activity decreases;
when AMP levels increase and ATP levels fall, enzyme activity increases.[9]
Why ADP isn’t a positive effector of PFK-1
There are two reasons.
First, when the cell’s energy charge decreases, ADP is used to regenerate ATP via the reaction catalyzed by adenylate kinase.
Second, as previously mentioned, even a small decrease in ATP leads to proportionally larger changes in ADP levels, and, especially, in AMP levels.[70]
Role of hydrogen ions
Hydrogen ions also inhibit PFK-1. This inhibition helps regulate the rate of glycolysis, preventing excessive lactate accumulation and a consequent drop in cellular pH.[71]
Role of citrate
Citrate is an allosteric inhibitor of PFK-1 that acts by enhancing the inhibitory effect of ATP.
It is the product of the first step of the citric acid cycle, a metabolic pathway that provides building blocks for biosynthetic processes and directs electrons into the mitochondrial electron transport chain for ATP synthesis via oxidative phosphorylation.
High cytosolic citrate levels indicate that energy demands are met and an abundance of biosynthetic precursors is available (exported from the mitochondria); in other words, the citric acid cycle is saturated.
Under such conditions, glycolysis, which feeds the cycle under aerobic conditions, can slow down, sparing glucose.
Therefore, it should be noted that PFK-1 functionally couples glycolysis to the citric acid cycle.[65][72]
Regulation of PFK-1 and FBPase-1 in the liver
In the liver, the central control point of glycolysis and gluconeogenesis is the substrate cycle between F6P and F1,6BP, catalyzed by PFK-1 and fructose 1,6-bisphosphatase.
The liver plays a pivotal role in maintaining blood glucose levels within the normal range.
When blood glucose levels drop, glucagon stimulates hepatic glucose production via both glycogenolysis and gluconeogenesis, while simultaneously signaling the liver to stop consuming glucose for its own energy needs.
Conversely, when blood glucose levels are high, insulin promotes glucose utilization for energy and the synthesis of glycogen and triglycerides.
In this context, the regulation of glycolysis and gluconeogenesis is mediated by fructose 2,6-bisphosphate (F2,6BP), a molecule that enables the liver to play a major role in regulating blood glucose levels and whose concentration is controlled by insulin and glucagon.[56]
Role of fructose 2,6-bisphosphate
By binding to its allosteric site on PFK-1, fructose 2,6-bisphosphate increases the affinity of the enzyme for fructose 6-phosphate, its substrate, while counteracting the inhibitory effect citrate and ATP.
It is remarkable that under physiological concentrations of substrates and both positive and negative allosteric effectors, PFK-1 would be virtually inactive in the absence of fructose 2,6-bisphosphate.
On the other hand, the binding of fructose 2,6-bisphosphate to fructose 1,6-bisphosphatase inhibits the enzyme, even in the absence of AMP, another of its allosteric inhibitors.
As a result of these effects, fructose 2,6-bisphosphate increases the net flux of glucose through glycolysis.[66][67][73]
Role of xylulose 5-phosphate
Xylulose 5-phosphate also plays a role in controlling carbon flux through glycolysis and gluconeogenesis. It is a product of the pentose phosphate pathway, whose levels in hepatocytes rise after the ingestion of a carbohydrate-rich meal. By activating protein phosphatase 2A, this metabolite induces the dephosphorylation of the bifunctional enzyme PFK-2/FBPase-2. This dephosphorylation activates its kinase domain, ultimately increasing fructose 2,6-bisphosphate levels, thereby enhancing glycolytic flux and reducing gluconeogenic flux.[74]
Regulation of pyruvate kinase activity
Another key control point for carbon flux through glycolysis and gluconeogenesis is the substrate cycle between phosphoenolpyruvate and pyruvate, catalyzed by pyruvate kinase in glycolysis and by the combined action of pyruvate carboxylase and phosphoenolpyruvate carboxykinase (EC 4.1.1.32) in gluconeogenesis.[9]
Allosteric regulation
All pyruvate kinase isozymes are allosterically inhibited by high concentrations of ATP, long-chain fatty acids, and acetyl-CoA, all indicators that the cell is in a high-energy state. Alanine, which can be synthesized from pyruvate through a transamination reaction, is also an allosteric inhibitor; its accumulation signals that biosynthetic precursors are abundant.
Regulation of Pyruvate Kinase Activity
Conversely, pyruvate kinase is allosterically activated by fructose 1,6-bisphosphate, the product of the first committed step of glycolysis. F1,6BP thus ensures that pyruvate kinase activity keeps pace with the upstream flux of intermediates. It should be emphasized that, at physiological concentrations of PEP, ATP, and alanine, the enzyme would be almost completely inhibited in the absence of F1,6BP activation.[27][75]
Covalent regulation in the liver
The hepatic isoenzyme, but not the muscle isoenzyme, is also regulated by phosphorylation through:
primarily protein kinase A (PKA), activated by glucagon (or epinephrine via β-adrenergic receptors);
calcium/calmodulin dependent protein kinase (CAMK), activated by epinephrine via α1-adrenergic receptors.
Phosphorylation of the enzyme decreases its activity by increasing the Km for PEP, thereby slowing glycolysis.
For example, when blood glucose levels are low, glucagon-induced phosphorylation reduces pyruvate kinase activity. The phosphorylated enzyme is also less responsive to activation by fructose 1,6-bisphosphate and more sensitive to inhibition by alanine and ATP.
Conversely, the dephosphorylated form of pyruvate kinase is more responsive to F1,6BP, that is, it requires a lower concentration for activation, and less sensitive to ATP and alanine. This mechanism allows the liver, under low-glucose conditions, to limit its own glucose utilization, preserving glucose for other organs such as the brain.
It should be noted, however, that the powerful activation by F1,6BP overrides the inhibitory effects of phosphorylation by PKA.
An increase in the insulin/glucagon ratio promotes dephosphorylation and enzyme activation.[9]
References
^ abcdefghijkl Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ Alberts B., Johnson A., Lewis J., Morgan D., Raff M., Roberts K., Walter P. Molecular Biology of the Cell. 7th Edition. Garland Science, Taylor & Francis Group, 2022.
^ abcdefgh Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
^ Fruton J.S. A History of Biochemistry. Harvard University Press, 1982.
^ Buchner E., Rapp R. Alkoholische Gärung ohne Hefezellen. Berichte der Deutschen Chemischen Gesellschaft 1899;32(1):127-137. doi:10.1002/cber.18990320124
^ Kohler R. The background to Eduard Buchner’s discovery of cell-free fermentation. J Hist Biol 1971;4:35-61. doi:10.1007/BF00356976
^ abcdefghijk Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 4th Edition. St. Louis: Elsevier, 2018.
^ Lu Z., Imlay J.A. When anaerobes encounter oxygen: mechanisms of oxygen toxicity, tolerance and defence. Nat Rev Microbiol 2021;19(12):774-785. doi:10.1038/s41579-021-00583-y
^ abcdefghijkl Berg J.M., Tymoczko J.L., Gregory J.G. Jr, Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
^ Plaxton W.C., Podestá F.E. The functional organization and control of plant respiration. Crit Rev Plant Sci 2006;25(2):159-198. doi:10.1080/07352680600563876
^ Hochachka P.W., Somero G.N. Biochemical adaptation: mechanism and process in physiological evolution. Oxford University Press, 2002. doi:10.1093/oso/9780195117028.001.0001
^ ab Naifeh N., Dimri M., Varacallo M.A. Biochemistry, Aerobic Glycolysis. [Updated 2023 Apr 9]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/sites/books/NBK470170/
^ Lunt S.Y., Vander Heiden M.G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 2011;27:441-64. doi:10.1146/annurev-cellbio-092910-154237
^ abcd Wilson J.E. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol 2003;206(Pt 12):2049-57. doi:10.1242/jeb.00241
^ abcdefghij Atkins P.W., Ratcliffe R.G., Wormald M., De Paula J. Physical chemistry for the life sciences. 3rd Edition. Oxford University Press, 2023.
^ Mertens E. Pyrophosphate-dependent phosphofructokinase, an anaerobic glycolytic enzyme? FEBS Lett 1991;285(1):1-5. doi:10.1016/0014-5793(91)80711-b
^ Knowles J.R. Enzyme catalysis: not different, just better. Nature 1991;350(6314):121-4. doi:10.1038/350121a0
^ Sirover M.A. On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase: biochemical mechanisms and regulatory control. Biochim Biophys Acta 2011;1810(8):741-51. doi:10.1016/j.bbagen.2011.05.010
^ Rojas-Pirela M., Andrade-Alviárez D., Rojas V., Kemmerling U., Cáceres A.J., Michels P.A., Concepción J.L., Quiñones W. Phosphoglycerate kinase: structural aspects and functions, with special emphasis on the enzyme from Kinetoplastea. Open Biol 2020;10(11):200302. doi:10.1098/rsob.200302
^ Levin S., Almo S.C., Satir B.H. Functional diversity of the phosphoglucomutase superfamily: structural implications. Protein Eng 1999;12(9):737-46. doi:10.1093/protein/12.9.737
^ Rapoport S., Luebering J. The formation of 2,3-diphosphoglycerate in rabbit erythrocytes: the existence of a diphosphoglycerate mutase. J Biol Chem 1950;183(2):507-516.
^ Webb K.L., Dominelli P.B., Baker S.E., Klassen S.A., Joyner M.J., Senefeld J.W., Wiggins C.C. Influence of high hemoglobin-oxygen affinity on humans during hypoxia. Front Physiol 2022;12:763933. doi:10.3389/fphys.2021.763933
^ Li M., Wu C., Yang Y., Zheng M., Yu S., Wang J., Chen L., Li H. 3-Phosphoglycerate dehydrogenase: a potential target for cancer treatment. Cell Oncol (Dordr) 2021;44(3):541-556. doi:10.1007/s13402-021-00599-9
^ Lebioda L., Stec B. Mechanism of enolase: the crystal structure of enolase-Mg2+-2-phosphoglycerate/phosphoenolpyruvate complex at 2.2-A resolution. Biochemistry 1991;30(11):2817-22. doi:10.1021/bi00225a012
^ ab Schormann N., Hayden K.L., Lee P., Banerjee S., Chattopadhyay D. An overview of structure, function, and regulation of pyruvate kinases. Protein Sci 2019;28(10):1771-1784. doi:10.1002/pro.3691
^ Yang Y., Sauve A.A. NAD+ metabolism: bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta 2016;1864(12):1787-1800. doi:10.1016/j.bbapap.2016.06.014
^ Gray L.R., Tompkins S.C., Taylor E.B. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-604. doi:10.1007/s00018-013-1539-2
^ Rogatzki M.J., Ferguson B.S., Goodwin M.L., Gladden L.B. Lactate is always the end product of glycolysis. Front Neurosci 2015;9:22. doi:10.3389/fnins.2015.00022
^ Geison G.L. The Private Science of Louis Pasteur. Princeton University Press, 1995.
^ Dong S., Qian L., Cheng Z., Chen C., Wang K., Hu S., Zhang X., Wu T. Lactate and myocardiac energy metabolism. Front Physiol 2021;12:715081. doi:10.3389/fphys.2021.715081
^ National Research Council (US) Panel on the Applications of Biotechnology to Traditional Fermented Foods. Applications of Biotechnology to Fermented Foods: Report of an Ad Hoc Panel of the Board on Science and Technology for International Development. Washington (DC): National Academies Press (US); 1992. 5, Lactic Acid Fermentations. Available from: https://www.ncbi.nlm.nih.gov/books/NBK234703/
^ Dzialo M.C., Park R., Steensels J., Lievens B., Verstrepen K.J. Physiology, ecology and industrial applications of aroma formation in yeast. FEMS Microbiol Rev 2017;41(Supp_1):S95-S128. doi:10.1093/femsre/fux031
^ Maicas S. The role of yeasts in fermentation processes. Microorganisms 2020;8(8):1142. doi:10.3390/microorganisms8081142
^ Patel M.S., Nemeria N.S., Furey W., and Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem 2014;289(24):16615-16623. doi:10.1074/jbc.R114.563148
^ Liu H., Wang S., Wang J., Guo X., Song Y., Fu K., Gao Z., Liu D., He W., Yang L.L. Energy metabolism in health and diseases. Signal Transduct Target Ther 2025;10(1):69. doi:10.1038/s41392-025-02141-x
^ Borst P. The malate-aspartate shuttle (Borst cycle): How it started and developed into a major metabolic pathway. IUBMB Life 2020;72(11):2241-2259. doi:10.1002/iub.2367
^ ab Ahmad M., Wolberg A., Kahwaji C.I. Biochemistry, Electron Transport Chain. [Updated 2023 Sep 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK526105/
^ Chaudhry R., Varacallo M.A. Biochemistry, Glycolysis. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482303/
^ DeBerardinis R.J., Chandel N.S. Fundamentals of cancer metabolism. Sci Adv 2016;2(5):e1600200. doi:10.1126/sciadv.1600200
^ Kiela P.R., Ghishan F.K. Physiology of intestinal absorption and secretion. Best Pract Res Clin Gastroenterol 2016;30(2):145-59. doi:10.1016/j.bpg.2016.02.007
^ ab Dholariya S.J., Orrick J.A. Biochemistry, Fructose Metabolism. [Updated 2022 Oct 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK576428/
^ ab Hannou S.A., Haslam D.E., McKeown N.M., Herman M.A. Fructose metabolism and metabolic disease. J Clin Invest 2018;128(2):545-555. doi:10.1172/JCI96702
^ Legeza B., Marcolongo P., Gamberucci A., Varga V., Bánhegyi G., Benedetti A., Odermatt A. Fructose, glucocorticoids and adipose tissue: implications for the metabolic syndrome. Nutrients 2017;9(5):426. doi:10.3390/nu9050426
^ Frey P.A. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J 1996;10(4):461-70. doi:10.1096/fasebj.10.4.8647345
^ Conte F., van Buuringen N., Voermans N.C., Lefeber D.J. Galactose in human metabolism, glycosylation and congenital metabolic diseases: time for a closer look. Biochim Biophys Acta Gen Subj 2021;1865(8):129898. doi:10.1016/j.bbagen.2021.129898
^ Succoio M., Sacchettini R., Rossi A., Parenti G., Ruoppolo M. Galactosemia: biochemistry, molecular genetics, newborn screening, and treatment. Biomolecules 2022;12(7):968. doi:10.3390/biom12070968
^ Sharma V., Ichikawa M., Freeze H.H. Mannose metabolism: more than meets the eye. Biochem Biophys Res Commun 2014;453(2):220-8. doi:10.1016/j.bbrc.2014.06.021
^ ab de la Iglesia N., Mukhtar M., Seoane J., Guinovart J.J., Agius L. The role of the regulatory protein of glucokinase in the glucose sensory mechanism of the hepatocyte. J Biol Chem 2000;275(14):10597-10603. doi: 10.1074/jbc.275.14.10597
^ Zhang Y., Lin Z., Wang M., Lin H. Selective usage of isozymes for stress response. ACS Chem Biol 2018;13(11):3059-3064. doi:10.1021/acschembio.8b00767
^ ab Han H.S., Kang G., Kim J.S., Choi B.H., Koo S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp Mol Med 2016;48(3):e218. doi:10.1038/emm.2015.122
^ Matschinsky F.M., Magnuson M.A., Zelent D., Jetton T.L., Doliba N., Han Y., Taub R., Grimsby J. The network of glucokinase-expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes 2006;55(1):1-12. doi:diabetes.55.01.06.db05-0926
^ Sun B., Chen H., Xue J., Li P., Fu X. The role of GLUT2 in glucose metabolism in multiple organs and tissues. Mol Biol Rep 2023;50(8):6963-6974. doi:10.1007/s11033-023-08535-w
^ ab Adeva-Andany M.M., Pérez-Felpete N., Fernández-Fernández C., Donapetry-García C., Pazos-García C. Liver glucose metabolism in humans. Biosci Rep 2016;36(6):e00416. doi:10.1042/BSR20160385
^ Abu Aqel Y., Alnesf A., Aigha I.I., Islam Z., Kolatkar P.R., Teo A., Abdelalim E.M. Glucokinase (GCK) in diabetes: from molecular mechanisms to disease pathogenesis. Cell Mol Biol Lett. 2024 Sep 8;29(1):120. doi:10.1186/s11658-024-00640-3
^ Kaminski M.T., Schultz J., Waterstradt R., Tiedge M., Lenzen S., Baltrusch S. Glucose-induced dissociation of glucokinase from its regulatory protein in the nucleus of hepatocytes prior to nuclear export. Biochim Biophys Acta 2014;1843(3):554-564. doi:10.1016/j.bbamcr.2013.12.002
^ Sternisha S.M., Miller B.G. Molecular and cellular regulation of human glucokinase. Arch Biochem Biophys 2019;663:199-213. doi:10.1016/j.abb.2019.01.011
^ Matschinsky F.M., Wilson D.F. The central role of glucokinase in glucose homeostasis: a perspective 50 years after demonstrating the presence of the enzyme in islets of Langerhans. Front Physiol 2019;10:148. doi:10.3389/fphys.2019.00148
^ ab Mor I., Cheung E.C., Vousden K.H. Control of glycolysis through regulation of PFK1: old friends and recent additions. Cold Spring Harb Symp Quant Biol 2011;76:211-6. doi:10.1101/sqb.2011.76.010868
^ ab Van Schaftingen E., and Hers H-G. Inhibition of fructose-1,6-bisphosphatase by fructose-2,6-bisphosphate. Proc Natl Acad Sci USA 1981;78(5):2861-2863. doi:10.1073/pnas.78.5.2861
^ ab Van Schaftingen E., Jett M-F., Hue L., and Hers H-G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA 1981;78(6):3483-3486. doi:10.1073/pnas.78.6.3483
^ Lynch E.M., Hansen H., Salay L., Cooper M., Timr S., Kollman J.M., Webb B.A. Structural basis for allosteric regulation of human phosphofructokinase-1. Nat Commun 2024;15(1):7323. doi:10.1038/s41467-024-51808-6
^ Jenkins C.M., Yang J., Sims H.F., Gross R.W. Reversible high affinity inhibition of phosphofructokinase-1 by acyl-CoA: a mechanism integrating glycolytic flux with lipid metabolism. J Biol Chem 2011;286(14):11937-50. doi:10.1074/jbc.M110.203661
^ Atkinson D.E. The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 1968;7(11):4030-4. doi:10.1021/bi00851a033
^ Lešnik S., Konc J., Vodopivec T., Čamernik K., Karolina Potokar U., Legiša M. Small-molecule inhibitors of 6-phosphofructo-1-kinase simultaneously suppress lactate and superoxide generation in cancer cells. PLoS One 2025;20(5):e0321998. doi:10.1371/journal.pone.0321998
^ Usenik A., Legiša M. Evolution of allosteric citrate binding sites on 6-phosphofructo-1-kinase. PLoS One 2010;5(11):e15447. doi:10.1371/journal.pone.0015447
^ Chaekal O.K., Boaz J.C., Sugano T., Harris R.A. Role of fructose 2,6-bisphosphate in the regulation of glycolysis and gluconeogenesis in chicken liver. Arch Biochem Biophys 1983;225(2):771-8. doi:10.1016/0003-9861(83)90088-7
^ Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003;100:5107-5112. doi:10.1073/pnas.0730817100
^ Jurica M.S., Mesecar A., Heath P.J., Shi W., Nowak T., Stoddard B.L. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure 1998;6(2):195-210. doi:10.1016/s0969-2126(98)00021-5
Gluconeogenesis is a metabolic pathway that leads to the synthesis of glucose (Glu) from pyruvate (Pyr), the conjugate base of pyruvic acid, and other non-carbohydrate precursors, even in non-photosynthetic organisms.[1]
It occurs in all microorganisms, fungi, plants, and animals, and the reactions are essentially the same, leading to the synthesis of one glucose molecule from two pyruvate molecules. It is essentially glycolysis in reverse, sharing seven enzymes with it, but proceeding from pyruvate to glucose.[2]
Glycogenolysis is clearly distinct from gluconeogenesis, as it does not result in the de novo synthesis of glucose from non-carbohydrate precursors, as illustrated by its overall reaction:[3]
Glycogen or (glucose)n → n glucose 1-phosphate molecules.
The following discussion will focus on gluconeogenesis in higher animals, particularly in the mammalian liver.
Summary: Key Points
Definition: the metabolic pathway responsible for synthesizing glucose from non-carbohydrate precursors such as pyruvate, lactate, glycerol, and glucogenic amino acids.
Biological function: essential for maintaining stable blood glucose levels, particularly to supply glucose to the brain and red blood cells during prolonged fasting.
Mechanism: not a simple reversal of glycolysis; instead, it bypasses three irreversible glycolytic steps through the action of specific enzymes, including pyruvate carboxylase, phosphoenolpyruvate carboxykinase, and fructose-1,6-bisphosphatase.
Main sites: occurs primarily in the liver (≈90%) and, to a lesser extent, in the renal cortex, involving the mitochondria, cytosol, and endoplasmic reticulum.
Hormonal control: regulated mainly by the insulin-to-glucagon ratio and by allosteric effectors such as fructose-2,6-bisphosphate.
Gluconeogenesis is essential for two main reasons: it maintains appropriate blood glucose levels when glycogen stores are low and no carbohydrates are available, and it preserves metabolic intermediates, such as pyruvate, needed for energy production.[4]
Maintaining blood glucose between 3.3 and 5.5 mmol/L (60–99 mg/dL) is critical, as many cells rely on glucose to meet their ATP needs. These include red blood cells, neurons, the renal medulla, skeletal muscle under low oxygen, the testes, the lens and cornea, and embryonic tissues.[5]
For example, the brain alone requires about 120 g/day of glucose, which represents:
over 50% of the total body glucose stores (≈210 g), of which about 190 g are stored as liver and muscle glycogen, and 20 g are found free in body fluids;
about 75% of the daily glucose requirement (≈160 g).[4]
During fasting, such as between meals or overnight, blood glucose levels are maintained within the normal range primarily through hepatic glycogenolysis, as well as the release of fatty acids from adipose tissue and ketone body production by the liver.
Fatty acids and ketone bodies are preferentially used by skeletal muscle, thereby sparing glucose for cells that depend entirely on it, especially red blood cells and neurons.
However, after approximately 18 hours of fasting, or during intense and prolonged exercise, glycogen stores become insufficient. At this point, in the absence of carbohydrate intake, gluconeogenesis becomes critical.[6]
Its importance is further highlighted by the fact that blood glucose levels below 2 mmol/L can lead to unconsciousness.[7]
Additionally, without gluconeogenesis, pyruvate would be lost, reducing the body’s capacity to produce ATP via aerobic respiration (more than 10 ATP molecules per pyruvate oxidized), making its conservation essential during energy stress.[1]
Sites of gluconeogenesis
In higher animals, gluconeogenesis occurs in the liver, kidney cortex, and enterocytes.[4]
Quantitatively, the liver is the major site, producing about 90% of synthesized glucose, followed by the kidney cortex with around 10%. The liver’s key role is largely due to its size; in fact, on a wet weight basis, the kidney cortex produces more glucose than the liver.[8]
In the kidney, gluconeogenesis takes place in the proximal tubule cells. Much of the glucose produced is consumed by the renal medulla, while the kidney’s role in maintaining blood glucose becomes more prominent during prolonged fasting or liver failure. Unlike the liver, the kidney lacks significant glycogen stores and contributes to glucose homeostasis only through gluconeogenesis, not glycogenolysis.
Only limited portions of the gluconeogenic pathway are active in skeletal muscle, cardiac muscle, and the brain, and at very low rates.
However, these tissues cannot release free glucose into the bloodstream because they lack glucose 6-phosphatase (EC 3.1.3.9), the enzyme responsible for the final step of gluconeogenesis. As a result, glucose 6-phosphate (G6P) produced, either via gluconeogenesis or glycogenolysis, remains trapped in the cell and is used only for internal energy needs or glycogen resynthesis. In the brain, this occurs mainly in astrocytes. The only direct contribution of these tissues to blood glucose maintenance, particularly skeletal muscle, due to its large mass (approximately 18 times that of the liver), comes from the limited release of free glucose via the debranching enzyme (EC 3.2.1.33) during glycogenolysis.[4]
Most gluconeogenic reactions occur in the cytosol, though some steps take place in the mitochondria, and the final step, catalyzed by glucose 6-phosphatase, occurs within the endoplasmic reticulum cisternae.[3]
Irreversible steps in the pathway
As previously mentioned, gluconeogenesis is essentially glycolysis in reverse. Of the ten reactions that constitute gluconeogenesis, seven are shared with glycolysis. These reactions have a ΔG close to zero and are therefore easily reversible. However, under intracellular conditions, the overall ΔG of glycolysis is approximately –63 kJ/mol (–15 kcal/mol), while that of gluconeogenesis is about –16 kJ/mol (–3.83 kcal/mol). This indicates that both pathways are irreversiblein vivo.
Glycolysis and Gluconeogenesis Pathways
The irreversibility of glycolysis arises from three strongly exergonic reactions that cannot be used in gluconeogenesis.
Phosphorylation of glucose to glucose 6-phosphate, catalyzed by hexokinase (EC 2.7.1.1) or glucokinase (EC 2.7.1.2).
Phosphorylation of fructose 6-phosphate to fructose 1,6-bisphosphate (F1,6BP), catalyzed by phosphofructokinase-1 (PFK-1; EC 2.7.1.11).
Conversion of phosphoenolpyruvate (PEP) to pyruvate, catalyzed by pyruvate kinase (EC 2.7.1.40).
Irreversible reactions of glycolysis bypassed in gluconeogenesis
Reaction
Catalyzing enzyme
ΔG (kJ/mol) (kcal/mol)
ΔG°’ (kJ/mol) (kcal/mol)
Glu → G6P
Hexokinase or Glucokinase
–33.4 (–8.0)
–16.7 (–4.0)
F6P → F1,6BP
PFK-1
–22.2 (–5.3)
–14.2 (–3.4)
PEP → Pyr
Pyruvate kinase
–16.7 (–4.0)
–31.4 (–7.5)
In gluconeogenesis, these three steps are bypassed by enzymes that catalyze irreversible reactions in the direction of glucose synthesis.
These bypass reactions ensure that gluconeogenesis, like glycolysis, remains a thermodynamically irreversible pathway.[2]
The next sections will analyze these reactions in detail.
From pyruvate to phosphoenolpyruvate
The first step of gluconeogenesis that bypasses an irreversible step of glycolysis, namely the reaction catalyzed by pyruvate kinase, is the conversion of pyruvate to phosphoenolpyruvate.
This occurs through a two-step process catalyzed by:
Pyruvate carboxylase catalyzes the carboxylation of pyruvate to oxaloacetate, consuming one molecule of ATP in the process. The enzyme requires magnesium or manganese ions.
Pyr + HCO3–+ ATP → Oxaloacetate + ADP + Pi
Discovered in 1960 by Merton Utter, pyruvate carboxylase is a mitochondrial enzyme composed of four identical subunits, each containing a biotin prosthetic group. The biotin is covalently attached by an amide bond to the ε-amino group of a lysine residue and serves as a carrier of activated CO2 during the reaction. Each subunit also contains an allosteric site for acetyl-CoA, an essential activator of the enzyme.
In addition to its role in gluconeogenesis, this reaction also provides intermediates for the citric acid cycle.[10][11]
Phosphoenolpyruvate carboxykinase and PEP formation
Phosphoenolpyruvate carboxykinase catalyzes the decarboxylation and phosphorylation of oxaloacetate to form PEP. This reaction requires GTP as a phosphate donor and the presence of magnesium and manganese ions.[12]
Oxaloacetate + GTP → PEP + CO2 + GDP
The CO2 removed in this step is the same one added to pyruvate by pyruvate carboxylase. This carboxylation–decarboxylation sequence is crucial, as it facilitates the formation of PEP by making the reaction energetically favorable.[1]
More generally, such sequences are used to drive otherwise endergonic reactions and are also found in other metabolic pathways like the citric acid cycle, the pentose phosphate pathway, and fatty acid synthesis.[2]
Energetics of the overall conversion
The net reaction catalyzed by pyruvate carboxylase and PEPCK is:
Pyr + ATP + GTP + HCO3– → PEP + ADP + GDP + Pi + CO2
The ΔG°’ of this combined reaction is +0.9 kJ/mol (0.2 kcal/mol).
In contrast, the ΔG°’ for the reverse glycolytic reaction catalyzed by pyruvate kinase is +31.4 kJ/mol (7.5 kcal/mol).
Under intracellular conditions, however, the actual ΔG is highly negative (–25 kJ/mol, or –6 kcal/mol), due to rapid consumption of PEP, which keeps its concentration low.
Thus, the formation of PEP from pyruvate is effectively irreversible in vivo.[1]
Phosphoenolpyruvate precursors: pyruvate or alanine
Gluconeogenesis can proceed from various glucogenic precursors. This section examines the specific steps involved when pyruvate or alanine serve as the starting materials, highlighting how the pathway adapts according to the metabolic conditions.
Mitochondrial transport and conversion of pyruvate and alanine
The reactions described below predominate when pyruvate or alanine is the glucogenic precursor.
Since pyruvate carboxylase is a mitochondrial enzyme, pyruvate must first be transported from the cytosol into the mitochondrial matrix. This transport is mediated by specific proteins located in the inner mitochondrial membrane, namely MPC1 and MPC2. These two proteins associate to form a hetero-oligomeric complex that facilitates pyruvate import into mitochondria.[13]
Alternatively, pyruvate can be generated inside the mitochondrial matrix via transamination of alanine, catalyzed by alanine aminotransferase (EC 2.6.1.2). In this reaction, the amino group of alanine is transferred to α-ketoglutarate, producing glutamate and pyruvate. The resulting amino group is subsequently excreted as urea through the urea cycle.[14]
Oxaloacetate transport and conversion to PEP
Since most gluconeogenic enzymes (with the exception of glucose-6-phosphatase) are cytosolic, the oxaloacetate produced in the mitochondrial matrix must be transferred to the cytosol. However, oxaloacetate itself cannot cross the inner mitochondrial membrane directly.
To overcome this, it is reduced to malate in a reaction catalyzed by mitochondrial malate dehydrogenase (EC 1.1.1.37), an enzyme also involved in the citric acid cycle. This reaction uses NADH as a reducing agent, which is oxidized to NAD+.
Oxaloacetate + NADH + H+ ⇄ Malate + NAD+
Although the ΔG°’ of this reaction is strongly positive, under physiological conditions the ΔG is close to zero, making the reaction reversible and efficient in vivo.[2]
Malate is then transported into the cytosol via the malate-α-ketoglutarate transporter, a component of the malate-aspartate shuttle. Once in the cytosol, malate is re-oxidized to oxaloacetate by cytosolic malate dehydrogenase, regenerating NADH.
Malate + NAD+ → Oxaloacetate + NADH + H+
Note: the malate-aspartate shuttle is the most active mechanism for transferring reducing equivalents (NADH) from the cytosol into mitochondria. It operates in tissues such as the liver, kidney, and heart.[3]
The reverse use of this shuttle in gluconeogenesis allows NADH to be generated in the cytosol, which is essential for the reaction catalyzed by glyceraldehyde 3-phosphate dehydrogenase (EC 1.2.1.12). In fact, cytosolic NADH is normally present at very low concentrations, with a [NADH]/[NAD+] ratio of about 8 × 10−4, roughly 100,000 times lower than in mitochondria.[15]
Finally, the cytosolic oxaloacetate is converted to phosphoenolpyruvate by PEP carboxykinase.
Phosphoenolpyruvate precursor: lactate
Lactate is one of the major gluconeogenic precursors. It is produced, for example, by:
red blood cells, which rely entirely on anaerobic glycolysis for ATP production;
skeletal muscle during intense exercise, that is, under low-oxygen conditions when the rate of glycolysis exceeds that of the citric acid cycle and oxidative phosphorylation.[9]
When lactate serves as the gluconeogenic precursor, PEP synthesis proceeds through a different pathway than the one described for pyruvate or alanine.
In the hepatocyte cytosol, where the NAD+ concentration is high, lactate is oxidized to pyruvate by the liver isoenzyme of lactate dehydrogenase (EC 1.1.1.27). During this reaction, NAD+ is reduced to NADH.
Lactate + NAD+ → Pyr + NADH + H+
The generation of cytosolic NADH makes the export of reducing equivalents from mitochondria unnecessary.
Pyruvate then enters the mitochondrial matrix, where it is converted to oxaloacetate by pyruvate carboxylase. In this case, oxaloacetate is directly converted to PEP by the mitochondrial isoform of PEP carboxykinase. PEP is then transported out of the mitochondria via an anion transporter located in the inner mitochondrial membrane and continues along the gluconeogenic pathway in the cytosol.[3]
Note: the synthesis of glucose from lactate represents the hepatic phase of the Cori cycle, which links peripheral tissues to the liver in the recycling of lactate.[16]
From fructose 1,6-bisphosphate to fructose 6-phosphate
The second step of gluconeogenesis that bypasses an irreversible reaction of the glycolytic pathway, specifically the one catalyzed by phosphofructokinase-1, is the dephosphorylation of fructose 1,6-bisphosphate to fructose 6-phosphate.[5]
This reaction is catalyzed by fructose 1,6-bisphosphatase (FBPase-1; EC 3.1.3.11), a Mg2+-dependent enzyme located in the cytosol. It involves the hydrolysis of the phosphate group at the C-1 position of fructose 1,6-bisphosphate and does not generate ATP.
F1,6BP + H2O → F6P + Pi
The ΔG°’ for the reaction is –16.3 kJ/mol (–3.9 kcal/mol), indicating that it is irreversible under cellular conditions.[2]
From glucose 6-phosphate to glucose
The third step of gluconeogenesis that bypasses an irreversible reaction of the glycolytic pathway, specifically the one catalyzed by hexokinase or glucokinase, is the dephosphorylation of glucose 6-phosphate to glucose.[5]
This reaction is catalyzed by the catalytic subunit of glucose 6-phosphatase, part of a protein complex located in the endoplasmic reticulum membrane of hepatocytes, enterocytes, and the proximal tubule cells of the kidney.[8] The glucose 6-phosphatase complex consists of the catalytic subunit and a glucose 6-phosphate transporter known as T1 (glucose 6-phosphate translocase).
Importantly, the enzyme’s active site faces the lumen of the endoplasmic reticulum. This means that glucose is released inside the endoplasmic reticulum, not in the cytosol.[17]
Mechanism and regulation of glucose release
Glucose 6-phosphate, derived either from gluconeogenesis via the action of glucose 6-phosphate isomerase (EC 5.3.1.9), or from glycogenolysis via phosphoglucomutase (EC 5.4.2.2), is found in the cytosol and must be transported into the endoplasmic reticulum lumen to be dephosphorylated. This transport is mediated by T1 translocase.
Once inside the endoplasmic reticulum, the Mg2+-dependent catalytic subunit catalyzes the hydrolysis of the phosphate ester bond.[17]
G6P + H2O → Glu + Pi
As with the reaction catalyzed by FBPase-1, this is an irreversible reaction. The ΔG°′ is –13.8 kJ/mol (–3.3 kcal/mol). In contrast, the reverse reaction, catalyzed by hexokinase or glucokinase, would require the transfer of a phosphate group from glucose 6-phosphate to ADP, which would be highly endergonic (ΔG = +33.4 kJ/mol, or +8 kcal/mol).[2]
To prevent interference with glycolysis, which occurs in the cytosol, the glucose 6-phosphatase activity is spatially restricted to the endoplasmic reticulum lumen, ensuring compartmental separation of opposing pathways.[4]
Glucose and inorganic phosphate are exported from the endoplasmic reticulum to the cytosol by distinct transporters, likely T2, for glucose, and T3, an anion transporter for inorganic phosphate. Finally, glucose leaves the hepatocyte via the GLUT2 transporter, enters the bloodstream, and is delivered to tissues in need.[17]
Energy cost of gluconeogenesis
Like glycolysis, gluconeogenesis involves several energetically costly steps, particularly those that bypass the irreversible reactions of glycolysis.
In total, six high-energy phosphate bonds are consumed in the synthesis of one molecule of glucose from two molecules of pyruvate: four ATP and two GTP.
In addition, two molecules of NADH are required for the reduction of 1,3-bisphosphoglycerate to glyceraldehyde 3-phosphate, in the reaction catalyzed by glyceraldehyde 3-phosphate dehydrogenase.
The oxidation of NADH used in gluconeogenesis implies a loss of potential ATP that would otherwise be produced via oxidative phosphorylation, approximately five molecules of ATP are not synthesized due to the diversion of NADH from the electron transport chain.
These energetic demands reinforce the concept that gluconeogenesis is not simply glycolysis in reverse. If it were, it would require only the energy equivalent of two ATP molecules, as shown in the overall glycolytic equation.
In the liver, this energy is primarily provided by the oxidation of fatty acids. Under certain metabolic conditions, especially during prolonged fasting or high-protein intake, the oxidation of amino acid carbon skeletons can also contribute significantly to ATP and GTP production. The predominant source of energy depends on the available metabolic fuels at the time.[6]
Coordinated regulation of gluconeogenesis and glycolysis
If glycolysis and gluconeogenesis were simultaneously active at high rates within the same cell, the net result would be the consumption of ATP and the generation of heat, particularly at the irreversible steps of both pathways, with no productive metabolic outcome.
For example, consider the opposing reactions catalyzed by phosphofructokinase-1 and fructose 1,6-bisphosphatase-1.
PFK-1 reaction:
These two reactions, if occurring simultaneously in opposite directions, result in a futile cycle (also called a substrate cycle), leading to a wasteful dissipation of energy.[18]
To avoid this, the cell tightly regulates these pathways through three main mechanisms:
allosteric regulation
covalent modification (e.g., phosphorylation and dephosphorylation);
changes in enzyme concentration, regulated at the transcriptional or degradative level.
Allosteric regulation is rapid and reversible, occurring within milliseconds. In contrast, covalent modifications are typically triggered by extracellular signals, such as hormones like insulin, glucagon, or epinephrine, and occur over seconds to minutes. Regulation via enzyme synthesis or degradation takes even longer, on the order of hours.
This multi-layered regulation ensures that when pyruvate is directed into gluconeogenesis, glycolytic flux is simultaneously reduced, and vice versa, allowing for metabolic coordination in response to the cell’s energy status and systemic hormonal cues.[1]
Regulation of gluconeogenesis
The regulation of gluconeogenesis, like that of glycolysis, primarily targets the enzymes that are unique to each pathway, rather than those that are shared between them.
In glycolysis, the major regulatory control points are the reactions catalyzed by PFK-1 and pyruvate kinase. In contrast, gluconeogenesis is mainly regulated at the steps catalyzed by fructose 1,6-bisphosphatase and pyruvate carboxylase.
The other two enzymes unique to gluconeogenesis, glucose 6-phosphatase and phosphoenolpyruvate carboxykinase, are regulated primarily at the transcriptional level, in response to hormonal and metabolic signals.[4]
Pyruvate carboxylase
In mitochondria, pyruvate can be directed toward two major metabolic fates:
carboxylation to oxaloacetate via pyruvate carboxylase, initiating the gluconeogenic pathway.
The fate of pyruvate depends largely on the availability of acetyl-CoA, which in turn reflects the supply of fatty acids in the mitochondrion.
When fatty acids are abundant, their β-oxidation generates acetyl-CoA, which enters the citric acid cycle and produces GTP and NADH. If the cell’s energy needs are met, oxidative phosphorylation slows down, leading to an increase in the [NADH]/[NAD+] ratio. This elevated ratio inhibits the citric acid cycle, causing acetyl-CoA to accumulate in the mitochondrial matrix.
Acetyl-CoA acts as a key metabolic signal:
it is a positive allosteric effector of pyruvate carboxylase, promoting gluconeogenesis;
it is a negative allosteric effector of pyruvate kinase, inhibiting glycolysis;
it also inhibits the pyruvate dehydrogenase complex through feedback inhibition and via phosphorylation, activating a specific PDC kinase.
Thus, when the cellular energy charge is high, the conversion of pyruvate to acetyl-CoA slows down, while its conversion to oxaloacetate, and ultimately glucose, is promoted. In this context, acetyl-CoA acts as a metabolic signal indicating that further glucose oxidation is unnecessary, and that glucogenic precursors should be directed toward glucose synthesis and storage.
Conversely, when acetyl-CoA levels are low, pyruvate is preferentially directed toward energy production:
pyruvate kinase and the pyruvate dehydrogenase complex are more active;
the flow of carbon into the citric acid cycle increases;
cellular energy needs are met through oxidative metabolism.[10][20]
In summary, pyruvate carboxylase represents the first major control point of gluconeogenesis, determining whether pyruvate is used for energy production or diverted toward glucose synthesis, based on the energetic status of the cell.[21]
Fructose 1,6-bisphosphatase
The second major control point in gluconeogenesis is the reaction catalyzed by fructose 1,6-bisphosphatase. This enzyme is allosterically inhibited by AMP, meaning that when AMP levels are high, and consequently ATP levels are low, gluconeogenesis slows down. Thus, as previously mentioned, FBPase-1 is active only when the cellular energy charge is sufficiently high to support de novo glucose synthesis.[22]
In contrast, phosphofructokinase-1, the glycolytic counterpart, is allosterically activated by AMP and ADP, and inhibited by ATP and citrate, the latter being a product of acetyl-CoA and oxaloacetate condensation.[23]
Therefore:
when AMP levels are high, glycolysis is favored, and gluconeogenesis is inhibited;
when ATP, acetyl-CoA, or citrate levels are high, gluconeogenesis is promoted, and glycolysis slows down.
The accumulation of citrate, in particular, signals a slowdown in the citric acid cycle and facilitates the diversion of pyruvate toward glucose synthesis.[21][24]
PFK-1, FBPase-1, and fructose 2,6-bisphosphate
The liver plays a central role in maintaining blood glucose homeostasis, requiring tight coordination between glucose consumption and production. Two hormones are primarily responsible for this regulation: insulin and glucagon.[21]
Their intracellular effects are largely mediated through fructose 2,6-bisphosphate (F2,6BP), a potent allosteric activator of PFK-1 and inhibitor of FBPase-1. F2,6BP is structurally similar to fructose 1,6-bisphosphate but is not an intermediate in either glycolysis or gluconeogenesis.[1]
Discovered in 1980 by Emile Van Schaftingen and Henri-Gery Hers, F2,6BP was first identified as a powerful activator of PFK-1. The following year, it was also shown to be a strong inhibitor of FBPase-1.
F2,6BP binds to an allosteric site on PFK-1, reducing its affinity for ATP and citrate (inhibitors) while increasing its affinity for fructose 6-phosphate (its substrate). Without F2,6BP, and under physiological conditions with ATP and citrate present, PFK-1 is nearly inactive. The presence of F2,6BP activates PFK-1 and stimulates glycolysis, while simultaneously inhibiting FBPase-1, even in the absence of AMP. The effects of AMP and F2,6BP on FBPase-1 are synergistic.[25][26]
Regulation of F2,6BP levels
The cellular concentration of F2,6BP is regulated by the balance between its synthesis and degradation.
Synthesis: catalyzed by phosphofructokinase-2 (PFK-2), from fructose 6-phosphate (EC 2.7.1.105).
Degradation: catalyzed by fructose 2,6-bisphosphatase (FBPase-2), converting F2,6BP back to fructose 6-phosphate (EC 3.1.3.46).
These two enzymatic activities reside on a single bifunctional protein, often called the PFK-2/FBPase-2 tandem enzyme. In the liver, the balance of these two functions is hormonally regulated.[27]
Glucagon
After binding to its specific membrane receptors, glucagon stimulates adenylate cyclase (EC 4.6.1.1) to synthesize 3′,5′-cyclic adenosine monophosphate (cAMP). This second messenger activates cAMP-dependent protein kinase, also known as protein kinase A (PKA) (EC 2.7.11.11).
PKA catalyzes the phosphorylation of a specific serine residue (Ser32) on the bifunctional enzyme PFK-2/FBPase-2, using one molecule of ATP. Phosphorylation increases the enzyme’s fructose 2,6-bisphosphatase activity while decreasing its phosphofructokinase-2 activity. This shift is partly due to an increase in the Km for fructose 6-phosphate, which reduces the enzyme’s affinity for its substrate.
As a result, the intracellular concentration of fructose 2,6-bisphosphate decreases. Since this molecule is a potent activator of PFK-1 and inhibitor of FBPase-1, its reduction inhibits glycolysis and stimulates gluconeogenesis. Therefore, in response to glucagon, hepatic glucose production increases, helping the liver counteract the drop in blood glucose levels.[28][29]
Note: like adrenaline, glucagon also promotes gluconeogenesis by increasing the availability of key substrates such as glycerol and amino acids.[30]
Insulin
Insulin has the opposite effect. Upon receptor binding, it activates phosphoprotein phosphatase 2A (PP2A), which dephosphorylates PFK-2/FBPase-2:
this activates PFK-2
and inhibits FBPase-2
Insulin also stimulates cAMP phosphodiesterase, which degrades cAMP into AMP, further reducing PKA activity. The result is an increase in F2,6BP levels, which inhibits gluconeogenesis and stimulates glycolysis.
Additionally:
fructose 6-phosphate allosterically activates PFK-2 and inhibits FBPase-2;
both PFK-2 and FBPase-2 are subject to product inhibition.
However, the main regulatory factors are the level of fructose 6-phosphate and the phosphorylation state of the bifunctional enzyme.[31]
Glucose 6-phosphatase
Unlike pyruvate carboxylase and fructose-1,6-bisphosphatase, the catalytic subunit of glucose 6-phosphatase is not regulated allosterically or through covalent modification. Instead, its activity is modulated at the transcriptional level.[32]
Conditions that promote glucose production, such as low blood glucose, glucagon, and glucocorticoids, stimulate the expression of the enzyme. In contrast, insulin acts as a negative regulator, inhibiting its synthesis.[33]
Additionally, the Km of glucose 6-phosphatase for glucose 6-phosphate is significantly higher than the physiological concentration of the substrate. This means the enzyme’s activity is almost linearly dependent on the substrate concentration, making it effectively regulated by substrate availability rather than by allosteric effectors.[34]
PEP carboxykinase
PEP carboxykinase is expressed in both the cytosol and mitochondria of hepatocytes, encoded by separate nuclear genes. Its expression is tightly regulated, primarily through changes in its synthesis and degradation rates. For example, enzyme levels are low before birth but rise markedly within hours after delivery, coinciding with the activation of gluconeogenesis in the newborn.[35][36]
High levels of glucagon or fasting promote enzyme production by stabilizing its mRNA and enhancing gene transcription. Conversely, elevated blood glucose or insulin suppress transcription, thereby reducing enzyme levels.[34]
Xylulose 5-phosphate
Xylulose 5-phosphate, a product of the pentose phosphate pathway, is a relatively recently identified regulatory molecule. It stimulates glycolysis and inhibits gluconeogenesis by modulating the concentration of fructose 2,6-bisphosphate in the liver.[37]
When blood glucose levels rise, such as after a carbohydrate-rich meal, both glycolysis and the pentose phosphate pathway are activated in hepatocytes. The resulting production of xylulose 5-phosphate activates protein phosphatase 2A. As previously described, PP2A dephosphorylates PFK-2/FBPase-2, thereby inhibiting FBPase-2 and activating PFK-2. This leads to a rise in fructose 2,6-bisphosphate levels, which in turn inhibits gluconeogenesis and stimulates glycolysis. The increased glycolytic flux results in the production of acetyl-CoA, a key precursor for lipid synthesis.[38]
Simultaneously, the enhanced activity of the pentose phosphate pathway produces NADPH, which provides reducing power for fatty acid biosynthesis. Additionally, PP2A dephosphorylates carbohydrate-responsive element-binding protein (ChREBP), a transcription factor that upregulates the expression of hepatic genes involved in lipid synthesis.[39]
Thus, in response to elevated blood glucose levels, xylulose 5-phosphate acts as a crucial regulator, promoting lipid synthesis and coordinating carbohydrate and fat metabolism.[38]
Precursors
Besides the aforementioned pyruvate, the major gluconeogenic precursors are lactate, glycerol, most amino acids, and, more generally, any compound that can be converted into pyruvate or oxaloacetate.[40]
Glycerol
Glycerol is released by lipolysis in adipose tissue. With the exception of propionyl-CoA, it is the only part of the lipid molecule that can be used for glucose synthesis in animals.[5]
Glycerol enters gluconeogenesis, or glycolysis, depending on the cellular energy charge, as dihydroxyacetone phosphate (DHAP). Its synthesis occurs in two steps.
In the first step, glycerol is phosphorylated to glycerol 3-phosphate by the enzyme glycerol kinase (EC 2.7.1.30), with the consumption of one ATP.
Glycerol + ATP → Glycerol 3-phosphate + ADP
This enzyme is absent in adipocytes, but present in the liver. This means that glycerol must reach the liver to be further metabolized.
Glycerol 3-phosphate is then oxidized to DHAP in a reaction catalyzed by glycerol 3-phosphate dehydrogenase (EC 1.1.1.8), during which NAD+ is reduced to NADH.[9]
Glycerol 3-phosphate + NAD+ ⇄ DHAP + NADH + H+
During prolonged fasting, glycerol becomes the major gluconeogenic precursor, accounting for approximately 20% of total glucose production.[41]
Glucogenic amino acids
Pyruvate and oxaloacetate are the entry points for the glucogenic amino acids, that is, those whose carbon skeleton, or part of it, can be used for de novo glucose synthesis.
Amino acids are derived from the catabolism of proteins, whether dietary or endogenous, such as skeletal muscle proteins during fasting or intense, prolonged exercise.
The catabolic pathways of the twenty amino acids that make up proteins converge to form seven major metabolic products:
Among these, only acetyl-CoA and acetoacetyl-CoA cannot be used for gluconeogenesis. Therefore, glucogenic amino acids can be defined as those whose carbon skeleton (or a part of it) can be converted into pyruvate, oxaloacetate, α-ketoglutarate, succinyl-CoA, or fumarate.
Note: only leucine and lysine are exclusively ketogenic, as their carbon skeletons are broken down into acetyl-CoA and/or acetoacetyl-CoA, which cannot serve as gluconeogenic substrates.
Below are the entry points of the gluconeogenic amino acids.
The intermediates α-ketoglutarate, succinyl-CoA, and fumarate, all part of the citric acid cycle, enter the gluconeogenic pathway after conversion to oxaloacetate.[42]
Propionate
Propionate, a short-chain fatty acid, is a gluconeogenic precursor because its active form, propionyl-CoA, can be converted into succinyl-CoA.
It originates from several sources.
β-Oxidation of odd-chain fatty acids, such as margaric acid (C17:0). In the final β-oxidation step, a five-carbon fatty acid is split into acetyl-CoA and propionyl-CoA. These fatty acids, though less common, are found in lipids of marine organisms, ruminants, and some plants.[43]
Oxidation of branched-chain fatty acids, like phytanic acid, derived from phytol, a chlorophyll breakdown product.[44]
In ruminants, microbial fermentation of cellulose-derived glucose in the rumen produces propionate. Once absorbed, it can be used for gluconeogenesis, fatty acid synthesis, or energy production. In these animals, where gluconeogenesis is nearly constant, propionate is the major gluconeogenic precursor.[45]
It also derives from the catabolism of the amino acids valine, isoleucine, methionine, and threonine.[46]
In the colon, fermentation of non-digestible carbohydrates (e.g., resistant starch and fibers) by the gut microbiota produces propionate. After absorption by enterocytes, the portion not metabolized locally reaches the liver via the portal vein.[47]
Conversion of propionyl-CoA to succinyl-CoA involves three reactions:
^ abcdefg Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ abcde Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
^ abcdef Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 4th Edition. Elsevier health sciences, 2018.
^ abcde Chourpiliadis C., Mohiuddin S.S. Biochemistry, Gluconeogenesis. [Updated 2023 Jun 5]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK544346/
^ ab Gerich J.E. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med 2010;27(2):136-42. doi:10.1111/j.1464-5491.2009.02894.x
^ abc Melkonian E.A., Asuka E., Schury M.P. Physiology, Gluconeogenesis. [Updated 2023 Nov 13]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK541119/
^ Adina-Zada A., Zeczycki T.N., Attwood P.V. Regulation of the structure and activity of pyruvate carboxylase by acetyl CoA. Arch Biochem Biophys 2012;519(2):118-30. doi:10.1016/j.abb.2011.11.015
^ Utter M.F., Keech D.B. Formation of oxaloacetate from pyruvate and carbon dioxide. J Biol Chem 1960;235:PC17-8. doi:10.1016/S0021-9258(18)69442-6
^ Case C.L., Mukhopadhyay B. Kinetic characterization of recombinant human cytosolic phosphoenolpyruvate carboxykinase with and without a His10-tag. Biochim Biophys Acta 2007;1770(11):1576-84. doi:10.1016/j.bbagen.2007.07.012
^ McCommis K.S. and Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J 2015;466(3):443-454. doi:10.1042/BJ20141171
^ Wu G. Amino acids: biochemistry and nutrition. CRC Press, 2010.
^ Holeček M. Roles of malate and aspartate in gluconeogenesis in various physiological and pathological states. Metabolism 2023;145:155614. doi:10.1016/j.metabol.2023.155614
^ abc van Schaftingen E., Gerin I. The glucose-6-phosphatase system. Biochem J 2002;362(Pt 3):513-32. doi:10.1042/0264-6021:3620513
^ Newsholme E.A. Substrate cycles: their metabolic, energetic and thermic consequences in man. Biochem Soc Symp 1978;43:183-205.
^ Patel M.S., Korotchkina L.G. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans 2006;34(2):217-22. doi:10.1042/bst0340217
^ Jitrapakdee S., St Maurice M., Rayment I., Cleland W.W., Wallace J.C., Attwood P.V. Structure, mechanism and regulation of pyruvate carboxylase. Biochem J 2008;413(3):369-87. doi:10.1042/BJ20080709
^ abcd Rosenthal M.D., Glew R.H. Medical biochemistry – Human metabolism in health and disease. John Wiley & Sons, Inc., Publication, 2009.
^ Villeret V., Huang S., Zhang Y., Lipscomb W.N. Structural aspects of the allosteric inhibition of fructose-1,6-bisphosphatase by AMP: the binding of both the substrate analogue 2,5-anhydro-D-glucitol 1,6-bisphosphate and catalytic metal ions monitored by X-ray crystallography. Biochemistry 1995;34(13):4307-15. doi:10.1021/bi00013a020
^ Lynch E.M., Hansen H., Salay L., Cooper M., Timr S., Kollman J.M., Webb B.A. Structural basis for allosteric regulation of human phosphofructokinase-1. Nat Commun 2024;15(1):7323. doi:10.1038/s41467-024-51808-6
^ Rui L. Energy metabolism in the liver. Compr Physiol 2014;4(1):177-97. doi:10.1002/cphy.c130024
^ Van Schaftingen E., and Hers H-G. Inhibition of fructose-1,6-bisphosphatase by fructose-2,6-bisphosphate. Proc Natl Acad Sci USA 1981;78(5):2861-2863. doi:https://doi.org/10.1073/pnas.78.5.2861
^ Van Schaftingen E., Jett M-F., Hue L., and Hers H-G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA 1981;78(6):3483-3486. doi:https://doi.org/10.1073/pnas.78.6.3483
^ Okar DA., Lange A.J. Fructose-2,6-bisphosphate and control of carbohydrate metabolism in eukaryotes. Biofactors 1999;10(1):1-14. doi:https://doi.org/10.1002/biof.5520100101
^ Payne V.A., Arden C., Wu C., Lange A.J., Agius L. Dual role of phosphofructokinase-2/fructose bisphosphatase-2 in regulating the compartmentation and expression of glucokinase in hepatocytes. Diabetes 2005;54(7):1949-57. doi:https://doi.org/10.2337/diabetes.54.7.1949
^ Winther-Sørensen M., Galsgaard K.D., Santos A., et al. Glucagon acutely regulates hepatic amino acid catabolism and the effect may be disturbed by steatosis. Mol Metab 2020;42:101080. doi:https://doi.org/10.1016/j.molmet.2020.101080
^ Hatting M., Tavares C.D.J., Sharabi K., Rines A.K., Puigserver P. Insulin regulation of gluconeogenesis. Ann N Y Acad Sci 2018;1411(1):21-35. doi:https://doi.org/10.1111/nyas.13435
^ Soty M., Chilloux J., Delalande F., Zitoun C., Bertile F., Mithieux G., and Gautier-Stein A. Post-Translational regulation of the glucose-6-phosphatase complex by cyclic adenosine monophosphate is a crucial determinant of endogenous glucose production and is controlled by the glucose-6-phosphate transporter. J Proteome Res 2016;15(4):1342-1349. doi:https://doi.org/10.1021/acs.jproteome.6b00110
^ Schmoll D., Walker K.S., Alessi D.R., Grempler R., Burchell A., Guo S., Walther R., Unterman T.G. Regulation of glucose-6-phosphatase gene expression by protein kinase Balpha and the forkhead transcription factor FKHR. Evidence for insulin response unit-dependent and -independent effects of insulin on promoter activity. J Biol Chem 2000;275(46):36324-33. doi:https://doi.org/10.1074/jbc.M003616200
^ ab Rodwell V.W., Bender D.A., Botham K.M., Kennelly P.J., Weil P.A. Harper’s Illustrated Biochemistry. 31st Edition. McGraw-Hill, 2018.
^ Yabaluri N., Bashyam M.D. Hormonal regulation of gluconeogenic gene transcription in the liver. J Biosci 2010;35(3):473-84. doi:https://doi.org/10.1007/s12038-010-0052-0
^ Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003;100:5107-5112. doi:https://doi.org/10.1073/pnas.0730817100
^ Iizuka K., Takao K., Yabe D. ChREBP-mediated regulation of lipid metabolism: involvement of the gut microbiota, liver, and adipose tissue. Front Endocrinol (Lausanne) 2020;11:587189. doi:https://doi.org/10.3389/fendo.2020.587189
^ Baba H., Zhang X.J., Wolfe R.R. Glycerol gluconeogenesis in fasting humans. Nutrition 1995;11(2):149-53.
^ Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ Ferrannini E., Barrett E.J., Bevilacqua S., DeFronzo R.A. Effect of fatty acids on glucose production and utilization in man. J Clin Invest 1983;72(5):1737-47. doi:https://doi.org/10.1172/JCI111133
^ Verhoeven N.M., Wanders R.J., Poll-The B.T., Saudubray J.M., Jakobs C. The metabolism of phytanic acid and pristanic acid in man: a review. J Inherit Metab Dis 1998;21(7):697-728. doi:https://doi.org/10.1023/a:1005476631419
^ Mann G., Mora S., Madu G., Adegoke O.A.J. Branched-chain amino acids: catabolism in skeletal muscle and implications for muscle and whole-body metabolism. Front Physiol 2021;12:702826. doi:https://doi.org/10.3389/fphys.2021.702826
^ Portincasa P., Bonfrate L., Vacca M., De Angelis M., Farella I., Lanza E., Khalil M., Wang D.Q.-H., Sperandio M., Di Ciaula A. Gut microbiota and short chain fatty acids: implications in glucose homeostasis. Int J Mol Sci 2022;23:1105. doi:https://doi.org/10.3390/ijms23031105
The glucose-alanine cycle, also known as the Cahill cycle, was first described independently by Mallette, Exton, and Park, and by Felig and colleagues between 1969 and 1970. It consists of a series of metabolic steps through which extrahepatic tissues export pyruvate and amino groups to the liver in the form of alanine, while receiving glucose from the liver via the bloodstream.[1][2]
Through this interorgan exchange, the glucose-alanine cycle provides a functional link between carbohydrate and amino acid metabolism, as schematically illustrated below.[3]
Glucose → Pyruvate → Alanine → Pyruvate → Glucose
Although originally described in skeletal muscle, the glucose-alanine cycle is not restricted to muscle–liver interactions, but also involves other extrahepatic tissues, including cells of the immune system.[4]
The glucose-alanine cycle shares functional similarities with the Cori cycle; however, unlike lactate, alanine transports both carbon and nitrogen, thereby directly linking energy metabolism with nitrogen metabolism.[3]
Summary: Key Points
Definition: a metabolic pathway in which extrahepatic tissues (mainly skeletal muscle) export nitrogen and the carbon skeleton of pyruvate, produced by glycolysis, to the liver in the form of alanine.
Physiological roles: transport of nitrogen in a non-toxic form and of the gluconeogenic substrate pyruvate to the liver, thereby shifting the metabolic burden associated with nitrogen disposal and glucose regeneration to the liver.
Energy cost: net energy cost to the liver of approximately 3–5 ATP per cycle, necessary to support gluconeogenesis and the urea cycle.
Comparison with the Cori cycle: unlike the Cori cycle, it requires oxygen and functional mitochondria and involves the transport of both carbon and nitrogen.
The principal steps of the glucose-alanine cycle can be outlined as follows.
When amino acids are oxidized for energy in extrahepatic tissues, pyruvate produced via glycolysis acts as an amino-group acceptor, leading to the formation of alanine, a nonessential amino acid.
Alanine is released into the bloodstream and transported to the liver.
In hepatocytes, the amino group of alanine is transferred to α-ketoglutarate, producing pyruvate and glutamate.
Most of the amino groups derived from glutamate enter the urea cycle, whereas a smaller fraction may serve as nitrogen donors in biosynthetic reactions.
The pyruvate generated in the liver enters gluconeogenesis and is used for glucose synthesis.
Newly synthesized glucose is released into the bloodstream and taken up by peripheral tissues, where it is converted back into pyruvate via glycolysis, thereby completing the cycle.
The Glucose-Alanine Cycle Between Muscle and Liver
Mechanisms and biochemical bases
The following section examines the biochemical basis of the glucose-alanine cycle, with particular emphasis on the metabolic interplay between skeletal muscle and the liver.
Under physiological conditions, both intracellular and extracellular proteins undergo continuous turnover, being hydrolyzed into their constituent amino acids and subsequently resynthesized. The rates of these opposing processes are tightly regulated, thereby preventing the loss of fat-free mass.[5]
Under catabolic conditions, such as prolonged fasting or intense and sustained exercise, muscle protein breakdown exceeds protein synthesis. This imbalance leads to the release of amino acids, some of which are oxidized for energy production, while others are diverted toward gluconeogenesis.[3]
The oxidation of amino acid carbon skeletons, particularly those of branched-chain amino acids (BCAAs) such as leucine, isoleucine, and valine, can represent a significant energy source for skeletal muscle. Depending on their catabolic fate, amino acids are classified as glucogenic or ketogenic, according to whether their carbon skeletons give rise to substrates for gluconeogenesis or ketone body synthesis, respectively.[6] For example, after approximately 90 minutes of strenuous exercise, amino acid oxidation may account for 10–15% of the total energy required for muscle contraction.[7]
The use of amino acid carbon skeletons for energy necessarily involves the removal of their amino groups, followed by the safe excretion of amino nitrogen in a non-toxic form.[8]
Removal of the α-amino group occurs primarily through transamination, which can be summarized by the following reversible reaction:
α-Keto acid + Amino acid ⇌ New amino acid + New α-keto acid
These reactions, catalyzed by enzymes known as aminotransferases or transaminases (EC 2.6.1.-), are freely reversible.[9]
In skeletal muscle, branched-chain amino acids commonly transfer their amino group to α-ketoglutarate (2-oxoglutarate), forming glutamate and the corresponding branched-chain α-keto acids. This reaction is catalyzed by branched-chain aminotransferase (BCAT, EC 2.6.1.42).[10]
Glucose-alanine cycle in skeletal muscle
In skeletal muscle, newly formed glutamate may react with ammonia to form glutamine, which serves as a major vehicle for the interorgan transport of nitrogen, particularly to tissues such as the brain.[11] This reaction is catalyzed by the cytosolic enzyme glutamine synthetase (EC 6.3.1.2) and requires the consumption of one molecule of ATP:
Glutamate + NH4+ + ATP → Glutamine + ADP + Pi
In this case, glutamate exits the Cahill cycle.[9]
Alternatively, and in contrast to what occurs in most other tissues, glutamate may transfer its amino group to pyruvate (derived from glycolysis) to form alanine and α-ketoglutarate. This transamination is catalyzed by alanine aminotransferase (ALT, EC 2.6.1.2), an enzyme widely distributed in both animal and plant tissues:[12]
Glutamate + Pyruvate ⇄ Alanine + α-Ketoglutarate
The alanine thus produced, along with that released directly from muscle protein breakdown (muscle proteins are relatively rich in alanine), can exit the cell and enter the bloodstream, ultimately reaching the liver. In this way, the amino group is efficiently transported to the liver.[13]
Moreover, the rate at which alanine formed by pyruvate transamination enters the circulation is proportional to intracellular pyruvate production.[14]
Note: alanine and glutamine are the major interorgan carriers of nitrogen and carbon derived from amino acid metabolism.[3]
Glucose-alanine cycle in the liver
Once in the liver, hepatic alanine aminotransferase catalyzes a transamination in which alanine, the major gluconeogenic amino acid, acts as an amino group donor, while α-ketoglutarate serves as the α-keto acid acceptor.[15] The products of this reaction are pyruvate (the carbon skeleton of alanine) and glutamate:
Alanine + α-Ketoglutarate ⇄ Glutamate + Pyruvate
In a subsequent reaction catalyzed by glutamate dehydrogenase (EC 1.4.1.2), an enzyme located in the mitochondrial matrix, glutamate is deaminated to produce an ammonium ion (NH4+), which enters the urea cycle, and α-ketoglutarate, which may re-enter the citric acid cycle. This reaction is considered anaplerotic, as it links amino acid metabolism with the citric acid cycle.[16]
Alternatively, glutamate can undergo transamination with oxaloacetate to form aspartate and α-ketoglutarate, in a reaction catalyzed by aspartate aminotransferase (EC 2.6.1.1).[17] Aspartate is not only involved in urea synthesis, but also plays a key role in the biosynthesis of purines and pyrimidines.[18]
The pyruvate generated from alanine may follow two alternative metabolic fates.
It can be oxidized for ATP production, in which case it exits the glucose-alanine cycle.
It can enter the gluconeogenesis pathway, continuing the cycle.[19]
The glucose synthesized in the liver is released into the bloodstream and delivered to peripheral tissues, such as skeletal muscle, where it is converted into pyruvate through glycolysis. This newly formed pyruvate may then accept an amino group from glutamate, thus completing the cycle.[20]
Transaminases
As noted above, the removal of amino groups from amino acids occurs via transamination. These reactions are catalyzed by enzymes known as aminotransferases or transaminases.[12]
Transaminases are cytosolic enzymes present in all cells, but are particularly abundant in the liver, kidney, intestine, and muscle. They require pyridoxal phosphate (PLP), the active form of vitamin B6 (pyridoxine), as a tightly bound coenzyme at their active site.[13]
During transamination, the amino group of most free amino acids (with the exceptions of threonine and lysine) is transferred to one of a small set of keto acids, primarily pyruvate, oxaloacetate, or α-ketoglutarate.[3]
Cells express multiple aminotransferases that share specificity for α-ketoglutarate as the amino-group acceptor but differ in their amino-acid substrates, from which they derive their names. Key examples include:
alanine aminotransferase, also known as alanine transaminase or glutamate-pyruvate transaminase (GPT);
aspartate aminotransferase (AST), also called glutamate-oxaloacetate transaminase (GOT).[9]
Importantly, transamination reactions do not result in net deamination; the amino group simply “swaps” from the amino acid to the keto acid, with no loss of amino groups overall.[12]
Physiological roles
The glucose-alanine cycle serves several important physiological functions.
It transports nitrogen in a non-toxic form from peripheral tissues to the liver in the form of alanine.[8]
It transports pyruvate, a key gluconeogenic substrate, to the liver for glucose synthesis.[8]
It removes pyruvate from peripheral tissues, promoting more efficient ATP production. By reducing cytosolic pyruvate, the NADH generated during glycolysis can be more effectively oxidized via mitochondrial oxidative phosphorylation.[13]
It helps maintain a relatively high concentration of alanine in hepatocytes, which may contribute to the inhibition of hepatic protein degradation.[15]
It may contribute to host defense mechanisms during infectious diseases, although the precise role is still under investigation.[21]
Summary: functions of the glucose-alanine cycle
Function
Description
Nitrogen transport
Safely carries nitrogen from peripheral tissues to the liver in the form of alanine.
Pyruvate transport
Delivers pyruvate (a gluconeogenic substrate) to the liver for glucose production.
ATP optimization
Enhances ATP yield in peripheral tissues by enabling full oxidation of glycolytic NADH via mitochondria.
Protein-sparing effect
Helps maintain high alanine levels in hepatocytes, potentially reducing hepatic protein degradation.
Support in host defense
May contribute to immune response during infections (mechanism under study).
No net glucose gain
The cycle does not result in net glucose synthesis.
Finally, it is important to emphasize that there is no net synthesis of glucose in the glucose-alanine cycle.[3]
Energy cost of the glucose-alanine cycle
Like the Cori cycle, the glucose-alanine cycle incurs an energy cost, estimated at approximately 3–5 ATP per cycle.[22]
The peripheral (extrahepatic) part of the cycle yields energy:
2 ATP are generated through glycolysis.
An additional 3–5 ATP are produced from the oxidation of NADH and FADH2.
In contrast, the hepatic part of the cycle (in the liver) consumes energy:
6 ATP are required for gluconeogenesis per molecule of glucose synthesized;
4 ATP are used in the urea cycle per molecule of urea formed.
Thus, the glucose-alanine cycle, like the Cori cycle, shifts part of the metabolic burden from extrahepatic tissues to the liver. However, the energy cost sustained by the liver is justified by the physiological benefits the cycle provides. It facilitates the breakdown of proteins in extrahepatic tissues, particularly skeletal muscle, under specific conditions (e.g., prolonged exercise or fasting). This, in turn, enables both the utilization of amino acids for energy and the supply of gluconeogenic substrates to the liver.[3]
Similarities with the Cori cycle
There are several similarities between the two cycles, summarized below.
The Cahill cycle partially overlaps with the Cori cycle when pyruvate is converted into glucose in the liver, and the resulting glucose is transported back to extrahepatic tissues, where it is reconverted into pyruvate via glycolysis.[3]
The entry point into the gluconeogenic pathway is similar in both cycles: both alanine and lactate are converted to pyruvate.[9]
Like the Cori cycle, the glucose-alanine cycle occurs between different cell types, unlike metabolic pathways such as glycolysis, the citric acid cycle, or gluconeogenesis, which take place within individual cells.[23]
Comparison Between the Glucose-Alanine and Cori Cycles
Differences from the Cori cycle
Here are some key differences between the two cycles.
The main difference lies in the three-carbon intermediate transported from peripheral tissues to the liver: lactate in the Cori cycle, and alanine in the glucose-alanine cycle.[15]
Another difference involves the fate of NADH produced by glycolysis in peripheral tissues.
In the Cori cycle, NADH is used to reduce pyruvate to lactate via the enzyme lactate dehydrogenase (EC 1.1.1.27).
In the glucose-alanine cycle, this reduction does not occur. Instead, electrons from NADH are transferred into the mitochondria via the malate–aspartate and glycerol-3-phosphate shuttles, generating NADH through the former and FADH2 through the latter. The ATP yields are approximately 2.5 per NADH and 1.5 per FADH2.[23]
As a result, unlike the Cori cycle, the glucose-alanine (Cahill) cycle requires the presence of both oxygen and functional mitochondria in peripheral tissues.[11]
Summary: Cori cycle vs. glucose-alanine cycle
Feature
Cori cycle
Glucose-alanine cycle
Three-carbon compound transported to liver
Lactate
Alanine
Source of 3-carbon compound
Reduction of pyruvate by NADH
Transamination of pyruvate with glutamate
NADH usage in peripheral tissues
Consumed to reduce pyruvate to lactate
Transferred to mitochondria via shuttles
Requirement for oxygen and mitochondria
Not required
Required
Nitrogen transport
None
Transports amino groups (as alanine) to liver
Occurs between different cell types
Yes
Yes
Entry into gluconeogenesis
Lactate → Pyruvate
Alanine → Pyruvate
Energy cost in liver
High (gluconeogenesis)
High (gluconeogenesis + urea cycle)
References
^ Felig P., Pozefsky T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
^ Mallette L.E., Exton J.H., and Park C.R. Control of gluconeogenesis from amino acids in the perfused rat liver. J Biol Chem 1969;244(20):5713-5723. doi:10.1016/S0021-9258(18)63618-X
^ abcdefgh Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ Zhao H., Raines L.N., Huang S.C. Carbohydrate and amino acid metabolism as hallmarks for innate immune cell activation and function. Cells 2020;9(3):562. doi:10.3390/cells9030562
^ Lecker S.H., Goldberg A.L. and Mitch W.E. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J Am Soc Nephrol 2006;17(7):1807-1819. doi:10.1681/ASN.2006010083
^ Rennie M.J., Tipton K.D. Protein and amino acid metabolism during and after exercise and the effects of nutrition. Annu Rev Nutr 2000;20:457-83. doi:10.1146/annurev.nutr.20.1.457
^ Witard O.C., Hearris M., Morgan P.T. Protein nutrition for endurance athletes: a metabolic focus on promoting recovery and training adaptation. Sports Med 2025;55(6):1361-1376. doi:10.1007/s40279-025-02203-8
^ abc Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ abcd Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
^ Nong X., Zhang C., Wang J., Ding P., Ji G., Wu T. The mechanism of branched-chain amino acid transferases in different diseases: research progress and future prospects. Front Oncol 2022;12:988290. doi:10.3389/fonc.2022.988290
^ ab Wu G. Amino acids: biochemistry and nutrition. CRC Press, 2010.
^ abc Berg J.M., Tymoczko J.L., Gatto J.G., Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
^ Kurmi K., Haigis M.C. Nitrogen metabolism in cancer and immunity. Trends Cell Biol 2020;30(5):408-424. doi:10.1016/j.tcb.2020.02.005
^ Gray L.R., Tompkins S.C., Taylor E.B. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-604. doi:10.1007/s00018-013-1539-2
^ Petersen K.F., Dufour S., Cline G.W., Shulman G.I. Regulation of hepatic mitochondrial oxidation by glucose-alanine cycling during starvation in humans. J Clin Invest 2019;129(11):4671-4675. doi:10.1172/JCI129913
^ Newsholme E.A., Parry-Billings M. Properties of glutamine release from muscle and its importance for the immune system. JPEN J Parenter Enteral Nutr 1990;14(4 Suppl):63S-67S. doi:10.1177/014860719001400406
The Cori cycle was discovered in the 1930s and 1940s by Carl Ferdinand Cori and Gerty Theresa Radnitz, a husband-and-wife research team. They demonstrated the existence of a metabolic cooperation between skeletal muscle, operating under hypoxic conditions, and the liver.[1]
The cycle enables the conversion of lactate, the conjugate base of lactic acid and its predominant form at physiological pH, back into glucose, thereby ensuring a continuous supply of this monosaccharide to peripheral tissues. Its physiological importance is underscored by the fact that it accounts for up approximately 36% of plasma glucose turnover.[2][3]
From a biochemical perspective, the Cori cycle connects gluconeogenesis and anaerobic glycolysis, using different tissues to compartmentalize these opposing metabolic pathways.[4]
This metabolic cooperation also occurs between the liver and other extrahepatic tissues. Indeed, like the glucose-alanine cycle, the Cori cycle is active between the liver and all tissues that do not fully oxidize glucose to CO2 and H2O.[5]
Summary: Key Points
Metabolic cooperation: it connects extrahepatic tissues, primarily hypoxic skeletal muscle and red blood cells, with the liver, ensuring a continuous energy supply to peripheral tissues.
Lactate recycling: it converts lactate, produced via extrahepatic anaerobic glycolysis, back into glucose through hepatic and renal gluconeogenesis.
Energy cost: the cycle cannot run indefinitely as it requires a net consumption of 4 high-energy bonds; specifically, 6 ATP/GTP are spent in the liver to yield only 2 ATP in muscles.
Physiological role: it prevents intracellular lactic acidosis during intense exercise and helps maintain blood glucose levels during fasting and starvation.
The steps of the Cori cycle are typically analyzed by considering the lactate produced by red blood cells and muscle fibers.
The Cori Cycle
The cycle begins with the conversion of glucose to lactate via anaerobic glycolysis, followed by the action of lactate dehydrogenase (LDH; EC 1.1.1.27), which catalyzes the reduction of pyruvate, the conjugate base of pyruvic acid.[6]
Lactate then diffuses out of the cell and into the bloodstream, through which it is transported to the liver, its primary site of utilization, and to the renal cortex, particularly the proximal tubules, another site where gluconeogenesis occurs.[6]
In both the liver and renal cortex, lactate is oxidized back to pyruvate in a reaction also catalyzed by lactate dehydrogenase. Pyruvate is subsequently converted into glucose via the gluconeogenic pathway.[7]
Finally, glucose enters the bloodstream and is delivered to red blood cells and muscle fibers, thereby completing the cycle.[8]
Red blood cells
Mature red blood cells, lacking a nucleus, ribosomes, and mitochondria, are smaller than most other cells. Their small size allows them to pass through narrow capillaries, but the absence of mitochondria makes them completely dependent on anaerobic glycolysis for ATP production. As a result, these cells continuously produce lactic acid.[8]
The availability of NAD+ is essential not only for glycolysis to proceed but also to sustain its rate. The oxidized form of this coenzyme is required for the oxidation of glyceraldehyde 3-phosphate to 1,3-bisphosphoglycerate, a reaction catalyzed by glyceraldehyde 3-phosphate dehydrogenase (EC 1.2.1.12).[3]
The accumulation of NADH is prevented by the reduction of pyruvate to lactate, in a reaction catalyzed by lactate dehydrogenase, in which NADH acts as the reducing agent, being oxidized back to NAD+.[9][10]
Muscle fibers
Fast-twitch muscle fibers contain relatively few mitochondria and, under hypoxic conditions, such as during intense exercise, produce significant amounts of lactic acid.[3] In these conditions:
the rate of pyruvate production via glycolysis exceeds the rate of its oxidation through the citric acid cycle, so that less than 10% of the pyruvate enters the cycle;[7]
the rate of oxygen uptake by the cells is insufficient to support the aerobic oxidation of all the NADH produced.[11]
As a result, anaerobic glycolysis leads to the production of 2 ATP per molecule of glucose (or 3 ATP if the glucose is derived from muscle glycogen), a much lower yield compared to the 29–30 ATP generated through complete oxidation of glucose.[10]
However, the rate of ATP production via anaerobic glycolysis is higher than that of complete aerobic oxidation.[8]
Finally, as in red blood cells, the reaction catalyzed by lactate dehydrogenase, which regenerates NAD+, enables glycolysis to continue, though it results in lactate production.[3]
Lactate
Lactic acid is a metabolic end product that must be converted back into pyruvate in order to be reutilized.[7]
The plasma membrane of most cells is freely permeable to both pyruvate and lactate, allowing them to enter the bloodstream.[12]
In skeletal muscle, for example, the amount of lactate that leaves the cell is greater than that of pyruvate. This is due to the high NADH/NAD+ ratio in the cytosol and the catalytic properties of the lactate dehydrogenase isozyme expressed in muscle fibers.[13]
Once in the bloodstream, lactate reaches, among other tissues, the liver and the renal cortex, where it is oxidized back to pyruvate by tissue-specific LDH isozymes.[14]
In the hepatocyte, this oxidation is favored by the low NADH/NAD+ ratio in the cytosol. The resulting pyruvate then enters gluconeogenesis, the next step in the Cori cycle, and is converted into glucose.[7]
Glucose subsequently enters the bloodstream and is delivered to muscle fibers and red blood cells, thus completing the cycle. Naturally, the monosaccharide also reaches all other tissues and cells that require it.[15]
Energy cost of the Cori cycle
The Cori cycle results in a net consumption of 4 ATP per cycle.
The gluconeogenic phase of the cycle consumes 2 GTP and 4 ATP per molecule of glucose synthesized, totaling 6 high-energy phosphate bonds. >The ATP- (or GTP-) consuming reactions are catalyzed by the following enzymes:
glyceraldehyde 3-phosphate dehydrogenase (EC 1.2.1.12): 1 ATP.
Since two molecules of lactate are needed to synthesize one molecule of glucose, the net energy cost is:
2 x 3 = 6 high energy bonds per molecule of glucose.
In contrast, the glycolytic phase of the cycle yields only 2 ATP per molecule of glucose.
Therefore, more energy is required to convert lactate back into glucose than is gained from the initial anaerobic glycolysis in extrahepatic tissues. This explains why the Cori cycle cannot be sustained indefinitely.[15]
Is the Cori cycle a futile cycle?
The continuous breakdown and resynthesis of glucose, characteristic of the Cori cycle, might initially appear to be a waste of energy. In reality, however, this cycle enables many extrahepatic cells to function effectively, at the energetic expense of the liver and renal cortex.
Therefore, it is more accurate to describe the Cori cycle as a substrate cycle, rather than a futile cycle.[8]
Below are some illustrative examples.
The Cori cycle contributes to the disposal of lactic acid produced continuously by red blood cells.[3]
During intense exercise, anaerobic glycolysis is a major source of ATP for muscle fibers. This process, however, could lead to the accumulation of lactate and a subsequent decrease in intracellular pH.
This accumulation is largely prevented by the Cori cycle, in which gluconeogenic tissues (mainly liver and renal cortex) bear the energy cost of removing excess muscle lactate.[16]
Furthermore, the oxygen debt incurred after intense exercise is largely due to the increased oxygen demand of hepatocytes, where the oxidation of fatty acids, their primary fuel, supplies the ATP needed for gluconeogenesis.[15][17]
In conditions such as trauma, sepsis, burns, or major surgery, intense cell proliferation occurs in the wound (a hypoxic environment) and in the bone marrow. This leads to increased lactic acid production, a greater flux through the Cori cycle, and thus increased ATP consumption by the liver.[18] As previously mentioned, this demand is met by enhanced fatty acid oxidation.
A similar situation seems to occur in cancer patients experiencing progressive weight loss.[19]
The Cori cycle also plays an important role during overnight fasting and starvation, helping maintain blood glucose levels.[5][17]
Similarities with the glucose-alanine cycle
These two metabolic pathways, which operate across different organs via the bloodstream, contribute to maintaining a continuous supply of glucose to peripheral tissues.
In both cycles, entry into gluconeogenesis involves the conversion of alanine or lactate into pyruvate.
Finally, in both cycles, the glucose produced is delivered to peripheral tissues, where it is metabolized via glycolysis, regenerating pyruvate.[15]
Differences from the glucose-alanine cycle
The main difference between the two cycles lies in the three-carbon intermediate that is recycled:
in the Cori cycle, carbon is returned to the liver as pyruvate.
in the glucose-alanine cycle, it is returned as alanine.[20]
Additionally, the metabolic fate of NADH also differs:
in the Cori cycle, NADH acts as a reducing agent in the reaction catalyzed by lactate dehydrogenase;
in the glucose-alanine cycle, the electrons from NADH are used to synthesize ATP in the mitochondria.[15]
As a result, another key difference is that the glucose-alanine cycle requires oxygen, whereas the Cori cycle can function under anaerobic conditions.[3]
Key differences between the Cori cycle and the glucose-alanine cycle.
^ Katz J., Tayek J.A. Gluconeogenesis and the Cori cycle in 12-, 20-, and 40-h-fasted humans. Am J Physiol 1998;275(3):E537-42. doi:10.1152/ajpendo.1998.275.3.E537
^ abcdef Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ ab Rui L. Energy metabolism in the liver. Compr Physiol 2014;4(1):177-97. doi:10.1002/cphy.c130024
^ ab Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012.
^ abcd Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002.
^ abcd Rosenthal M.D., Glew R.H. Medical biochemistry – Human metabolism in health and disease. John Wiley J. & Sons, Inc., Publication, 2009.
^ Spinelli S., Marino A., Morabito R., Remigante A. Interplay between metabolic pathways and increased oxidative stress in human red blood cells. Cells 2024;13(23):2026. doi:10.3390/cells13232026
^ Rich P.R. The molecular machinery of Keilin’s respiratory chain. Biochem Soc Trans 2003;31(Pt 6):1095-105. doi:10.1042/bst0311095
^ Halestrap A.P. The monocarboxylate transporter family — Structure and functional characterization. IUBMB Life 2012;64(1):1-9. doi:10.1002/iub.573
^ Gleeson T.T. Post-exercise lactate metabolism: a comparative review of sites, pathways, and regulation. Annu Rev Physiol 1996;58:565-81. doi:10.1146/annurev.ph.58.030196.003025
^ Gladden L.B. Lactate metabolism: a new paradigm for the third millennium. J Physiol 2004;558(Pt 1):5-30. doi:10.1113/jphysiol.2003.058701
^ abcde Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
^ van Hall G. Lactate kinetics in human tissues at rest and during exercise. Acta Physiol (Oxf) 2010;199(4):499-508. doi:10.1111/j.1748-1716.2010.02122.x
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ Liu S., Yang T., Jiang Q., Zhang L., Shi X., Liu X., Li X. Lactate and lactylation in sepsis: a comprehensive review. J Inflamm Res 2024;17:4405-4417. doi:10.2147/JIR.S459185
^ Li X., Yang Y., Zhang B., Lin X., Fu X., An Y., Zou Y., Wang J.X., Wang Z., Yu T. Lactate metabolism in human health and disease. Signal Transduct Target Ther 2022;7(1):305. doi:10.1038/s41392-022-01151-3
^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
Bile salts have similarities and differences with cholesterol molecule.
Like the steroid, they have a nucleus composed of four fused rings: three cyclohexane rings, labeled A, B and C, and a cyclopentane ring, labeled D. This structure is the perhydrocyclopentanophenanthrene, more commonly known as steroid nucleus.
Bile Acids and Their Conjugates
In higher vertebrates, they have 24 carbon atoms, as the side chain is three carbons shorter than the original. In lower vertebrates, bile acids have 25, 26, or 27 carbon atoms. The side chain ends with a carboxyl group, ionized at pH 7, that can be linked to the amino acid glycine or taurine.
In addition to the hydroxyl group at position 3, they have hydroxyl groups at positions 7 and/or 12.
All this makes them much more polar than cholesterol.
Since A and B rings are fused in cis configuration, the planar structure of the steroid nucleus is curved, and it is possible to identify:
a concave side, which is hydrophilic because the hydroxyl groups and the carboxyl group of the side chain, with or without the linked amino acid, are oriented towards it;
a convex side, which is hydrophobic because the methyl groups present at position 18 and 19 are orientated towards it.
Cholic Acid Structure
Therefore, having both polar and nonpolar groups, they are amphiphilic molecules and excellent surfactants. However, their chemical structure makes them different from many other surfactants, often composed of a polar head region and a nonpolar tail.
Primary, conjugated and secondary bile salts
Primary bile acids are those synthesized directly from cholesterol in the hepatocytes. In humans, the most important are cholic acid and chenodeoxycholic acid, which make up 80 percent of all bile acids.
Before being secreted into the biliary tree, they are almost completely conjugated, up to 98 percent, with the glycine or taurine, to form glycoconjugates and tauroconjugates, respectively. In particular, approximately 75 percent of cholic acid and chenodeoxycholic acid are conjugated with glycine, to form glycocholic acid and glycochenodeoxycholic acid, the remaining 25 percent with taurine, to form taurocholic acid and taurochenodeoxycholic.
Synthesis of Conjugated Bile Acids
Conjugated bile acids are molecules with more hydrophilic groups than unconjugated bile acids, therefore they are much more effective emulsifiers. In fact, conjugation decreases the pKa of bile acids, from about 6, a value typical of non-conjugated molecules, to about 4 for glycocholic acid, and about 2 for taurocholic acid. This makes that conjugated bile acids are ionized in a broader range of pH to form the corresponding salts.
The hydrophilicity of the common acid and bile salts decreases in the following order: glycine-conjugated < taurine-conjugated < lithocholic acid < deoxycholic acid < chenodeoxycholic acid < cholic acid < ursodeoxycholic acid.
Finally, conjugation also decreases the cytotoxicity of primary bile acids.
Secondary bile acids are formed from primary bile acids which have not been reabsorbed from the small intestine. Once they reach the colon, they can undergo several modifications by bacteria of the gut microbiota, which is part of the larger human microbiota, to form secondary bile acids, which, along with short-chain fatty acids, are two major type of bacterial metabolites produced in the gut. They make up the remaining 20 percent of the body’s bile acid pool.
Another way of categorizing bile salts is based on their conjugation with glycine and taurine and their degree of hydroxylation. On this basis, three categories are identified.
Trihydroxy conjugates, such as taurocholic acid and glycocholic acid.
Dihydroxy conjugates, such as glycodeoxycholic acid, glycochenodeoxycholic acid, taurochenodeoxycholic acid, and taurodeoxycholic acid. They account for about 60 percent of bile salts present in the bile.
Unconjugated forms, such as cholic acid, deoxycholic acid, chenodeoxycholic acid, and lithocholic acid.
Function of bile acids
All their physiological functions are performed in the conjugated form.
They are the major route for the elimination of cholesterol from human body.
Indeed, humans do not have the enzymes to break open the cyclohexane rings or the cyclopentane ring of the steroid nucleus, nor to oxidize cholesterol to CO2 and water.
The other mechanism to eliminate the steroid from the body is as cholesterol per se in the bile.
Bile salts are strong surfactants. And in particular, di- and trihydroxy conjugates are the best surfactants among bile acids, much more effective than unconjugated counterparts, since they have more polar groups.
Once in contact with apolar lipids in the lumen of the small intestine, the convex apolar surface interacts with the apolar lipids, such as triglycerides, cholesterol esters, and ester of fat-soluble vitamins, whereas the concave polar surface interacts with the surrounding aqueous medium. This increases the dispersion of apolar lipids in the aqueous medium, as it allows the formation of tiny lipid droplets, increasing the surface area for lipase activity, mainly pancreatic lipase, (bile salts also play a direct role in the activation of this enzyme), andintestinal esterase activity. Subsequently, they facilitate lipid absorption, as well as absorption of fat soluble vitamins by the intestinal mucosa, thanks to the formation of mixed micelles.
Bile acids perform a similar function in the gallbladder where, forming mixed micelles with phospholipids, they prevent the precipitation of cholesterol.
Note: as a consequence of the arrangement of polar and nonpolar groups, bile acids form micelles in aqueous solution, usually made up of less than 10 monomers, as long as their concentration is above the so-called critical micellar concentration or CMC.
At the intestinal level, they modulate the secretion of pancreatic enzymes and cholecystokinin.
In the small and large intestine, they have a potent antimicrobial activity, mainly deoxycholic acid, in particular against Gram-positive bacteria. This activity may be due to oxidative DNA damage, and/or to the damage of the cell membrane. Therefore, they play an important role in the prevention of bacterial overgrowth, but also in the regulation of gut microbiota composition.
In the last few years, it becomes apparent their regulatory role in the control of energy metabolism, and in particular for the hepatic glucose handling.
Enterohepatic circulation
After fat intake, enteroendocrine cells of the duodenum secrete cholecystokinin into the blood stream. Hormone binding to receptors on smooth muscle cells of the gallbladder promotes their contraction; the hormone also causes the relaxation of the sphincter of Oddi. All this results in the secretion of the bile, and therefore of bile acids into the duodenum.
Under physiological conditions, human bile salt pool is constant, and equal to about 3-5 g. This is made possible by two processes:
their intestinal reabsorption;
their de novo synthesis.
Up to 95 percent of the secreted bile salts is reabsorbed from the gut, not together with the products of lipid digestion, but through a process called enterohepatic circulation.
It is an extremely efficient recycling system, which seems to occur at least two times for each meal, and includes the liver, the biliary tree, the small intestine, the colon, and the portal circulation through which reabsorbed molecules return to the liver. Such recirculation is necessary since liver’s capacity to synthesize bile acids is limited and insufficient to satisfy intestinal needs if the bile salts were excreted in the feces in high amounts.
Most of the bile salts are reabsorbed into the distal ileum, the lower part of the small intestine, by a sodium-dependent transporter within the brush border of the enterocytes, called sodium-dependent bile acid transporter or ASBT, which carries out the cotransport of a molecule of bile acid and two sodium ions.
Within the enterocyte, it is thought that bile acids are transported across the cytosol to the basolateral membrane by the ileal bile acid-binding protein or IBABP. They cross the basolateral membrane by the organic solute transporter alpha-beta or OSTalpha/OSTbeta, pass into the portal circulation, and, bound to albumin, reach the liver.
It should be noted that a small percentage of bile acids reach the liver through the hepatic artery.
A hepatic level, their extraction is very efficient, with a first-pass extraction fraction ranging from 50 to 90 percent, a percentage that depends on bile acid structure. The uptake of conjugated bile acids is mainly mediated by a Na+-dependent active transport system, that is, the sodium-dependent taurocholate cotransporting polypeptide or NTCP. However, a sodium-independent uptake can also occur, carried out by proteins of the family of organic anion transporting polypeptides or OATP, mainly OATP1B1 and OATP1B3.
The rate limiting step in the enterohepatic circulation is their canalicular secretion, largely mediated by the bile salt export pump or BSEP, in an ATP-dependent process. This pump carries monoanionic bile salts, which are the most abundant. Bile acids conjugated with glucuronic acid or sulfate, which are dianionic, are transported by different carriers, such as MRP2 and BCRP.
Note: Serum levels of bile acids vary on the basis of the rate of their reabsorption, and therefore they are higher during meals, when the enterohepatic circulation is more active.
Intestinal metabolism
Bile acids which escape ileal absorption pass into the colon where they partly undergo modifications by intestinal microbiota and are converted to secondary bile acids.
The main reactions are listed below.
Deconjugation
On the side chain, hydrolysis of the C24 N-acyl amide bond can occur, with release of unconjugated bile acids and glycine or taurine. This reaction is catalyzed by bacterial hydrolases present both in the small intestine and in the colon.
7alpha-Dehydroxylation
Quantitatively, it is the most important reaction, carried out by colonic bacterial dehydratases that remove the hydroxyl group at position 7 to form 7-deoxy bile acids. In particular, deoxycholic acid is formed from cholic acid, and lithocholic acid, a toxic secondary bile acid, from chenodeoxycholic acid.
It should be noted that 7alpha-dehydroxylation, unlike oxidation and epimerization, can only occur on unconjugated bile acids, and therefore, deconjugation is an essential prerequisite.
Oxidation and epimerization
They are reactions involving the hydroxyl groups at positions 3, 7 and 12, catalyzed by bacterial hydroxysteroid dehydrogenases. For example, ursodeoxycholic acid derives from the epimerization of chenodeoxycholic acid.
Intestinal Metabolism of Bile Acids
Some of the secondary bile acids are then reabsorbed from the colon and return to the liver. In the hepatocytes, they are reconjugated, if necessary, and resecreted. Those that are not reabsorbed, are excreted in the feces.
While oxidations and deconjugations are carried out by a broad spectrum of anaerobic bacteria, 7alpha-dehydroxylations is carried out by a limited number of colonic anaerobes.
7alpha-Dehydroxylations and deconjugations increase the pKa of the bile acids, and therefore their hydrophobicity, allowing a certain degree of passive absorption across the colonic wall.
The increase of hydrophobicity is also associated with an increased toxicity of these molecules. And a high concentration of secondary bile acids in the bile, blood, and feces has been associated to the pathogenesis of colon cancer.
Soluble fibers and reabsorption
The reabsorption of bile salts can be reduced by chelating action of soluble fibers, such as those found in fresh fruits, legumes, oats and oat bran, which bind them, decreasing their uptake. In turn, this increases bile acid de novo synthesis, up-regulating the expression of the 7alpha-hydroxylase and sterol 12alpha-hydroxylase, and thereby reduces hepatocyte cholesterol concentration.
The depletion of hepatic cholesterol increases the expression of the LDL receptor, and thus reduces plasma concentration of LDL cholesterol. On the other hand, it also stimulates the synthesis of HMG-CoA reductase, the key enzyme in cholesterol biosynthesis.
Note: Some anti-cholesterol drugs act by binding bile acids in the intestine, thereby preventing their reabsorption.
Synthesis
Quantitatively, bile acids are the major product of cholesterol metabolism.
As previously said, enterohepatic circulation and their de novo synthesis maintain a constant bile acid pool size. In particular, de novo synthesis allows the replacement of bile salts excreted in the faces, about 5-10 percent of the body pool, namely ~ 0.5 g/day.
Below, the synthesis of cholic acid and chenodeoxycholic acid, and their conjugation with the amino acids taurine and glycine, is described.
There are two main pathways for bile acid synthesis: the classical pathway and the alternative pathway. In addition, some other minor pathways will also be described.
De Novo Synthesis of Primary Bile Acids and Their Conjugates
The classical or neutral pathway
In humans, up to 90 percent of bile salts are produced via the classical pathway, also referred to as “neutral” pathway since intermediates are neutral molecules.
It is a metabolic pathway present only in the liver, that consists of reactions catalyzed by enzymes localized in the cytosol, endoplasmic reticulum, peroxisomes, and mitochondria, and whose end products are the conjugates of cholic acid and chenodeoxycholic acid.
The first reaction is the hydroxylation at position 7 of cholesterol, to form 7alpha-hydroxycholesterol. The reaction is catalyzed by cholesterol 7alpha-hydroxylase or CYP7A1 (E.C. 1.14.14.23). It is an enzyme localized in the endoplasmic reticulum, and catalyzes the rate-limiting step of the pathway.
7alpha-Hydroxycholesterol undergoes oxidation of the 3beta-hydroxyl group and the shift of the double bond from the 5,6 position to the 4,5 position, to form 7alpha-hydroxy-4-cholesten-3-one. The reaction is catalyzed by 3beta-hydroxy-Δ5-C27-steroid oxidoreductase or HSD3B7 (E.C. 1.1.1.181), an enzyme localized in the endoplasmic reticulum.
7alpha-Hydroxy-4-cholesten-3-one can follow two routes:
to enter the pathway that leads to the synthesis of cholic acid, through the reaction catalyzed by 7alpha-hydroxy-4-cholesten-3-one12alpha-monooxygenase or sterol 12alpha-hydroxylase or CYP8B1 (E.C. 1.14.18.8), an enzyme localized in the endoplasmic reticulum;
to enter the pathway that leads the synthesis of chenodeoxycholic acid, through the reaction catalyzed by 3-oxo-Δ4-steroid 5beta-reductase or AKR1D1 (E.C. 1.3.1.3), a cytosolic enzyme.
It should be underlined that the activity of sterol 12alpha-hydroxylase determines the ratio of cholic acid to chenodeoxycholic acid, and, ultimately, the detergent capacity of bile acid pool. And in fact, the regulation of sterol 12alpha-hydroxylase gene transcription is one of the main regulatory step of the classical pathway.
Therefore, if 7alpha-hydroxy-4-cholesten-3-one proceeds via the reaction catalyzed by sterol 12alpha-hydroxylase, the following reactions will occur.
7alpha-Hydroxy-4-cholesten-3-one is hydroxylated at position 12 by sterol 12alpha-hydroxylase, to form 7alpha,12alpha-dihydroxy-4-cholesten-3-one.
7alpha,12alpha-Dihydroxy-4-cholesten-3-one undergoes reduction of the double bond at 4,5 position, in the reaction catalyzed by 3-oxo-Δ4-steroid5beta-reductase, to form 5beta-cholestan-7alpha,12alpha-diol-3-one.
5beta-Cholestan-7alpha,12alpha-diol-3-one undergoes reduction of the hydroxyl group at position 4, in the reaction catalyzed by 3 alpha-hydroxysteroid dehydrogenase or AKR1C4 (EC 1.1.1.213), a cytosolic enzyme, to form 5beta-cholestan-3 alpha,7alpha,12alpha-triol.
5beta-Cholestan-3alpha,7alpha,12alpha-triol undergoes oxidation of the side chain via three reactions catalyzed by sterol 27-hydroxylase or CYP27A1 (EC 1.14.15.15). It is a mitochondrial enzyme also present in extrahepatic tissues and macrophages, which introduces a hydroxyl group at position 27. The hydroxyl group is oxidized to aldehyde, and then to carboxylic acid, to form 3 alpha,7 alpha,12 alpha-trihydroxy-5 beta-cholestanoic acid.
3alpha,7alpha,12alpha-Trihydroxy-5beta-cholestanoic acid is activated to its coenzyme A ester, 3alpha,7alpha,12alpha-trihydroxy-5beta-cholestanoyl-CoA, in the reaction catalyzed by either very long chain acyl-CoA synthetase or VLCS (EC 6.2.1.-), or bile acid CoA synthetase or BACS (EC 6.2.1.7), both localized in the endoplasmic reticulum.
3alpha,7alpha,12alpha-Trihydroxy-5beta-cholestanoyl-CoA is transported to peroxisomes where it undergoes five successive reactions, each catalyzed by a different enzyme. In the last two reactions, the side chain is shortened to four carbon atoms, and finally cholylCoA is formed.
In the last step, the conjugation, via amide bond, of the carboxylic acid group of the side chain with the amino acid glycine or taurine occurs. The reaction is catalyzed by bile acid-CoA:amino acid N-acyltransferase or the BAAT (EC 2.3.1.65), which is predominantly localized in peroxisomes.
The reaction products are thus the conjugated bile acids: glycocholic acid and taurocholic acid.
If 7alpha-hydroxy-4-cholesten-3-one does not proceed via the reaction catalyzed by sterol 12alpha-hydroxylase, it enters the pathway that leads to the synthesis of chenodeoxycholic acid conjugates, through the reactions described below.
7alpha-Hydroxy-4-cholesten-3-one is converted to 7alpha-hydroxy-5beta-cholestan-3-one in the reaction catalyzed by 3-oxo-Δ4-steroid 5 beta-reductase.
7alpha-Hydroxy-5beta-cholestan-3-one is converted to 5beta-cholestan-3alpha,7alpha-diol in the reaction catalyzed by 3alpha-hydroxysteroid dehydrogenase.
Then, the conjugated bile acids glycochenodeoxycholic acid and taurochenodeoxycholic acid are formed by modifications similar to those seen for the conjugation of cholic acid, and catalyzed mostly by the same enzymes.
Note: Unconjugated bile acids formed in the intestine must reach the liver to be reconjugated.
The alternative or acidic pathway
It is prevalent in the fetus and neonate, whereas in adults it leads to the synthesis of less than 10 percent of the bile salts.
This pathway differs from the classical pathway in that:
the intermediate products are acidic molecules, from which the alternative name “acidic pathway”;
the oxidation of the side chain is followed by modifications of the steroid nucleus, and not vice versa;
the final products are conjugates of chenodeoxycholic acid.
The first step involves the conversion of cholesterol into 27-hydroxycholesterol in the reaction catalyzed by sterol 27-hydroxylase.
27-Hydroxycholesterol can follow two routes.
Route A
27-hydroxycholesterol is converted to 3beta-hydroxy-5-cholestenoic acid in a reaction catalyzed by sterol 27-hydroxylase.
3beta-Hydroxy-5-cholestenoic acid is hydroxylated at position 7 in the reaction catalyzed by oxysterol 7alpha-hydroxylase or CYP7B1 (EC 1.14.13.100), an enzyme localized in the endoplasmic reticulum, to form 3beta-7alpha-dihydroxy-5-colestenoic acid.
3beta-7alpha-Dihydroxy-5-cholestenoic acid is converted to 3-oxo-7alpha-hydroxy-4-cholestenoic acid, in the reaction catalyzed by 3beta-hydroxy-Δ5-C27-steroid oxidoreductase.
3-Oxo-7alpha-hydroxy-4-cholestenoic acid, as a result of side chain modifications, forms chenodeoxycholic acid, and then its conjugates.
Route B
27-Hydroxycholesterol is converted to 7alpha,27-dihydroxycholesterol in the reaction catalyzed by oxysterol 7alpha-hydroxylase and cholesterol 7alpha-hydroxylase.
7alpha,27-Dihydroxycholesterol is converted to 7alpha,26-dihydroxy-4-cholesten-3-one in the reaction catalyzed by 3beta-hydroxy-Δ5-C27-steroid oxidoreductase;
7alpha, 26-Dihydroxy-4-cholesten-3-one can be transformed directly to conjugates of chenodeoxycholic acid, or can be converted to 3-oxo-7alpha-hydroxy-4-colestenoic acid, and then undergo side chain modifications and other reactions that lead to the synthesis of the conjugates of chenodeoxycholic acid.
Minor pathways
There are also minor pathways that contribute to bile salt synthesis, although to a lesser extent than classical and alternative pathways.
For example:
A cholesterol 25-hydroxylase (EC 1.14.99.38) is expressed in the liver.
A cholesterol 24-hydroxylase or CYP46A1 (EC 1.14.14.25) is expressed in the brain, and therefore, although the organ cannot export cholesterol, it exports oxysterols.
A nonspecific 7alpha-hydroxylase has also been discovered. It is expressed in all tissues and appears to be involved in the generation of oxysterols, which may be transported to hepatocytes to be converted to chenodeoxycholic acid.
Additionally, sterol 27-hydroxylase is expressed in various tissues, and therefore its reaction products must be transported to the liver to be converted to bile salts.
Bile salts: regulation of synthesis
Regulation of bile acid synthesis occurs via a negative feedback mechanism, particularly on the expression of cholesterol 7alpha-hydroxylase and sterol 12alpha-hydroxylase.
When an excess of bile acids, both free and conjugated, occurs, these molecules bind to the nuclear receptor farnesoid X receptor or FRX, activating it: the most efficacious bile acid is chenodeoxycholic acid, while others, such as ursodeoxycholic acid, do not activate it.
FRX induces the expression of the transcriptional repressor small heterodimer partner or SHP, which in turn interacts with other transcription factors, such as liver receptor homolog-1 or LRH-1, and hepatocyte nuclear factor-4alpha or HNF-4alpha. These transcription factors bind to a sequence in the promoter region of 7alpha-hydroxylase and 12alpha-hydroxylase genes, region called bile acid response elements or BAREs, inhibiting their transcription.
One of the reasons why bile salt synthesis is tightly regulated is because many of their metabolites are toxic.
References
Chiang J.Y.L. Bile acids: regulation of synthesis. J Lipid Res 2009;50(10):1955-1966. doi:10.1194/jlr.R900010-JLR200
Gropper S.S., Smith J.L. Advanced nutrition and human metabolism. 6th Edition. Cengage Learning, 2012
Moghimipour E., Ameri A., and Handali S. Absorption-enhancing effects of bile salts. Molecules 2015;20(8); 14451-14473. doi:10.3390/molecules200814451
Monte M.J., Marin J.J.G., Antelo A., Vazquez-Tato J. Bile acids: Chemistry, physiology, and pathophysiology. World J Gastroenterol 2009;15(7):804-816. doi:10.3748/wjg.15.804
Rosenthal M.D., Glew R.H. Medical biochemistry – Human metabolism in health and disease. John Wiley J. & Sons, Inc., Publication, 2009
Sundaram S.S., Bove K.E., Lovell M.A. and Sokol R.J. Mechanisms of Disease: inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol 2008;5(8):456-468. doi:10.1038/ncpgasthep1179
Alpha-amylase (α-amylase; EC 3.2.1.1) is an enzyme that catalyzes the hydrolysis of α-(1→4) glycosidic bonds present in the linear chains of amylose and amylopectin, the two major components of starch granules.[1][2]
It is widely distributed in animals, plants, fungi (including Ascomycota and Basidiomycota), bacteria (notably species of Bacillus), and Archaea. This broad distribution highlights its central role in carbohydrate metabolism.[3][4]
In humans, alpha-amylase is encoded by two genes and is synthesized mainly in the parotid glands and the exocrine pancreas.[5] In plants, the alpha-amylase gene family consists of six members.[6]
In animals, it initiates starch digestion, producing the disaccharide maltose, the oligosaccharides maltotriose and alpha-limit dextrins, and small amounts of glucose.[7]
Since starch is the primary dietary source of carbohydrates, and thus of glucose and energy, alpha-amylase activity may influence postprandial blood glucose levels.[8]
α-Amylase also has applications in various industrial sectors as well as in clinical diagnostics.[9][10]
In plants, particularly in grasses such as wheat, barley, and rice, it plays a crucial role in seed germination and grain maturation.[6][11]
The first evidence of amylase activity dates back to 1811, when the Russian chemist Konstantin Sigizmundovich Kirchhoff, working with wheat, discovered an enzymatic activity capable of breaking down starch.[12]
In 1831, the German chemist Erhard Friedrich Leuchs observed starch degradation when mixed with human saliva, and named the responsible agent “ptyalin”.
In 1833, French chemists Anselme Payen and Jean-François Persoz, working in a sugar factory, isolated from barley an enzyme able to degrade starch, which they named diastase.[7]
The term alpha-amylase was introduced by Richard Kuhn in 1925, after he discovered that the hydrolysis products were in the alpha configuration.[13]
In 1894, alpha-amylase became the first enzyme to be produced industrially. Initially extracted from a fungal source, it was used to treat digestive disorders. Starting in 1917, it was also extracted from Bacillus subtilis and Bacillus mesentericus. Today, it is mainly produced using genetically modified organisms.[3][14]
Amylases in general, including α-amylase, beta-amylase (EC 3.2.1.2), and gamma-amylase (EC 3.2.1.3), are widely applied in industries such as textiles, detergents, animal feed, food processing, brewing (beer and whiskey), cosmetics, pharmaceuticals, and clinical diagnostics.[9]
Alpha-amylases can be classified according to the degree of substrate hydrolysis: as liquefying enzymes, when they cleave 30–40% of starch glycosidic bonds, or saccharifying enzymes, when they cleave 50–60% of the bonds.[3]
Human alpha-amylase genes
In many mammals, including humans, alpha-amylase is encoded by two distinct but closely related genes located on the short arm of chromosome 1:
AMY1, which encodes the enzyme present in saliva, the mammary gland, and certain tumors, such as lung cancer;[15]
AMY2, which encodes the pancreatic enzyme. AMY2 occurs in two forms, AMY2A and AMY2B, whose copy number varies among populations, although two copies of each gene appear to be common. Copy numbers of AMY2 may also be associated with those of AMY1.[5][16]
Activity of alpha-amylase
Alpha-amylase catalyzes the first step of starch digestion, the hydrolysis of α-(1→4) glycosidic bonds in the linear chains of amylose and amylopectin, by acting at random sites along α-glucans. As an endoglycosidase, it requires calcium ions, has an optimal pH around 7, and produces disaccharides and oligosaccharides in the alpha configuration. Specifically, it generates maltose and maltotriose from amylose, and maltose, alpha-limit dextrins, and small amounts of glucose from amylopectin.[17][18]
Alpha-amylase
Like other enzymes involved in starch metabolism, such as pullulanase (EC 3.2.1.41), isoamylase (EC 3.2.1.68), starch-branching enzymes (EC 2.4.1.18), and starch-debranching enzymes, alpha-amylase belongs to glycoside hydrolase family 13 (GH13), a group of enzymes that act on substrates containing alpha-glycosidic bonds.[4]
Two enzymes involved in trehalose metabolism also fall within this family: trehalose synthase (EC 5.4.99.16), which catalyzes the interconversion of maltose and trehalose, and trehalose 6-phosphate hydrolase (EC 3.2.1.93), which hydrolyzes trehalose 6-phosphate to yield one molecule of glucose and one molecule of glucose 6-phosphate.[7][19]
Salivary alpha-amylase
Salivary alpha-amylase, also known as ptyalin, is synthesized mainly by the parotid glands and, to a lesser extent, by the submandibular glands.[20]
It occurs in both glycosylated and non-glycosylated forms and contributes to the initial, limited phase of starch digestion in the oral cavity.[21]
This enzyme is among the most abundant salivary proteins, accounting for 40–50% of total salivary protein content. Along with lingual lipase (EC 3.1.1.3), which hydrolyzes triglycerides and cholesterol esters during lipid digestion, salivary alpha-amylase is one of the principal enzymes in saliva.[22]
Pancreatic alpha-amylase
Pancreatic alpha-amylase is synthesized by the acinar cells of the pancreas.[23][24]
It is one of the major components of pancreatic juice and, upon entering the duodenum, hydrolyzes amylose and amylopectin, completing the first stage of amylolysis. The resulting products are subsequently cleaved by alpha-glucosidases located in the brush border of enterocytes, namely sucrase-isomaltase (EC 3.2.1.48) and maltase-glucoamylase (EC 3.2.1.20), which can also hydrolyze the α-(1→2) glycosidic bond of sucrose.[25]
Mammalian pancreatic α-amylase can bind to N-linked glycans on brush-border membrane glycoproteins.[26] This interaction enhances cooperation with sucrase-isomaltase, leading to a substantial increase in glucose production, up to 240%. However, high concentrations of alpha-amylase in the intestinal lumen have been observed to inhibit glucose uptake via SGLT1, an intestinal monosaccharide transporter involved in the absorption of monosaccharides. This inhibition has been hypothesized to represent an innate regulatory mechanism that prevents sudden spikes in glucose uptake and contributes to the regulation of postprandial blood glucose levels.[27]
Blood sugar control
Postprandial glycemic control is considered an effective strategy to prevent the development of type II diabetes. Among non-pharmacological approaches are interventions that reduce the ability of alpha-amylase to hydrolyze starch, either through food processing that induces starch retrogradation or through molecules naturally present in foods that, via different mechanisms, inhibit its activity.[8]
Food processing
Starch-rich foods are often subjected to hydrothermal treatments, such as boiling, which cause starch granule gelatinization: the granules absorb water, swell, and lose most of their crystalline structures, which are replaced by amorphous forms that are more easily hydrolyzed by alpha-amylase.[1] If gelatinized starch is subsequently cooled, recrystallization may occur, leading to the formation of retrograded starch, a type of starch resistant to α-amylase activity. This starch reaches the colon largely intact, where it is fermented by gut microbiota into short-chain fatty acids, similarly to soluble fiber.[28]
Role of polyphenols
Starch digestion by alpha-amylase is influenced by various food components, including non-starch cell wall polysaccharides, proteins, lipids, and, among phytochemicals, different classes of polyphenols.[1] These compounds can limit the action of alpha-amylase through at least four mechanisms.
Proteins and non-starch polysaccharides can limit the loss of the ordered structure of native starch during the hydrothermal processing of foods.
Lipids, polyphenols, proteins and non-starch polysaccharides can form ordered complexes with starch.
Proteins, non-starch polysaccharides, and polyphenols can create physical barriers that reduce the accessibility of hydrolytic enzymes to alpha-glucan chains.
Polyphenols and non-starch polysaccharides can directly reduce the activity of alpha-amylases and alpha-glucosidases.[8]
Among polyphenols, flavonoids, particularly proanthocyanidins (condensed tannins), appear to play a key role. For this inhibitory effect to be significant, the consumption of polyphenol-rich foods, such as black tea or green tea, should coincide with a carbohydrate-rich meal. Gluten however, may interfere with the interaction between polyphenols and starch granules, thereby reducing their inhibitory capacity.[8][29]
Amylase activity in clinical biochemistry
The measurement of serum α-amylase activity is a commonly used diagnostic laboratory test.
Increased serum amylase activity may result from:
acute pancreatitis or an exacerbation of chronic pancreatitis;
inflammation of the parotid glands;
obstruction of the secretory ducts of the parotid glands or the pancreatic duct.
Current assays do not distinguish between salivary and pancreatic isoforms. However, when elevated amylase activity is accompanied by increased lipase activity, the underlying pathology is most likely of pancreatic origin.[10][30][31]
Wheat alpha-amylase
In plants, particularly in wheat, alpha-amylase plays a key role in germination and in malting, a controlled germination process that converts grain into malt, used for the production of beer and whiskey.[32]
In wheat grains, the enzyme is mainly synthesized during the early stages of germination, which are heterotrophic and rely heavily on stored resources, especially starch in the endosperm. Its synthesis occurs in the aleurone layer and initiates the mobilization of starch reserves.[11]
Alpha-amylase is also implicated in one of the major quality defects of wheat: pre-harvest sprouting.[33]
References
^ abc Butterworth P.J., Bajka B.H., Edwards C.H., Warren F.J., Ellis P.R. Enzyme kinetic approach for mechanistic insight and predictions of in vivo starch digestibility and the glycaemic index of foods. Trends Food Sci Technol 2022;120:254-264. doi:10.1016/j.tifs.2021.11.015
^ Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
^ abc Tiwari S.P., Srivastava R., Singh C.S., Shukla K., Singh R.K., Singh P., Singh R., Singh N.L., and Sharma R. Amylases: an overview with special reference to alpha amylase. Journal of Global Biosciences 2015;4(SI1):1886-1901.
^ ab Janeček Š., Svensson B., MacGregor E.A. α-Amylase: an enzyme specificity found in various families of glycoside hydrolases. Cell Mol Life Sci 2014;71(7):1149-70. doi:10.1007/s00018-013-1388-z
^ ab Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W, Lee C. Detection of large-scale variation in the human genome. Nat Genet 2004;36:949–951. doi:10.1038/ng1416
^ ab Ju L., Pan Z., Zhang H., Li Q., Liang J., Deng G., Yu M., Long H. New insights into the origin and evolution of α-amylase genes in green plants. Sci Rep 2019;9(1):4929. doi:10.1038/s41598-019-41420-w
^ abc Butterworth P.J., Warren F.J. and Ellis P.R. Human α-amylase and starch digestion: an interesting marriage. Starch/Stärke 2011;63:395-405. doi:10.1002/star.201000150
^ abcd Gong L., Feng D., Wang T., Ren Y., Liu Y., Wang J. Inhibitors of α-amylase and α-glucosidase: potential linkage for whole cereal foods on prevention of hyperglycemia. Food Sci Nutr 2020;8(12):6320-6337. doi:10.1002/fsn3.1987
^ ab de Souza P.M., de Oliveira Magalhães P. Application of microbial α-amylase in industry – A review. Braz J Microbiol 2010;41(4):850-61. doi:10.1590/S1517-83822010000400004
^ ab Erta G., Gersone G., Jurka A., Tretjakovs P. Salivary α-amylase as a metabolic biomarker: analytical tools, challenges, and clinical perspectives. Int J Mol Sci 2025;26(15):7365. doi:10.3390/ijms26157365
^ Zhang Q., Pritchard J., Mieog J., Byrne K., Colgrave M.L., Wang J.R., Ral J.F. Over-expression of a wheat late maturity alpha-amylase type 1 impact on starch properties during grain development and germination. Front Plant Sci 2022;13:811728. doi:10.3389/fpls.2022.811728
^ Zakowski J.J., Bruns D.E. Biochemistry of human alpha amylase isoenzymes. Crit Rev Clin Lab Sci 1985;21(4):283-322. doi:10.3109/10408368509165786
^ Kuhn R. Mechanism of the action of amylases. Constitution of starch. Justus Liebigs Annalen der Chemie 1925;443:1-71.
^ Pandey A., Nigam P., Soccol C.R., Soccol V.T., Singh D., Mohan R. Advances in microbial amylases. Biotechnol Appl Biochem 2000;31(2):135-52.
^ Perry G.H., Dominy N.J., Claw K.G., et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet 2007;39(10):1256-60. doi:10.1038/ng2123
^ Carpenter D., Mitchell L.M., Armour J.A. Copy number variation of human AMY1 is a minor contributor to variation in salivary amylase expression and activity. Hum Genomics 2017;11(1):2. doi:10.1186/s40246-017-0097-3^
^ Benjamin S., Smitha R.B., Jisha V.N., Pradeep S., Sajith S., Sreedevi S., Priji P., Unni K.N., Sarath Josh M.K. A monograph on amylases from Bacillus spp. Adv biosci biotechnol 2013;4:No.2. doi:10.4236/abb.2013.42032
^ van der Maarel M.J., van der Veen B., Uitdehaag J.C., Leemhuis H., Dijkhuizen L. Properties and applications of starch-converting enzymes of the alpha-amylase family. J Biotechnol 2002;94(2):137-55. doi:10.1016/s0168-1656(01)00407-2
^ Agarwal N., Singh S.P. A novel trehalose synthase for the production of trehalose and trehalulose. Microbiol Spectr 2021;9(3):e0133321. doi:10.1128/Spectrum.01333-21
^ Schneyer L.H. Amylase content of separate salivary gland secretions of man. J Appl Physiol 1956;9(3):453-5. doi:10.1152/jappl.1956.9.3.453
^ Yang Z.M., Chen L.H., Zhang M., Lin J., Zhang J., Chen W.W., Yang X.R. Age differences of salivary alpha-amylase levels of basal and acute responses to citric acid stimulation between chinese children and adults. Front Physiol 2015;6:340. doi:10.3389/fphys.2015.00340
^Boehlke C., Zierau O., Hannig C. Salivary amylase – The enzyme of unspecialized euryphagous animals. Arch Oral Biol 2015;60(8):1162-76. doi:10.1016/j.archoralbio.2015.05.008
^ Keller P.J., Allan B.J. The protein composition of human pancreatic juice. J Biol Chem 1967;242(2):281-7. doi:10.1016/S0021-9258(19)81461-8
^ Basnet S., Ghimire M.P., Lamichhane T.R., Adhikari R., Adhikari A. Identification of potential human pancreatic α-amylase inhibitors from natural products by molecular docking, MM/GBSA calculations, MD simulations, and ADMET analysis. PLoS One 2023;18(3):e0275765. doi:10.1371/journal.pone.0275765
^ Nichols B.L., Avery S., Sen P., Swallow D.M., Hahn D., Sterchi E. The maltase-glucoamylase gene: common ancestry to sucrase-isomaltase with complementary starch digestion activities. Proc Natl Acad Sci U S A. 2003 Feb 4;100(3):1432-7. doi:10.1073/pnas.0237170100
^ Asanuma-Date K., Hirano Y., Le N., et al. Functional regulation of sugar assimilation by N-glycan-specific interaction of pancreatic α-amylase with glycoproteins of duodenal brush border membrane. J Biol Chem 2012;287(27):23104-18. doi:10.1074/jbc.m111.314658
^ Date K., Satoh A., Lida K., Ogawa H. Pancreatic α-amylase controls glucose assimilation by duodenal retrieval through N-glycan-specific binding, endocytosis, and degradation. J Biol Chem 2015;290(28):17439-50. doi:10.1074/jbc.M114.594937
^ Holtug K., Clausen M.R., Hove H., Christiansen J., Mortensen P.B. The colon in carbohydrate malabsorption: short-chain fatty acids, pH, and osmotic diarrhoea. Scand J Gastroenterol 1992;27(7):545-52. doi:10.3109/00365529209000118
^ Forester S.C., Gu Y., Lambert J.D. Inhibition of starch digestion by the green tea polyphenol, (-)-epigallocatechin-3-gallate. Mol Nutr Food Res 2012;56(11):1647-54. doi:10.1002/mnfr.201200206
^ Ismail O.Z., Bhayana V. Lipase or amylase for the diagnosis of acute pancreatitis? Clin Biochem 2017;50(18):1275-1280. doi:10.1016/j.clinbiochem.2017.07.003
^ Ekka N.M., Kujur A.D., Guria R., Mundu M., Mishra B., Sekhar S., Kumar A., Prakash J., Birua H. Serum lipase amylase ratio as an indicator to differentiate alcoholic from non-alcoholic acute pancreatitis: a systematic review and meta-analysis. Cureus 2023;15(2):e35618. doi:10.7759/cureus.35618
^ Fincher G.B. Molecular and cellular biology associated with endosperm mobilization in germinating cereal grains. Annu Rev Plant Physiol Plant Mol Biol 1989;40:305-46. doi:10.1146/annurev.pp.40.060189.001513
^ Gubler F., Millar A.A., Jacobsen J.V. Dormancy release, ABA and pre-harvest sprouting. Curr Opin Plant Biol 2005;8(2):183-7. doi:10.1016/j.pbi.2005.01.011
One of the main functions of the liver is to participate in the maintaining of blood glucose levels within well defined range (in the healthy state before meals 60-100 mg/dL or 3.33-5.56 mmol/L). To do it the liver releases glucose into the bloodstream in:
fasting state;
between meals;
during physical activity.
Blood glucose levels and hepatic glucose 6-phosphatase
In the liver, glycogen is the storage form of glucose which is released from the molecule not as such, but in the phosphorylated form, i.e., with charge, the glucose 1-phosphate. This process is called glycogenolysis.
The phosphorylated molecule can’t freely diffuse from the cell, but in the liver it is present the enzyme glucose-6-phosphatase that hydrolyzes glucose 6-phosphate, produced from glucose 1-phosphate in the reaction catalyzed by phosphoglucomutase, to glucose (an irreversible dephosphorylation).
Then, glucose can diffuse from the hepatocyte, via a transporter into the plasma membrane called GLUT2, into the bloodstream to be delivered to extra-hepatic cells, in primis neurons and red blood cells for which it is the main, and for red blood cells the only energy source. Neurons, with the exception of those in some brain areas that can use only glucose as energy source, can derive energy from another source, the ketone bodies, which becomes predominant during periods of prolonged fasting.
The liver obtains most of the energy required from the oxidation of fatty acids, a class of lipids, and not from glucose.
Glucose-6-phosphatase is present also in the kidney and gut but not in the muscle and brain. Therefore in these tissues glucose-6-phosphate can’t be released from the cell.
Glucose 6-phosphatase plays an important role also in gluconeogenesis.
Glucose 6-phosphatase is present into the membrane of endoplasmic reticulum and the hydrolysis of glucose-6-phosphate occurs into its lumen; therefore this reaction is separated from the process of glycolysis. The presence of a specific transporter, the glucose 6-phosphate translocase, is required to transport the phosphorylated molecule from citosol into the lumen of endoplasmic reticulum. Although a glucose transporter is present into the membrane of endoplasmic reticulum, most of the released glucose is not transported back into the cytosol of the cell but is secreted into the bloodstream. Finally, an ion transporter transports back into the cytosol the inorganic phosphate released into the endoplasmic reticulum.
References
Nelson D.L., M. M. Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
Roach P.J., Depaoli-Roach A.A., Hurley T.D and Tagliabracci V.C. Glycogen and its metabolism: some new developments and old themes. Biochem J 2012;441:763-787. doi:10.1042/BJ20111416
Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
When excess calories are consumed from carbohydrates or proteins, such surplus is used to synthesize fatty acids and then triacylglycerols, while it doesn’t occur if the excess come from fats.
De novo fatty acid synthesis in plants and animals
De novofatty acid synthesis is largely similar among plants and animals. It occurs in chloroplasts of photosynthetic cells of higher plants, and in cytosol of animal cells by the concerted action of two enzymes: acetyl-CoA carboxylase (EC 6.4.1.2) and fatty acid synthase (EC 2.3.1.85).
Fatty acid synthase catalyzes a repeating four-step sequence by which the fatty acyl chain is extended by two carbons, at the carboxyl end, every each passage through the cycle; this four-step process is the same in all organisms.
In animals, the primary site for lipid metabolism is liver, not the adipose tissue. However, adipose tissue is a major organ system in which fatty acid synthesis occurs, though in humans it is less active than in many other animal species.
It should be noted that in certain plants, such as palm and coconut, chain termination occurs earlier than palmitic acid release: up to 90 percent of the fatty acids produced and then present in the oils of these plants are between 8 (caprylic acid) and 14 (myristic acid) carbons long (palmitic acid: 16 carbon atoms).
Synthesis of long chain saturated and unsaturated fatty acids
Palmitic acid is the commonest saturated fatty acid in plant and animal lipids. Generally it is not present in very large proportions because it may be undergo into several metabolic pathways.
In fact:
it is the precursor of stearic acid;
it may be desaturated to palmitoleic acid, the precursor of all fatty acids of omega-7 or n-7 family, in a reaction catalyzed by Δ9-desaturase (EC 1.14.19.1), an ubiquitous enzyme in both plant and animal kingdoms and the most active lipid enzyme in mammalian tissues, the same enzyme that catalyzes the desaturation of stearic acid to oleic acid. Note: Δ9-desaturase inserts double bounds in the 9-10 position of the fatty acid carbon chain, position numbered from the carboxyl end of the molecule, and:
if the substrate is palmitic acid, the double bond is inserted between n-7 and n-8 position of the chain, so producing palmitoleic acid, the founder of omega-7 series;
if the substrate is stearic acid, the double bond is inserted between n-9 and n-10 position of the chain and oleic acid will be produced.
It may be esterified into complex lipids.
Of course, in plants and animals there are fatty acids longer and/or more unsaturated than these just seen thanks to modification systems that catalyze reactions of fatty acid synthesis that are organism- tissue- and cell- specific.
For example, stearic acid may be:
elongated to arachidic acid, behenic acid and lignoceric acid, all saturated fatty acids, in reactions catalyzed by elongases. Again, chain elongation occurs, both in mitochondria and in the smooth endoplasmic reticulum, by the addition of two carbon atom units at a time at the carboxylic end of the fatty acid through the action of fatty acid elongation systems (particularly long and very long saturated fatty acids, from 18 to 24 carbon atoms, are synthesized only on cytosolic face of the smooth endoplasmic reticulum);
desaturated, as seen, to oleic acid, an omega-9 or n-9 fatty acid, in a reaction catalyzed by Δ9-desaturase.
Several researchers have postulated that the reason for which stearic acid does not concur to hypercholesterolemia is its rapid conversion to oleic acid.
Oleic acid is the start point for the synthesis of many other unsaturated fatty acids by reactions of elongation and/or desaturation.
In fact:
gadoleic acid, erucic acid and nervonic acid, all monounsaturated fatty acids. Saturated fatty acids, and unsaturated fatty acids of the omega-9 series, usually oleic acid (but also palmitoleic acid and other omega-7 fatty acids) are the only fatty acids produced de novo in mammal systems.
Thanks to the consecutive action of the enzymes Δ12-desaturase (1.14.19.6) and Δ15-desaturase (EC 1.14.19.25), that insert a double bond respectively in the 12-13 and 15-16 position of the carbon chain of the fatty acid, oleic acid is converted first to linoleic acid, founder of all the omega-6 polyunsaturated fatty acids, and then to alpha-linolenic acid, founder of all the omega-3 polyunsaturated fatty acids (omega-3 and omega-6 PUFA that will be produced from these precursors through repeating reactions of elongation and desaturation).
In the case of essential fatty acid (EFA) deficiency, oleic acid may be desaturated and elongated to omega-9 polyunsaturated fatty acids, with accumulation especially of Mead acid.
Omega-3 and omega-6 PUFA synthesis
Animal tissues can desaturate fatty acids in the 9-10 position of the chain, thanks to the presence of Δ9 desaturase; as previously seen, if the substrate of the reaction is palmitic acid, the double bond will appear between n-7 and n-8 position, with stearic acid between n-9 and n-10 position, so leading to formation respectively of palmitoleic acid and oleic acid.
Animals lack Δ12- and Δ15-desaturases, enzymes able to desaturate carbon carbon bonds beyond the 9-10 position of the chain. For these reason, they can’t produce de novo omega-3 and omega-6 PUFA (which have double bonds also beyond the 9-10 position), that are so essential fatty acids.
Δ12- and Δ15-desaturases are present in plants; though many land plants lack Δ15-desaturase, also called omega-3 desaturase, planktons and aquatic plants in colder water possess it and produce abundant amounts of the omega-3 fatty acids.
References
Akoh C.C. and Min D.B. “Food lipids: chemistry, nutrition, and biotechnology”. CRC Press Taylor & Francis Group, 2008 3th ed. 2008
Bender D.A. “Benders’ dictionary of nutrition and food technology”. 2006, 8th Edition. Woodhead Publishing. Oxford
Burr G.O. and Burr M.M. A new deficiency disease produced by the rigid exclusion of fat from the diet. Nutr Rev 1973;31(8):148-149. doi:10.1111/j.1753-4887.1973.tb06008.x
Chow Ching K. “Fatty acids in foods and their health implication”. 3rd Edition. CRC Press Taylor & Francis Group, 2008
Rosenthal M.D., Glew R.H. Mediacal biochemistry. Human metabolism in health and disease. John Wiley & Sons, Inc. 2009
Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
Biochemistry and Metabolism
Utilizziamo cookie tecnici necessari per il funzionamento del sito. Vorremmo anche utilizzare, ma con il tuo consenso, cookie diversi da quelli tecnici, quali cookie analitici non equiparabili ai cookie tecnici, cookie di marketing e di terze parti, al fine di analisi, di miglioramento della tua esperienza di navigazione, presentare specifici annunci pubblicitari.
Puoi esprimere il consenso all’utilizzo di tali cookie cliccando sul tasto “Accetto tutti i cookie” oppure selezionando le tue preferenze cliccando sull’apposito tasto.
Cliccando su “Rifiuta” verranno installati solo i cookie tecnici o questi equiparabili.
Potrai revocare il tuo consenso in qualsiasi momento.
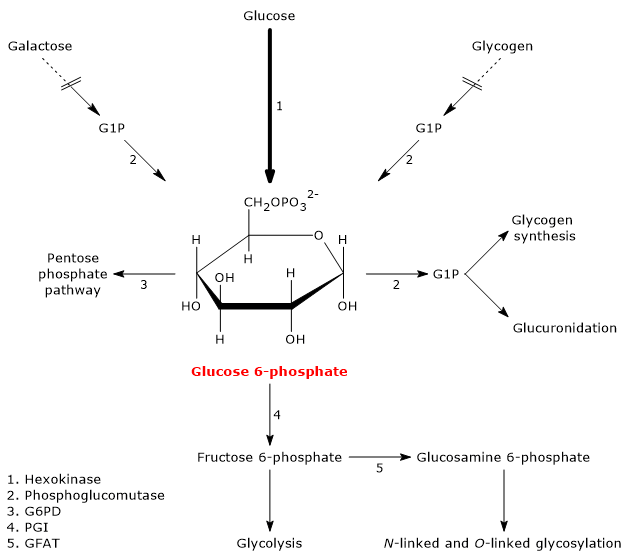
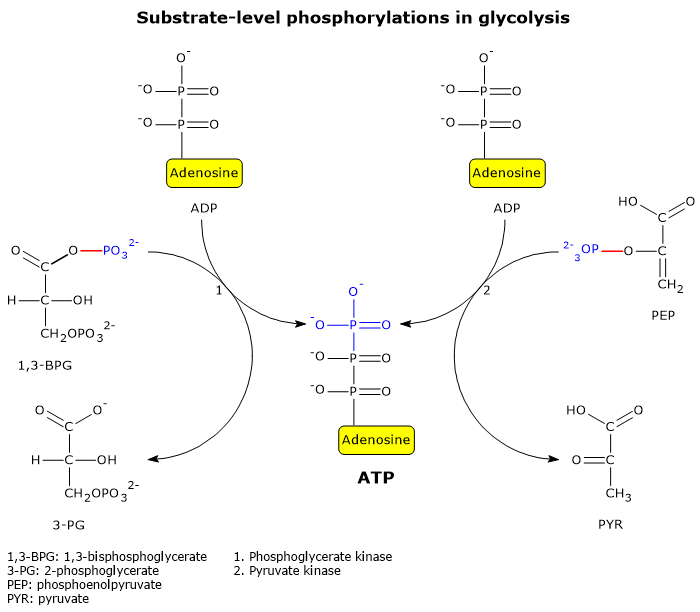 These reactions are catalyzed by phosphoglycerate kinase (EC 2.7.2.3) and pyruvate kinase (EC 2.7.1.40).
These reactions are catalyzed by phosphoglycerate kinase (EC 2.7.2.3) and pyruvate kinase (EC 2.7.1.40).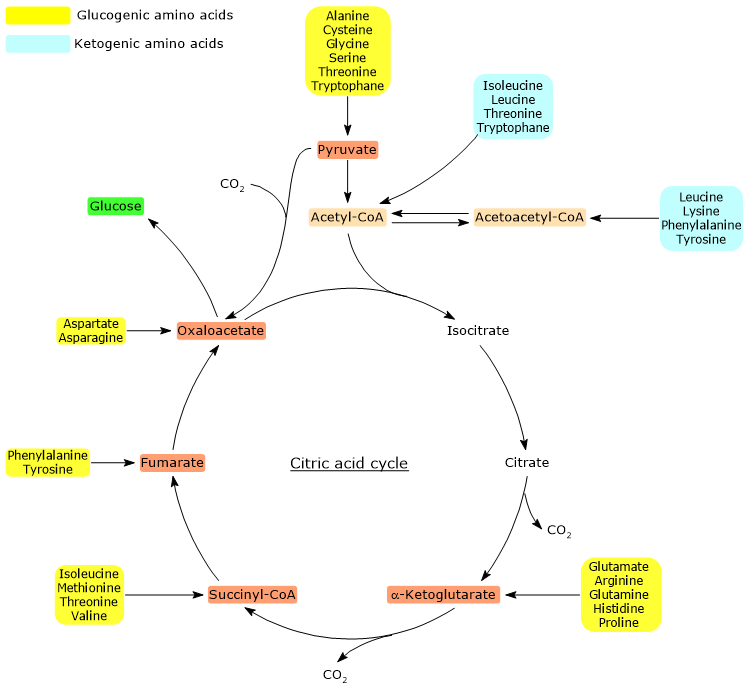
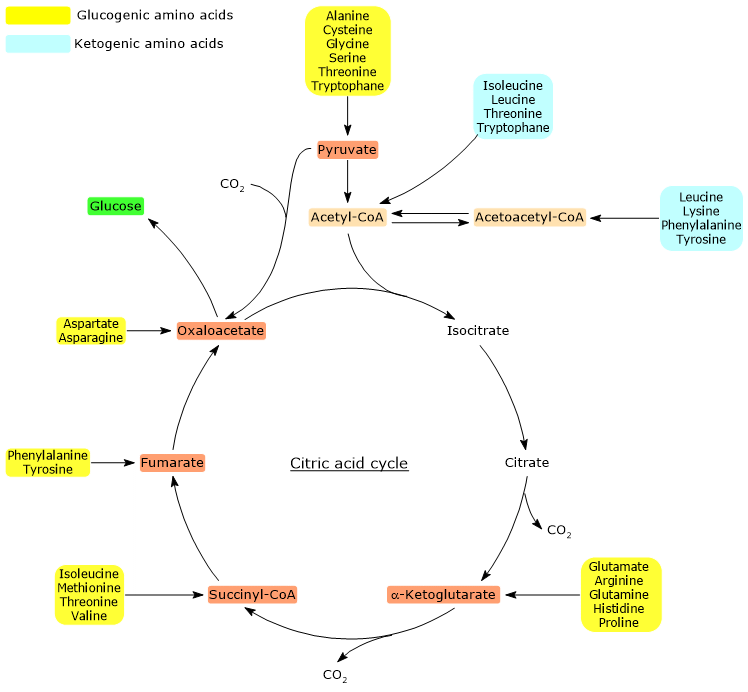
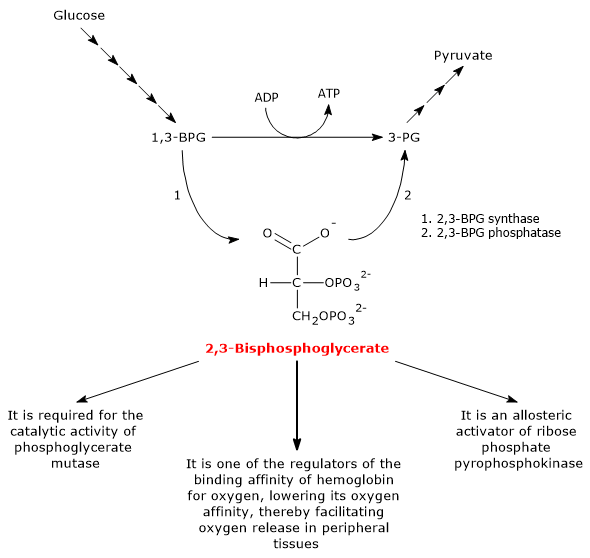
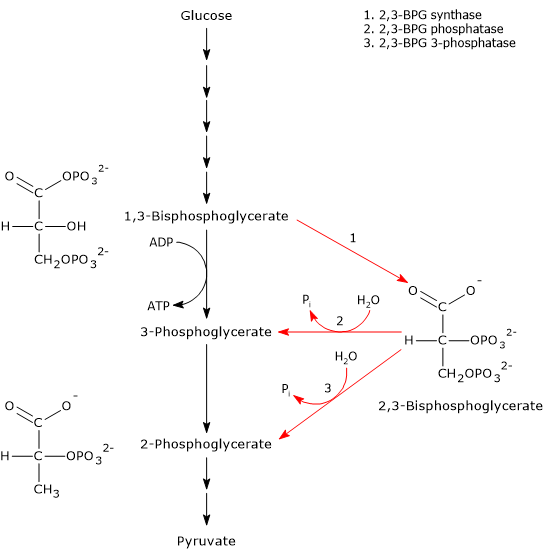
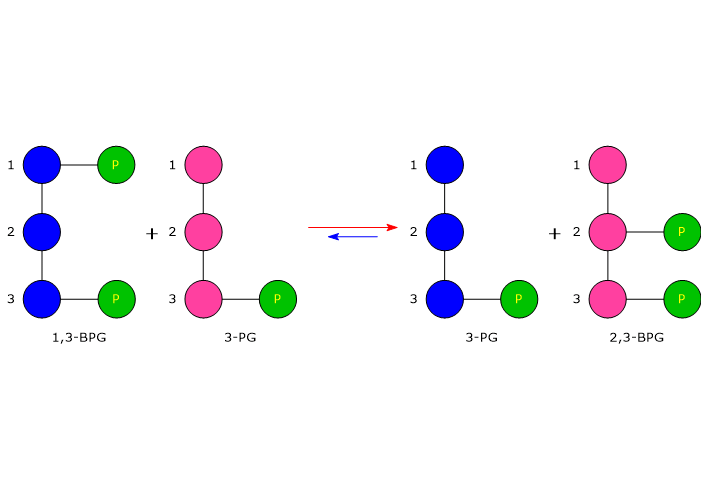 In the second step, the phosphatase activity of bisphosphoglycerate mutase catalyzes the synthesis of 3-phosphoglycerate, a glycolytic intermediate, following the hydrolysis of the phosphate group at position C-2 of 2,3-BPG. The 3-phosphoglycerate produced can then re-enter the glycolytic pathway at the reaction catalyzed by phosphoglycerate mutase, i.e., the eighth step of the pathway.
In the second step, the phosphatase activity of bisphosphoglycerate mutase catalyzes the synthesis of 3-phosphoglycerate, a glycolytic intermediate, following the hydrolysis of the phosphate group at position C-2 of 2,3-BPG. The 3-phosphoglycerate produced can then re-enter the glycolytic pathway at the reaction catalyzed by phosphoglycerate mutase, i.e., the eighth step of the pathway.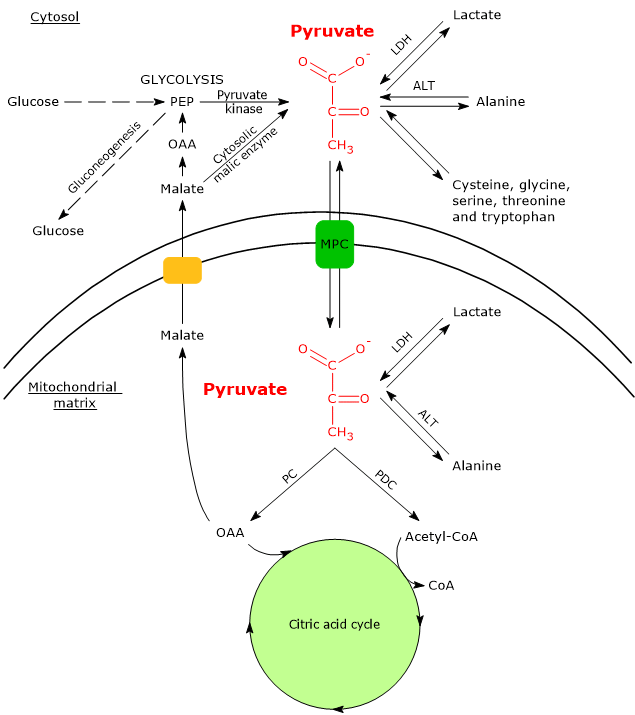
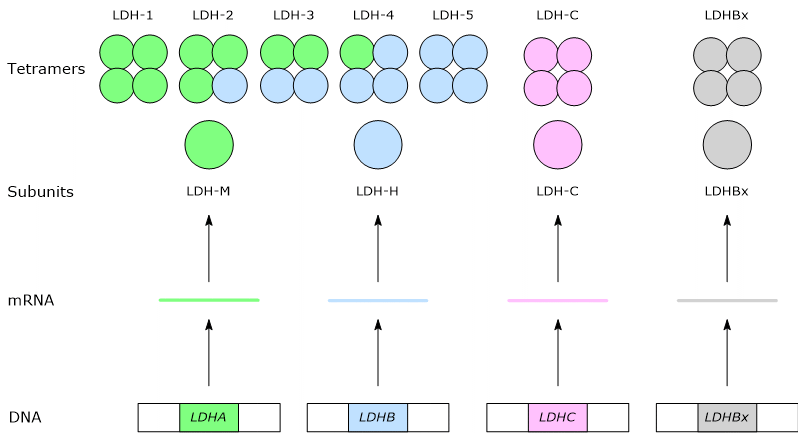
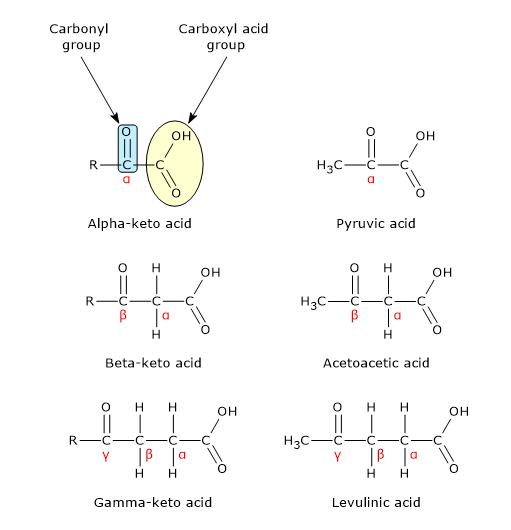
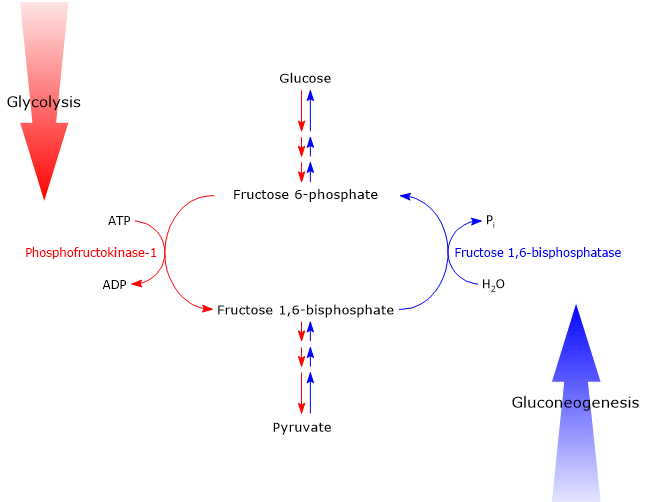
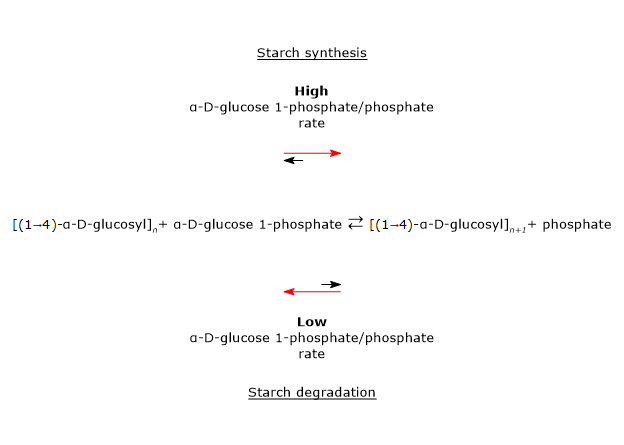 The enzyme belongs to the family of glucosyltransferases (EC 2.4), along with other enzymes such as
The enzyme belongs to the family of glucosyltransferases (EC 2.4), along with other enzymes such as 
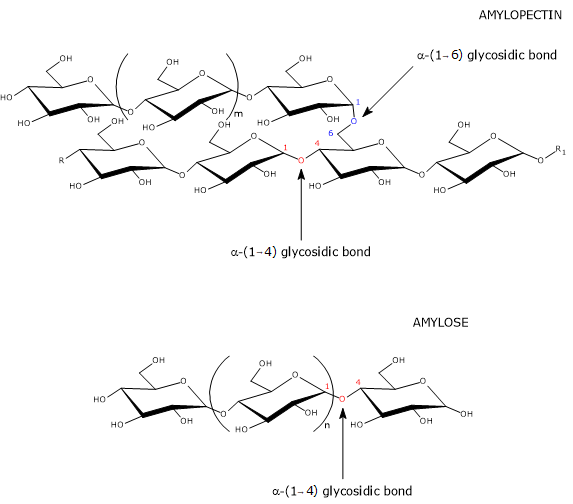
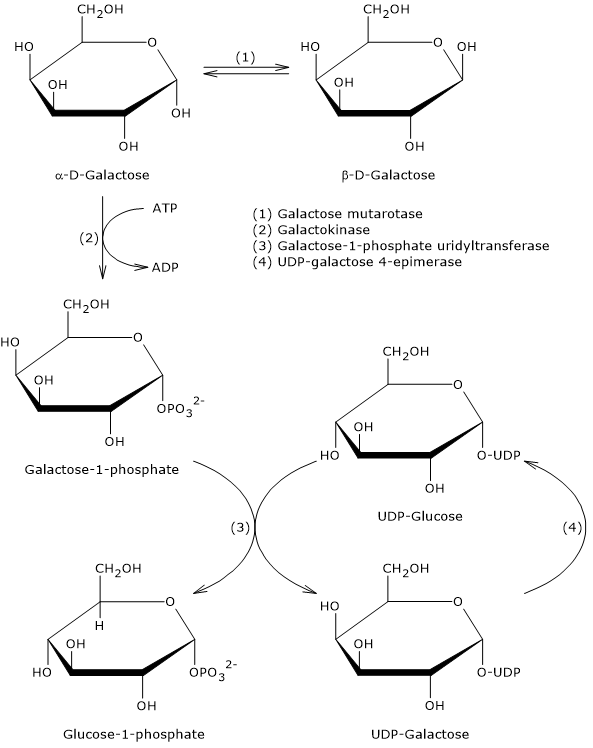
 For a solution containing n solutes, the equation that describes osmotic pressure is the sum of the contributions of each solute:
For a solution containing n solutes, the equation that describes osmotic pressure is the sum of the contributions of each solute: Metabolic channeling enhances catalytic efficiency, and therefore the reaction rate, in several ways, outlined below.
Metabolic channeling enhances catalytic efficiency, and therefore the reaction rate, in several ways, outlined below.
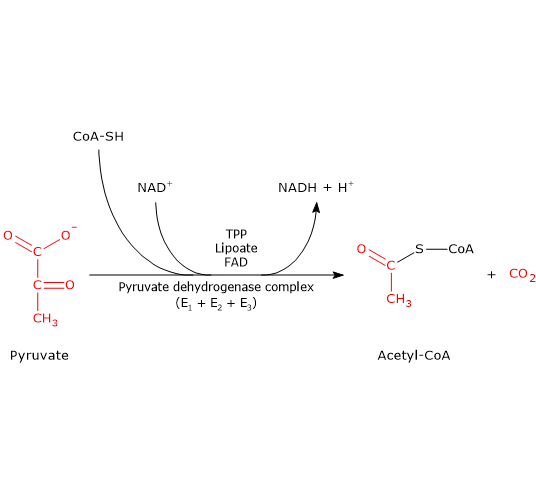
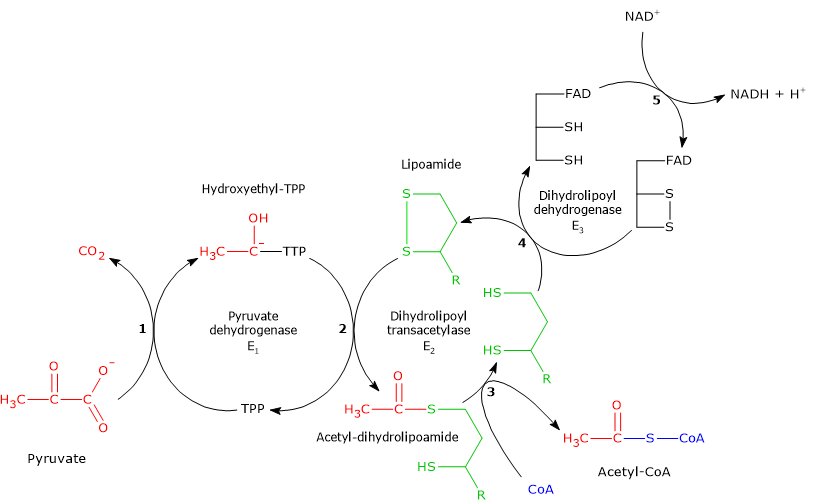
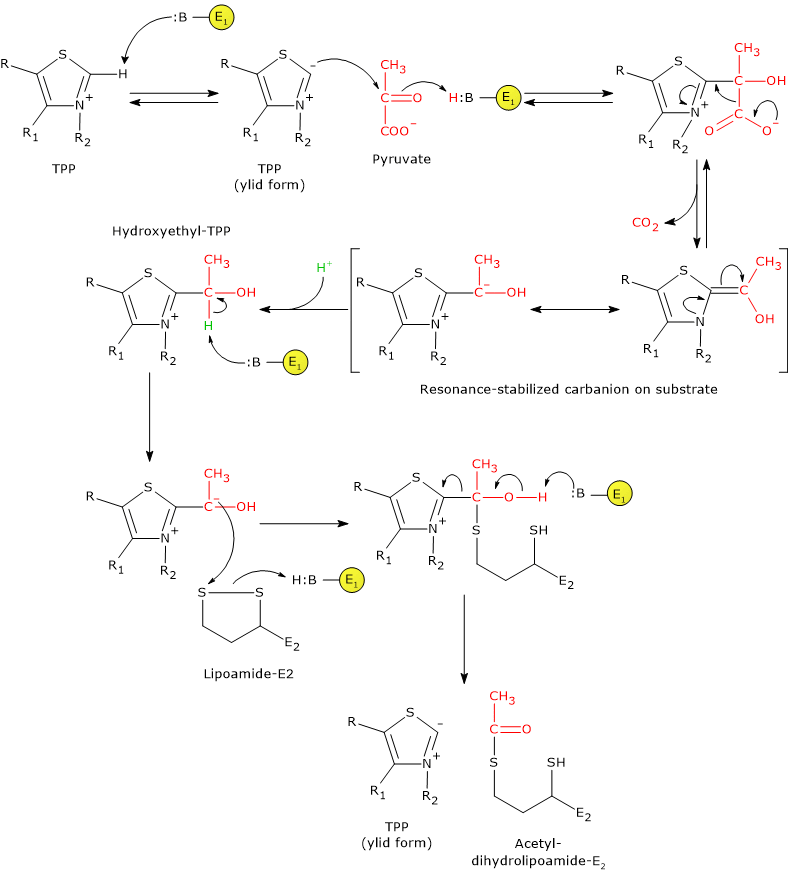
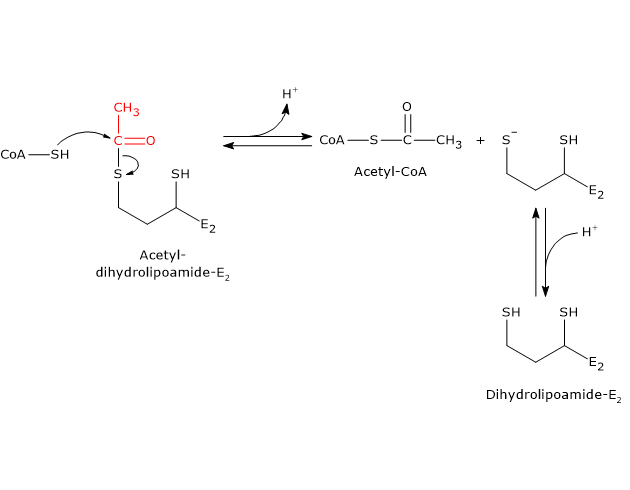
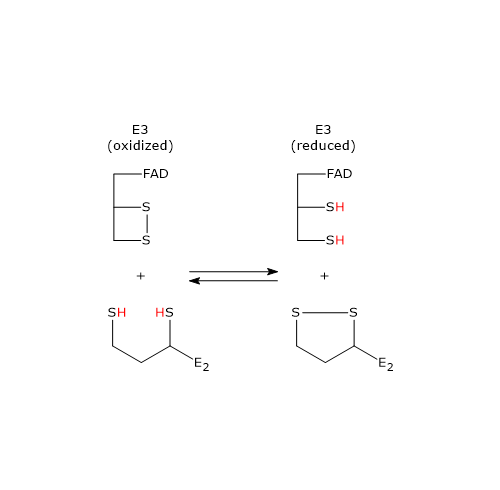
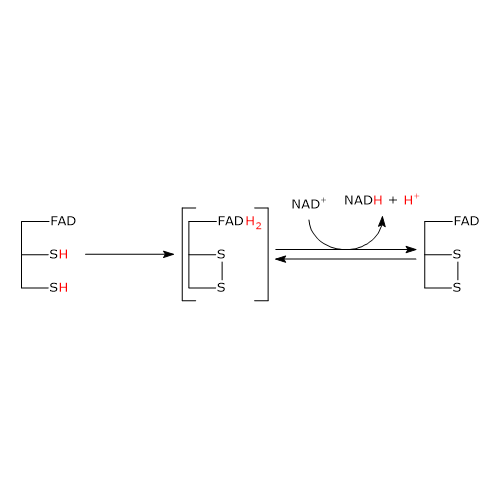
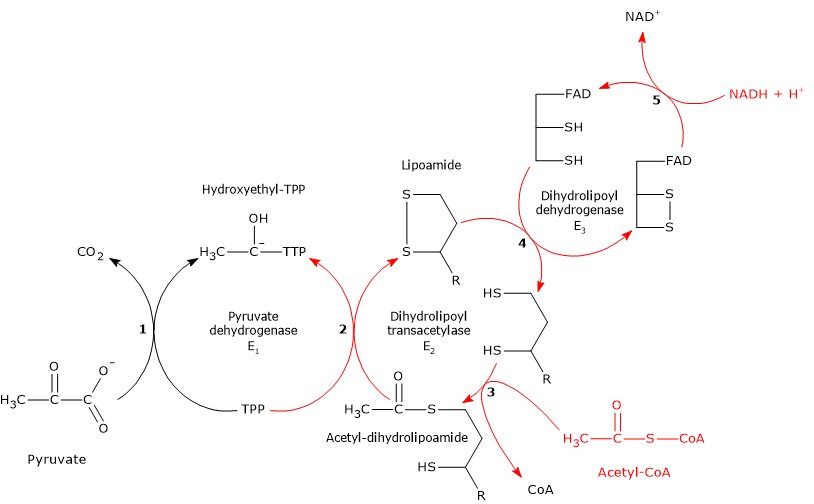
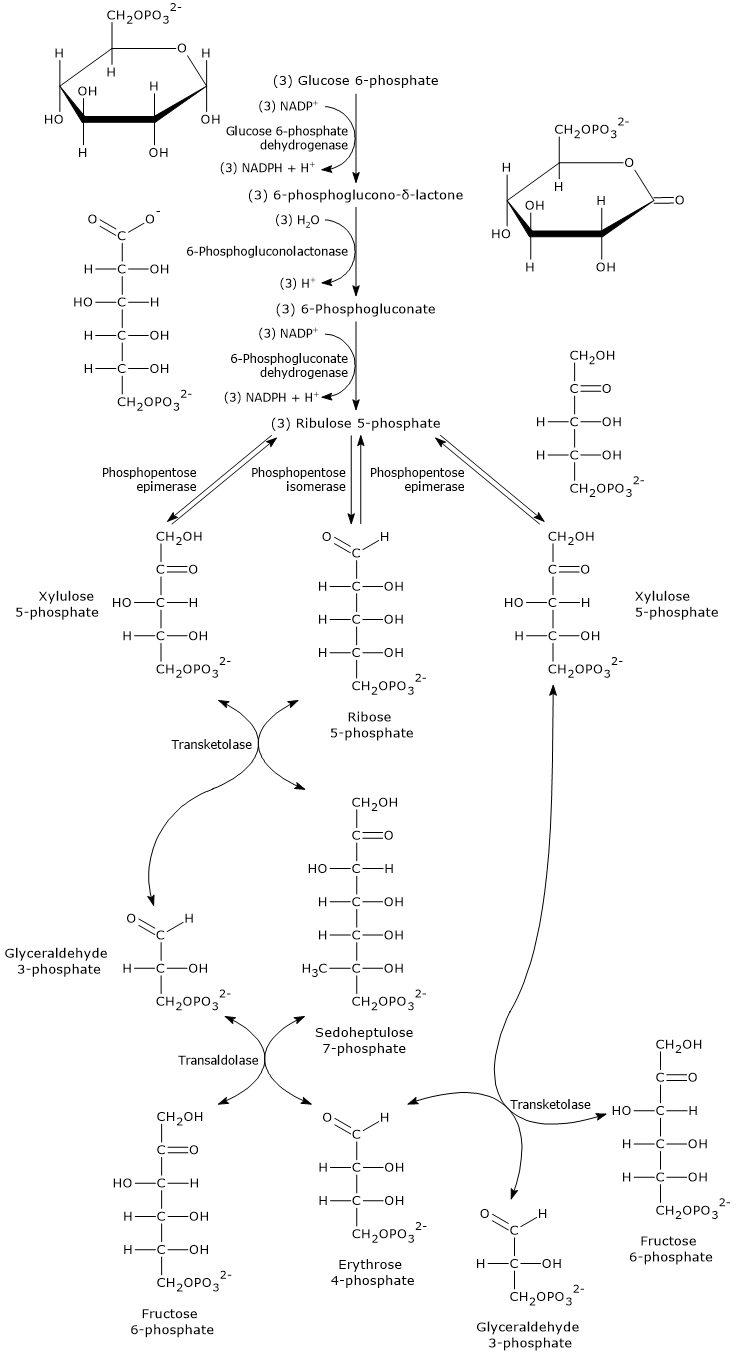
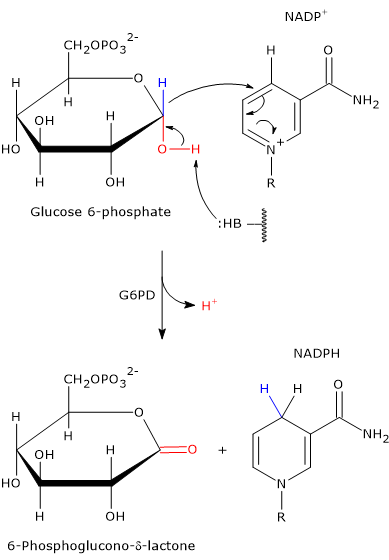
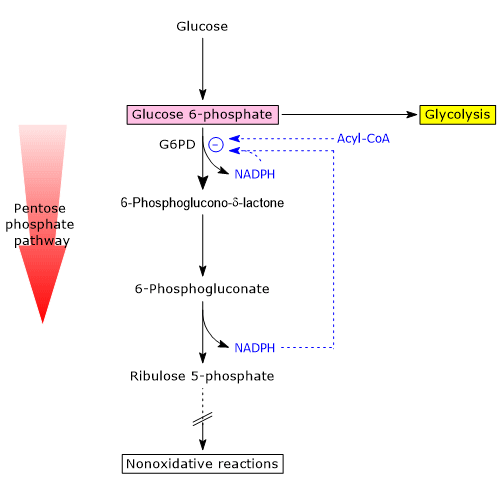
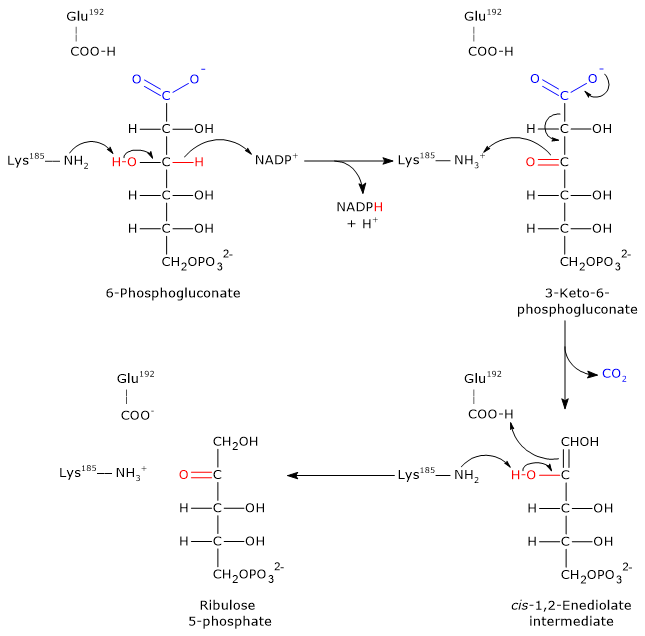
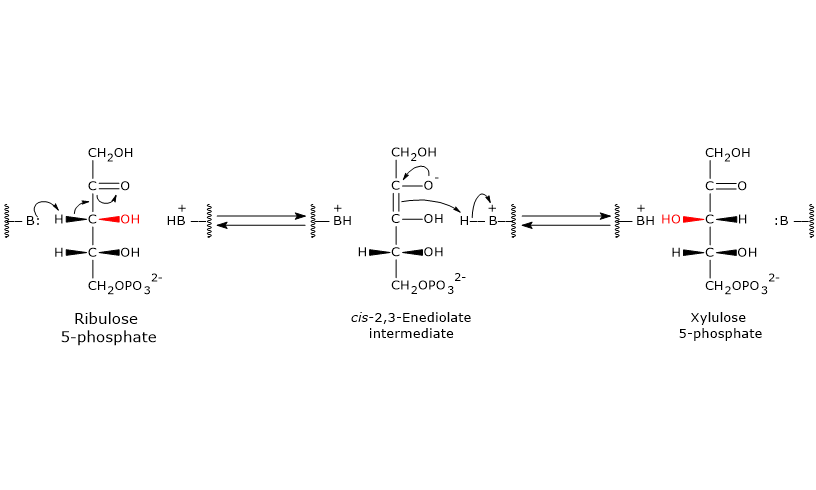
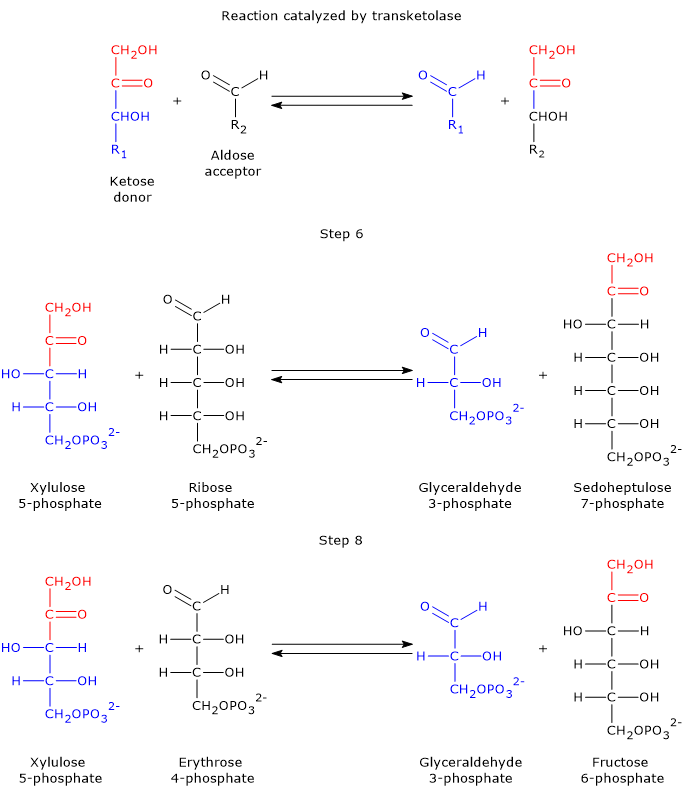
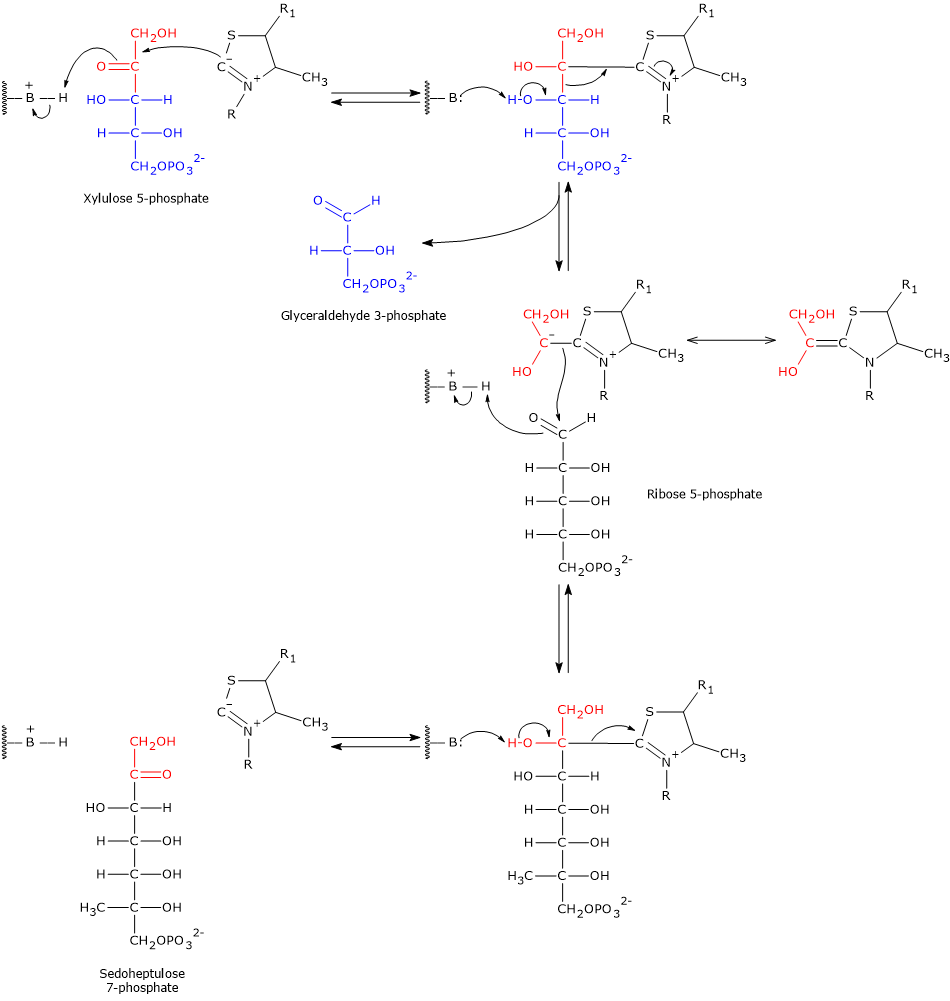
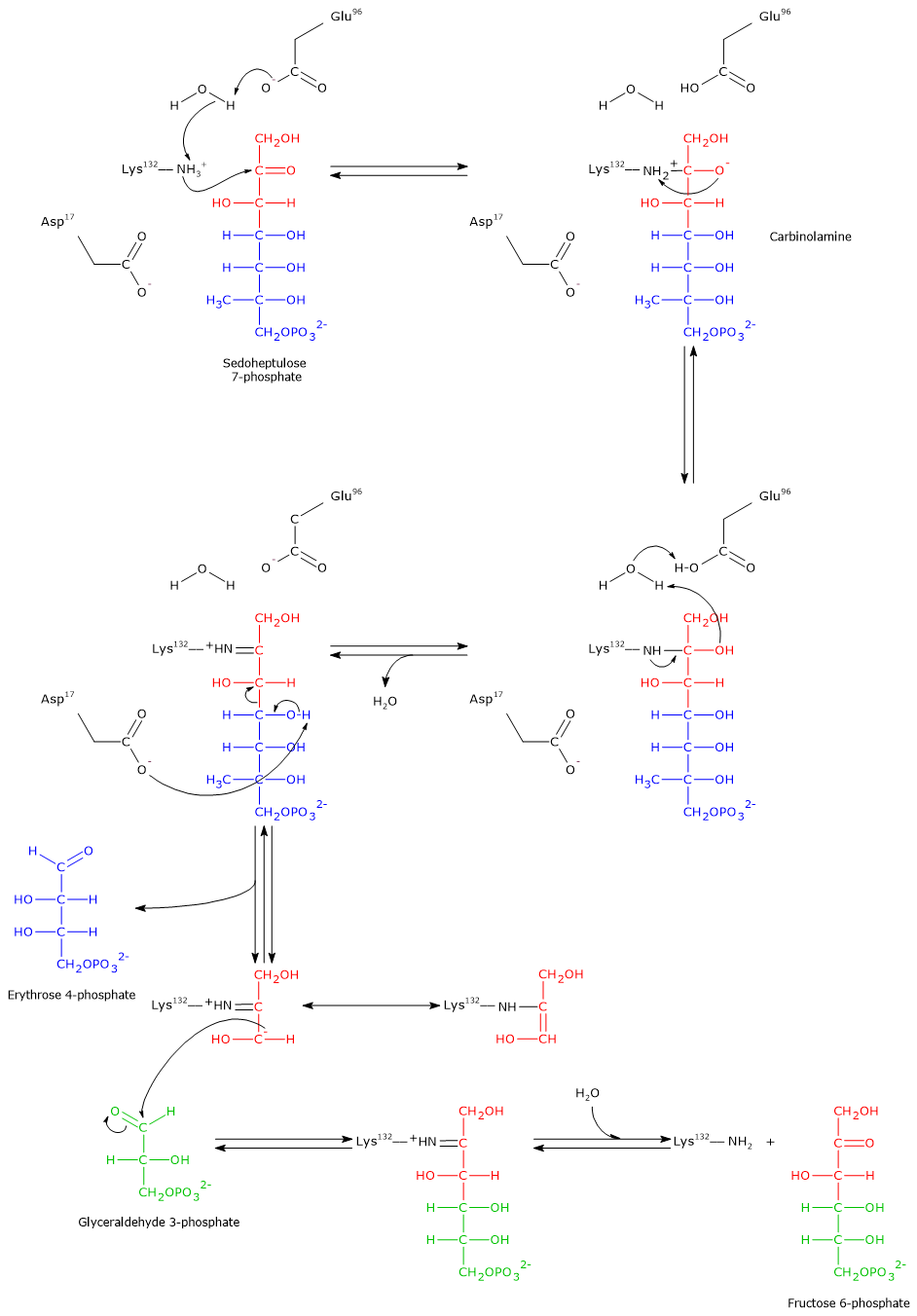
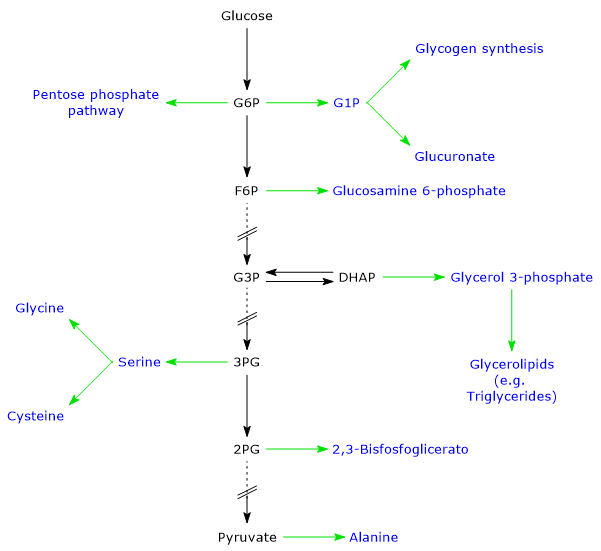
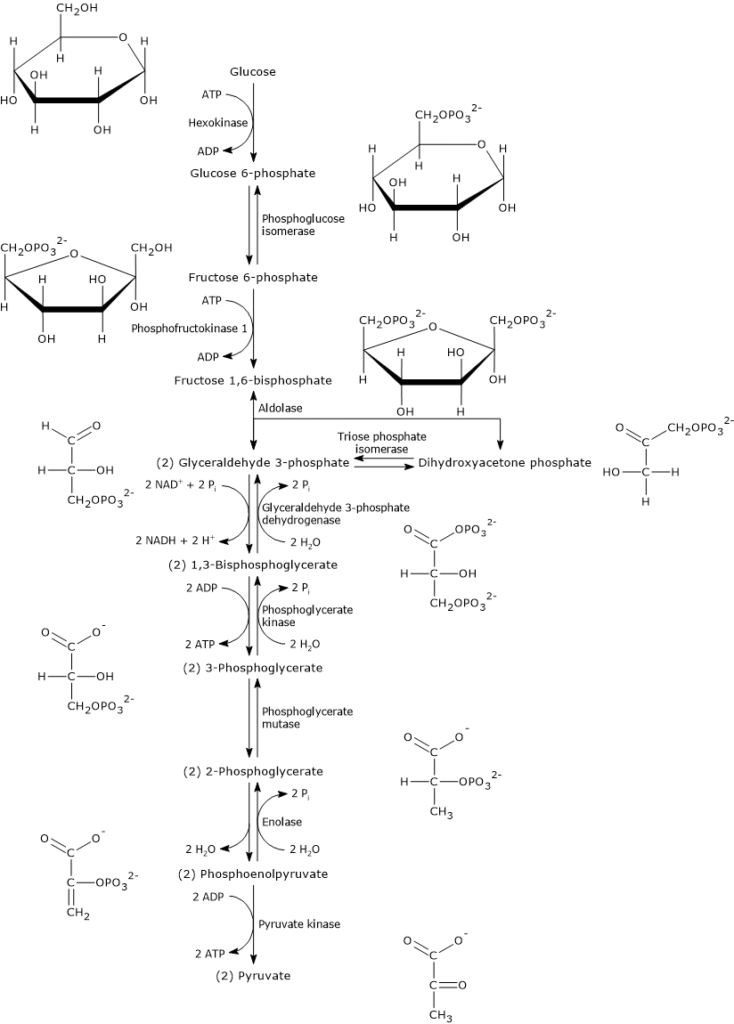
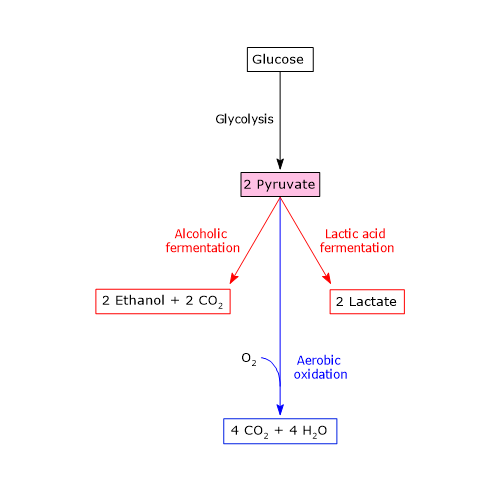
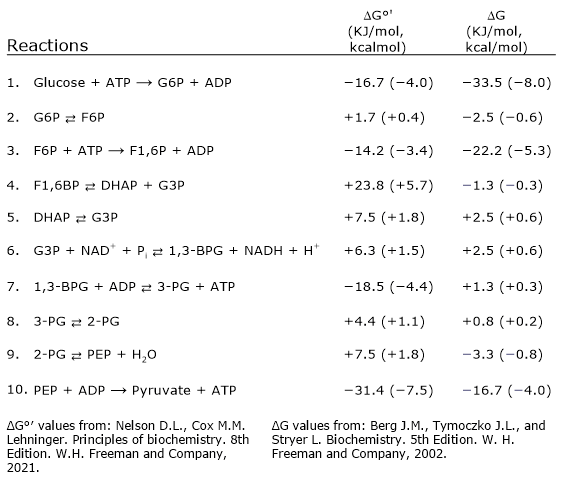
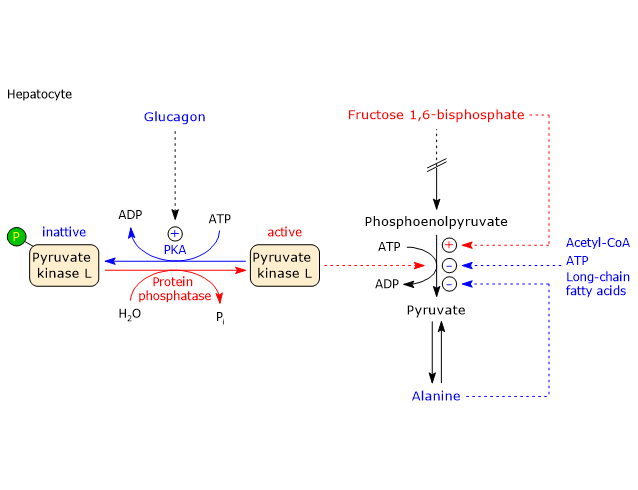
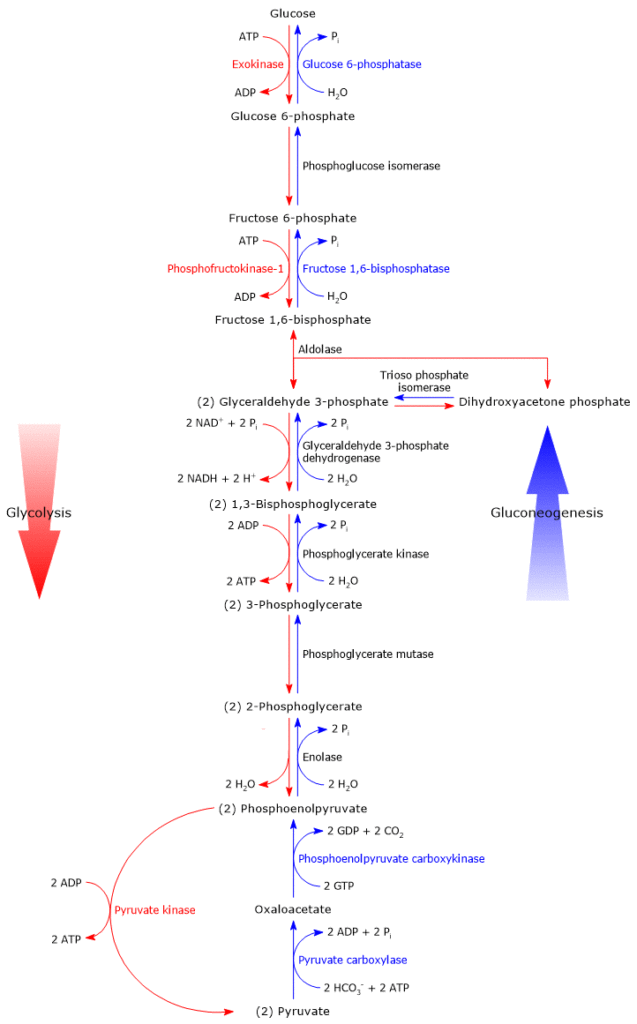
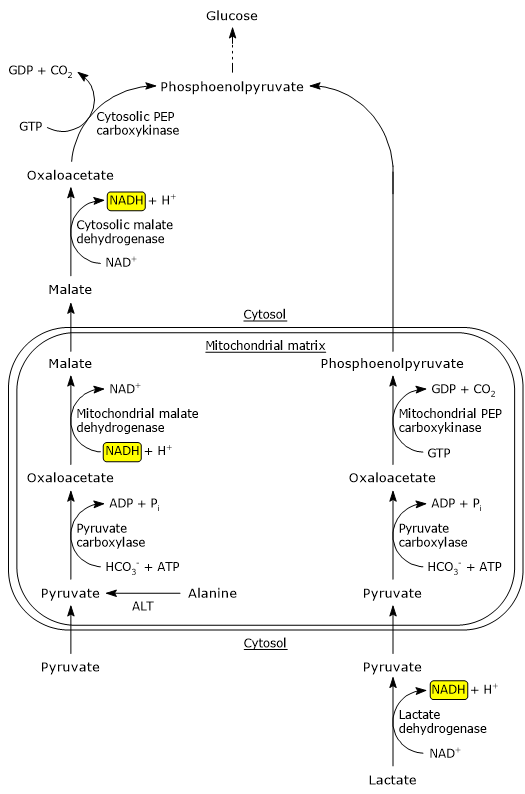
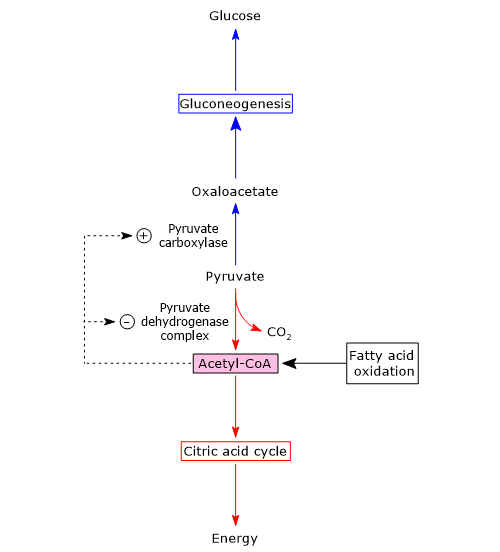
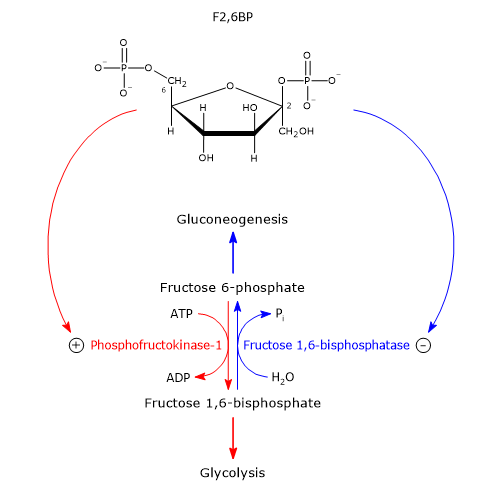
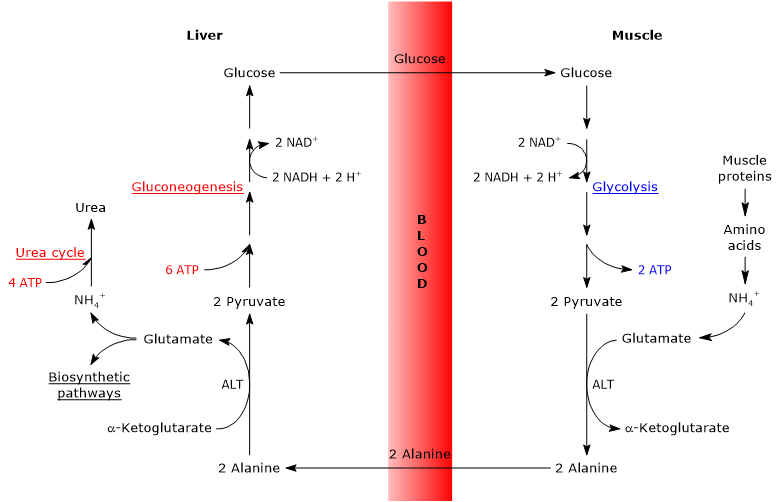
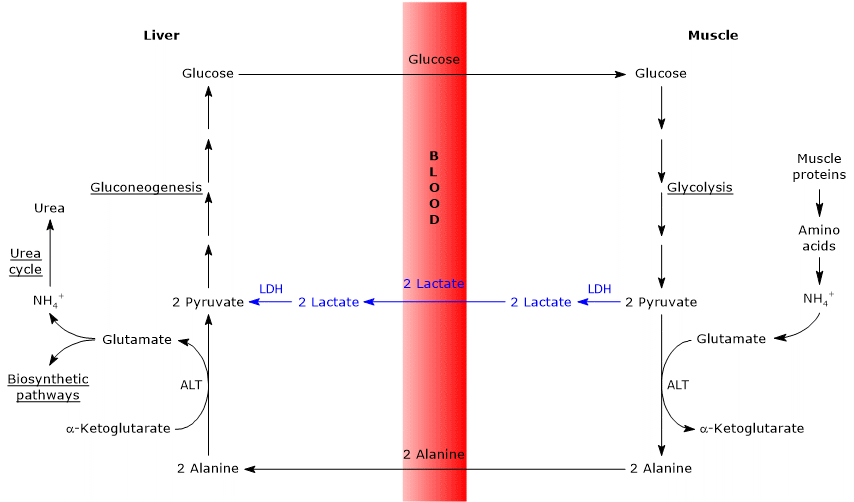
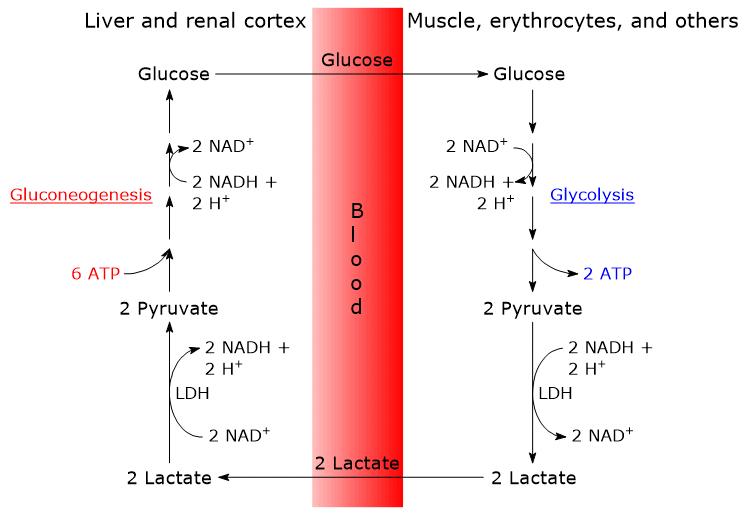
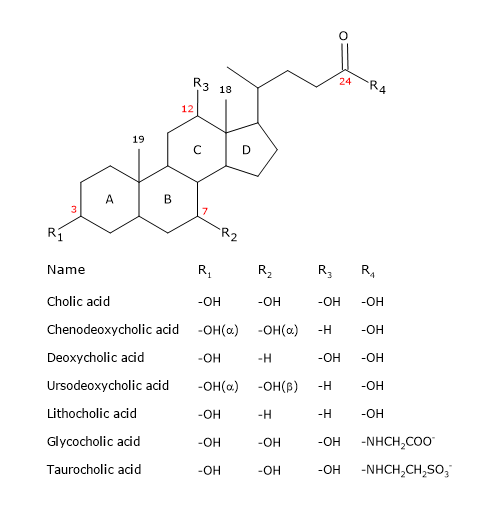
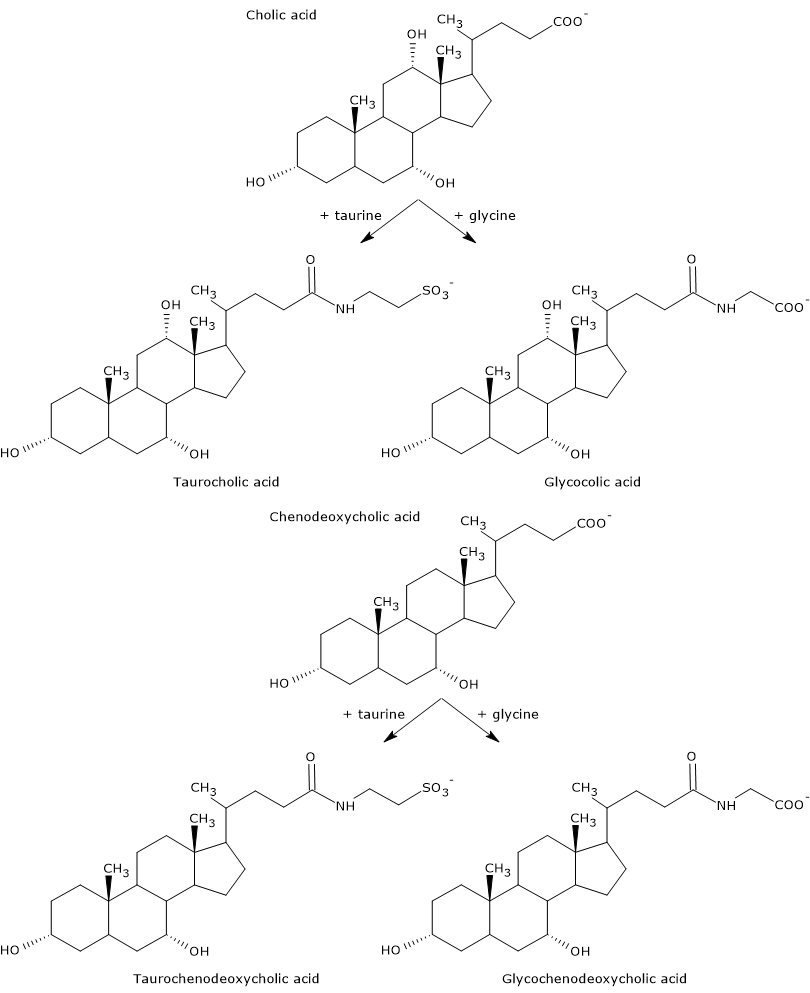
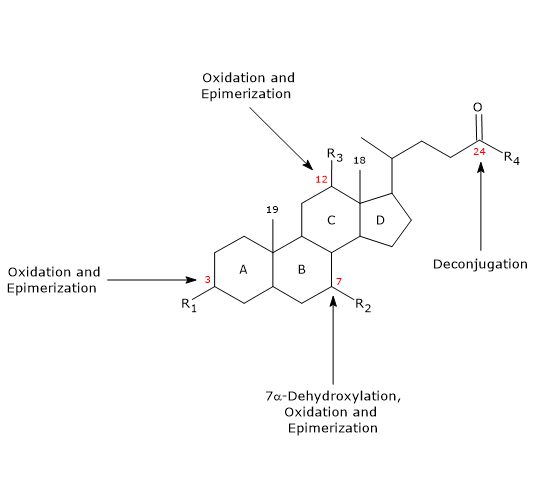
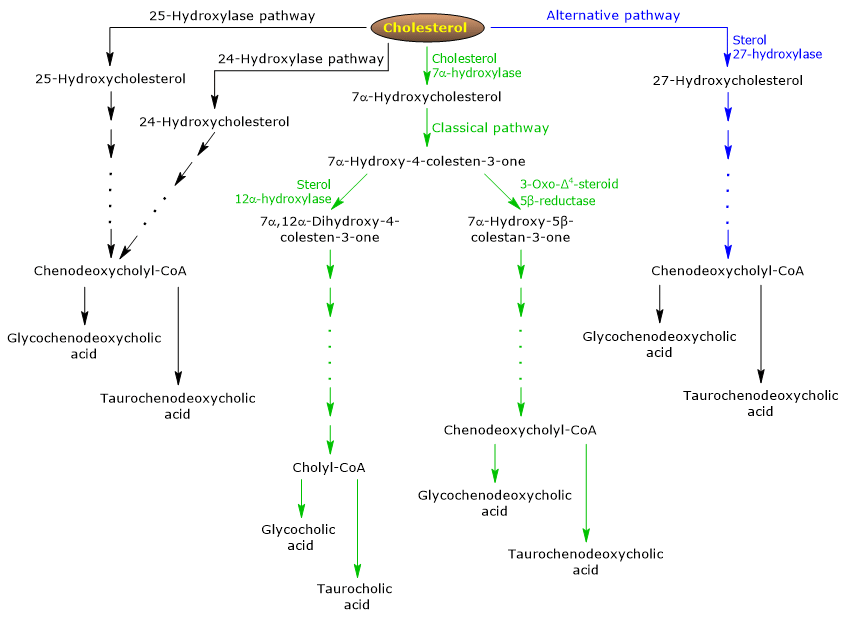
 Then, glucose can diffuse from the hepatocyte, via a transporter into the plasma membrane called GLUT2, into the bloodstream to be delivered to extra-hepatic cells, in primis neurons and red blood cells for which it is the main, and for red blood cells the only energy source. Neurons, with the exception of those in some brain areas that can use only glucose as energy source, can derive energy from another source, the ketone bodies, which becomes predominant during periods of prolonged fasting.
Then, glucose can diffuse from the hepatocyte, via a transporter into the plasma membrane called GLUT2, into the bloodstream to be delivered to extra-hepatic cells, in primis neurons and red blood cells for which it is the main, and for red blood cells the only energy source. Neurons, with the exception of those in some brain areas that can use only glucose as energy source, can derive energy from another source, the ketone bodies, which becomes predominant during periods of prolonged fasting.