La biochimica o chimica biologica si occupa dello studio dei processi chimici che avvengono negli animali, piante e microorganismi.
La biochimica è, con la chimica inorganica, la chimica organica, la chimica fisica e la chimica analitica una delle cinque branche principali della chimica. E, come suggerisce il suo nome, può essere considerata anche una branca della biologia.
Emerge come disciplina a se stante verso gli inizi del 1900 quando gli scienziati hanno iniziato a combinare biologia, fisiologia e chimica per studiare gli esseri viventi, sfruttando le conoscenze accumulate nel secolo precedente, grazie al lavori di scienziati quali Justus von Liebig, Eduard Buchner, Louis Pasteur o Emil Fischer.
A partire dalla metà del 1900 i biochimici, sfruttando anche l’avanzamento delle conoscenze in campi quali la chimica, la fisica e la matematica, sono riusciti ad analizzare e chiarire le vie metaboliche che operano all’interno delle cellule, vie che permettono di ricavare energia dagli alimenti o a catturarla dal sole, come la glicolisi, il ciclo dell’acido citrico, la lipolisi, la fosforilazione ossidativa, o la fotosintesi clorofilliana. E come le cellule utilizzano questa energia per produrre ciò di cui necessitano per vivere e riprodursi. Sono state cioè chiarite le vie metaboliche, anaboliche e cataboliche, che permettono la sintesi e la degradazione di molecole e macromolecole quali carboidrati, proteine, lipidi e acidi nucleici. Dunque si è fatta luce su quello che è il metabolismo cellulare. E stata anche chiarità la struttura di queste macromolecole, come ad esempio quella del DNA grazie al lavoro di Rosalind Elsie Franklin, James Dewey Watson e Francis Harry Compton Crick. E con il metabolismo e enzimologia e la biologia strutturale è una delle tre branche in cui può essere suddivisa la biochimica.
Viene definita fosforilazione a livello del substrato la produzione di ATP o GTP a seguito del trasferimento di un gruppo fosfato da un substrato all’ADP o al GDP.[1]
L’energia rilasciata dalla rottura del legame ad alta energia del substrato rende possibile la formazione del legame fosfoanidridico.[2] Parte di questa energia agisce come una sorta di forza trainante che spinge la reazione verso la formazione dei nucleotidi trifosfato, mentre parte è immagazzinata nel legame neoformato.[3]
E’ un processo che coinvolge enzimi solubili e intermedi chimici, differenziandosi in questo dalla fosforilazione ossidativa e dalla fotofosforilazione, processi in cui la sintesi dei nucleotidi trifosfati coinvolge enzimi di membrana e gradienti protonici transmembrana.[2]
Esempi di fosforilazione a livello del substrato si hanno nella via glicolitica e nel ciclo dell’acido citrico.[3]
Sebbene la maggior parte dell’ATP prodotto in condizioni aerobiche derivi dalla fosforilazione ossidativa e, negli organismi fotosintetizzanti, anche dalla fotofosforilazione, la fosforilazione a livello del substrato è comunque essenziale nel metabolismo energetico, specialmente in condizioni anaerobiche.[4][5]
Due reazioni della glicolisi sono esempi ben noti di fosforilazione a livello del substrato.
Nella seconda fase della via glicolitica, la fase di recupero energetico, due delle cinque reazioni che la compongono permettono l’estrazione diretta e l’immagazzinamento nella molecola di ATP di parte dell’energia chimica della molecola di glucosio.[5]
Le reazioni coinvolte sono quelle catalizzate dalla fosfoglicerato chinasi (EC 2.7.2.3), e dalla piruvato chinasi (EC 2.7.1.40).
Nelle reazione un gruppo fosfato ad alta energia è trasferito all’ADP:
dall’1,3-bisfosfoglicerato, con produzione di una molecola di ATP e una molecola di 3-fosfoglicerato;
dal fosfoenolpiruvato, con produzione di una seconda molecola di ATP e una molecola di piruvato.[6]
Queste due reazioni di fosforilazione a livello del substrato consentono la sintesi di ATP anche in condizioni anaerobiche.[3]
Ciclo dell’acido citrico e fosforilazione del GDP
Nel ciclo dell’acido citrico si trova un altro esempio di fosforilazione a livello del substrato nella reazione catalizzata dalla succinil-CoA sintetasi, anche detta succinato-CoA ligasi (EC 6.2.1.4). L’enzima catalizza la scissione del legame tioestere ad alta energia del succinil-CoA, analogo a quello presente nell’acetil-CoA. L’energia rilasciata rende possibile la formazione di un legame fosfoanidridico, quindi la sintesi del GTP dal GDP.[5]
Bibliografia
^ Ha C.E., Bhagavan N.V.. Chapter 11 – Carbohydrate metabolism I: glycolysis and the tricarboxylic acid cycle. Editor(s): Chung Eun Ha, N.V. Bhagavan. Essentials of Medical Biochemistry. 3rd Edition. Academic Press, 2023, pages 203-227. doi:10.1016/B978-0-323-88541-6.00030-2
Gli amminoacidi chetogenici sono amminoacidi il cui scheletro carbonioso può essere catabolizzato per intero o almeno in parte ad acetoacetil-CoA o acetil-CoA, molecole che possono essere utilizzate come precursori per la sintesi dei corpi chetonici, da cui il nome, o degli acidi grassi.[1]
Gli amminoacidi sono i costituenti fondamentali delle proteine. Una volta che il loro pool disponibile è sufficiente a soddisfare le necessità della sintesi proteica, non disponendo l’organismo di riserve specifiche di amminoacidi paragonabili a quelle del glicogeno per il glucosio o dei trigliceridi per gli acidi grassi, ne converte l’eccesso in intermedi del ciclo dell’acido citrico, impiegandoli nella produzione di energia.[2]
Quando il fabbisogno energetico cellulare è stato soddisfatto, i residui carboniosi derivanti dalla catabolismo amminoacidico vengono reindirizzati verso la sintesi del glucosio, degli acidi grassi, o dei corpi chetonici.[3]
Gli amminoacidi proteinogenici possono essere classificati come amminoacidi chetogenici o amminoacidi glucogenici sulla base del destino metabolico del loro scheletro carbonioso.[4] Lo scheletro carbonioso degli amminoacidi chetogenici viene catabolizzato ad acetoacetil-CoA e/o acetil-CoA, mentre quello degli amminoacidi glucogenici in uno o più dei seguenti cinque metaboliti precursori glucogenici: piruvato, ossalacetato, α-chetoglutarato, succinil-CoA e fumarato.[5]
Questa modalità di classificazione non è però univoca in quanto cinque amminoacidi sono sia chetogenici che glucogenici, poiché dal loro catabolismo si origina almeno un precursore glucogenico e uno chetogenico.[6]
Solamente due dei venti amminoacidi che compongono le proteine sono esclusivamente chetogenici: la leucina e la lisina. Dal catabolismo del loro scheletro carbonioso si origina acetoacetil-CoA e acetil-CoA.[6]
Amminoacido
Catabolita
chetogenico
glucogenico
Fenilalanina
Acetoacetil-CoA
Fumarato
Isoleucina
Acetil-CoA
Succinil-CoA
Leucina *
Acetil-CoA
Acetoacetil-CoA
Lisina *
Acetoacetil-CoA
Tirosina
Acetoacetil-CoA
Fumarato
Treonina
Acetil-CoA
Succinil-CoA
Triptofano
Acetil-CoA
Acetoacetil-CoA
Piruvato
* Amminoacidi esclusivamente chetogenici
Altri cinque amminoacidi, ossia isoleucina, fenilalanina, treonina, triptofano, e tirosina, sono invece sia chetogenici che glucogenici in quanto il catabolismo dello scheletro carbonioso porta alla formazione di acetil-CoA e/o acetoacetil-CoA e un precursore glucogenico.[5]
L’utilizzo degli scheletri carboniosi degli amminoacidi è preceduto dalla rimozione del gruppo amminico. Alanina e glutammato, amminoacidi glucogenici, svolgono un ruolo chiave nel trasporto dei gruppi amminici dai tessuti extraepatici al fegato. In particolare, l’alanina viene trasportata dal muscolo e da altri tessuti periferici al fegato attraverso il ciclo glucosio-alanina.[7]
Basi biochimiche
Acetil-CoA e acetoacetil-CoA non sono precursori glucogenici. La spiegazione risiede nella stechiometria del ciclo dell’acido citrico e nell’incapacità degli animali di convertire l’acetil-CoA in piruvato.[8]
L’acetil-CoA entra nel ciclo dell’acido citrico a livello della reazione catalizzata dalla citrato sintasi (EC 2.3.3.1). L’enzima catalizza la condensazione dell’acetil-CoA con l’ossalacetato a formare il citrato. Dunque si passa da un composto a quattro atomi di carbonio a uno a sei atomi di carbonio, con un guadagno netto di due atomi carboni.
Tuttavia, nelle due successive decarbossilazioni ossidative del ciclo, catalizzate dalla isocitrico deidrogenasi (EC 1.1.1.42) e dal complesso multienzimatico dell’α-chetoglutarato deidrogenasi, sono perduti due atomi di carbonio.[4]
Quindi l’ingresso dell’acetil-CoA nel ciclo non comporta alcun guadagno netto di carbonio.[6]
La discriminante è quindi rappresentata dal punto d’ingresso delle unità carboniose a monte o a valle delle decarbossilazioni ossidative del ciclo dell’acido citrico.[8]
A tutto ciò va aggiunto che negli animali non esiste una via metabolica che permetta di produrre piruvato dall’acetil-CoA, data anche l’irreversibilità della reazione catalizzata dal complesso della piruvato deidrogenasi, la decarbossilazione ossidativa del piruvato ad acetil-CoA.[8]
Amminoacidi chetogenici nel digiuno prolungato
A differenza degli amminoacidi glucogenici, gli amminoacidi chetogenici non hanno un ruolo cruciale durante il digiuno prolungato o nelle diete con forte restrizione dei carboidrati.
Durante il digiuno prolungato, la principale fonte di acetil-CoA per la chetogenesi è la beta-ossidazione degli acidi grassi, mentre il contributo degli amminoacidi chetogenici è marginale. In queste condizioni, il ruolo centrale è svolto dagli amminoacidi glucogenici, che, attraverso la gluconeogenesi, garantiscono il mantenimento dell’omeostasi glicemica quando le riserve di glicogeno epatico sono esaurite.[5]
Ciclo del gliossilato
Piante, lieviti, e molti batteri possono utilizzare l’acetil-CoA per la sintesi del glucosio grazie alla via metabolica chiamata ciclo del gliossilato. Tale ciclo ha alcune reazioni in comune con il ciclo dell’acido citrico, due esclusive catalizzate dalla isocitrato liasi (EC 4.1.3.1) e malato sintasi (EC 2.3.3.9), ma non ha reazioni di decarbossilazione. Quindi gli organismi che possiedono il ciclo del gliossilato sono in grado di utilizzare gli acidi grassi e gli amminoacidi chetogenici per la sintesi del glucosio.[9]
Bibliografia
^ D’Andrea G. Classifying amino acids as gluco(glyco)genic, ketogenic, or both. Biochem Educ 2000;28(1):27-28. doi:10.1016/s0307-4412(98)00271-4
^ Brosnan J.T. Interorgan amino acid transport and its regulation. J Nutr 2003;133(6 Suppl 1):2068S-2072S. doi:10.1093/jn/133.6.2068S
^ Litwack G. Human biochemistry. 2nd Edition. Academic Pr, 2021.
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ abc Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009.
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012.
^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
^ abc Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
^ Kondrashov F.A., Koonin E.V., Morgunov I.G., Finogenova T.V., Kondrashova M.N. Evolution of glyoxylate cycle enzymes in Metazoa: evidence of multiple horizontal transfer events and pseudogene formation. Biol Direct 2006;1:31. doi:10.1186/1745-6150-1-31
Sono definiti amminoacidi glucogenici quegli amminoacidi il cui scheletro carbonioso può essere catabolizzato in toto o in parte in precursori della sintesi del glucosio o gluconeogenesi, da cui il nome.[1]
Gli amminoacidi sono i mattoni costituenti le proteine. Quando il loro pool soddisfa le esigenze cellulari per la sintesi proteica, non esistendo nell’organismo depositi di amminoacidi analoghi al glicogeno per il glucosio o ai trigliceridi per gli acidi grassi, l’eccesso amminoacidico viene catabolizzato a precursori del ciclo dell’acido citrico e utilizzato per la produzione di energia.[2]
Tuttavia, nel momento in cui le esigenze energetiche della cellula sono soddisfatte, gli scheletri carboniosi derivanti dal catabolismo amminoacidico sono utilizzati per la sintesi di acidi grassi, corpi chetonici, o glucosio.[3]
Sulla base del destino metabolico dello scheletro carbonioso si individuano amminoacidi glucogenici e amminoacidi chetogenici. Tale classificazione non è però netta in quanto, dei venti amminoacidi standard che si ritrovano nelle proteine, cinque sono sia glucogenici che chetogenici.[4]
La sintesi del glucosio dagli amminoacidi glucogenici è resa possibile dal fatto che il loro scheletro carbonioso viene catabolizzato, per intero o in parte, in piruvato, ossalacetato, α-chetoglutarato, succinil-CoA, o fumarato.[5]
Piruvato e ossalacetato sono intermedi della gluconeogenesi, mentre l’ingresso degli altri tre metaboliti nel ciclo dell’acido citrico permette un guadagno netto di unità carboniose per il ciclo medesimo, unità che quindi potranno entrare nella gluconeogenesi come ossalacetato.[6]
Gli amminoacidi glucogenici sono particolarmente importanti nel digiuno prolungato, rappresentando una fonte rilevante di precursori per la gluconeogenesi, secondi solo al glicerolo derivante dalla lipolisi nel tessuto adiposo.[7]
Dei venti amminoacidi costituenti le proteine tredici sono esclusivamente glucogenici, ossia dal catabolismo del loro scheletro carbonioso si originano solo precursori del glucosio.[4]
Amminoacido
Catabolita
chetogenico
glucogenico
Alanina
Piruvato
Arginina
α-Chetoglutarato
Asparagina
Fumarato
Aspartato
Ossalacetato
Cisteina
Piruvato
Fenilalanina *
Acetoacetil-CoA
Fumarato
Glicina
Piruvato
Glutammato
α-Chetoglutarato
Glutammina
α-Chetoglutarato
Isoleucina *
Acetil-CoA
Succinil-CoA
Istidina
α-Chetoglutarato
Metionina
Succinil-CoA
Prolina
α-Chetoglutarato
Serina
Piruvato
Tirosina *
Acetoacetil-CoA
Fumarato
Treonina *
Acetil-CoA
Succinil-CoA
Triptofano *
Acetil-CoA
Acetoacetil-CoA
Piruvato
Valina
Succinil-CoA
* Amminoacidi sia glucogenici che chetogenici
Cinque amminoacidi, ossia isoleucina, fenilalanina, treonina, triptofano, e tirosina, sono invece sia glucogenici che chetogenici in quanto parte del loro scheletro carbonioso viene catabolizzato a precursori glucogenici e parte ad acetil-CoA e/o acetoacetil-CoA.[8]
L’utilizzo degli scheletri carboniosi degli amminoacidi deve essere preceduto dalla rimozione del loro gruppo amminico. Alanina e glutammato, le principali molecole responsabili del trasporto dei gruppi amminici dai tessuti extraepatici al fegato, sono amminoacidi glucogenici particolarmente importanti nei mammiferi. L’alanina è il principale substrato gluconeogenetico per il fegato, e arriva all’organo dal muscolo e da altri tessuti periferici seguendo la via del ciclo glucosio-alanina.[9]
Basi biochimiche
Come con gli amminoacidi chetogenici, anche con gli amminoacidi glucogenici l’analisi della stechiometria del ciclo dell’acido citrico chiarisce perché il loro scheletro carbonioso sia un precursore per la sintesi del glucosio. La discriminante è rappresentata dal punto d’ingresso nel ciclo dell’acido citrico.[6]
L’ingresso nel ciclo dei carboni di derivazione amminoacidica in forma di α-chetoglutarato, succinil-CoA, e fumarato comporta un guadagno netto di unità carboniose. Infatti, con l’esclusione dell’α-chetoglutarato, gli altri metaboliti vi entrano a valle delle due reazioni di decarbossilazione ossidativa catalizzate dalla isocitrico deidrogenasi (EC 1.1.1.42) e dal complesso multienzimatico dell’α-chetoglutarato deidrogenasi.[5]
Questo permette al ciclo un guadagno netto di un’unità carboniosa in caso di ingresso a livello dell’α-chetoglutarato, o due se l’ingresso avviene come succinil-CoA, e fumarato. Le unità carboniose potranno quindi entrare nella gluconeogenesi in forma di ossalacetato.[4]
Inoltre, anche l’irreversibilità della reazione catalizzata dal complesso della piruvato deidrogenasi, ossia la decarbossilazione ossidativa del piruvato ad acetil-CoA, e l’assenza negli animali di altre vie metaboliche che permettano la sintesi di piruvato dall’acetil-CoA, non permettono all’acetil-CoA di essere un substrato glucogenico.[6]
Amminoacidi glucogenici nel digiuno prolungato
Gli amminoacidi glucogenici sono particolarmente importanti nel digiuno prolungato e nelle diete con forte restrizione dell’apporto di carboidrati. In queste condizioni sono tra i più importanti precursori per la sintesi del glucosio.[6] L’utilizzo del loro scheletro carbonioso, assieme al glicerolo e al propionato, un acido grasso a catena corta, concorre infatti al mantenimento dell’omeostasi glicemica, via gluconeogenesi, quando le riserve epatiche di glicogeno sono esaurite.[7]
Tuttavia, anche in condizioni fisiologiche, ad esempio nel turn-over delle proteine cellulari, o a seguito di un pasto ricco in proteine, gli amminoacidi in eccesso rispetto alle necessità della sintesi proteica saranno catabolizzati e utilizzati, sulla base dello stato metabolico, a fini energetici o anabolici, per la sintesi del glucosio e di seguito per la glicogenosintesi, la sintesi dei corpi chetonici o degli acidi grassi, a seconda dell’amminoacido in questione.[2]
Bibliografia
^ D’Andrea G. Classifying amino acids as gluco(glyco)genic, ketogenic, or both. Biochem Educ 2000;28(1):27-28. doi:10.1016/s0307-4412(98)00271-4
^ ab Brosnan J.T. Interorgan amino acid transport and its regulation. J Nutr 2003;133(6 Suppl 1):2068S-2072S. doi:10.1093/jn/133.6.2068S
^ Litwack G. Human biochemistry. 2nd Edition. Academic Pr, 2021.
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012.
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ abcd Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
^ ab Kuriyama H., Shimomura I., Kishida K., Kondo H., Furuyama N., Nishizawa H., Maeda N., Matsuda M., Nagaretani H., Kihara S., Nakamura T., Tochino Y., Funahashi T., Matsuzawa Y. Coordinated regulation of fat-specific and liver-specific glycerol channels, aquaporin adipose and aquaporin 9. Diabetes 2002;51(10):2915-2921. 10.2337/diabetes.51.10.2915
^ Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009.
^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
Il 2,3-bisfosfoglicerato (2,3-BPG) è la base coniugata dell’acido 2,3-bisfosfoglicerico.[1]
Nel globulo rosso, a livello del mare, è presente in concentrazioni vicine a quella dell’emoglobina (Hb), circa 5 mM. Negli altri tipi cellulari è invece presente in tracce.[2]
Nell’eritrocita viene sintetizzato a partire dall’1,3-bisfosfoglicerato (1,3-BPG) nella prima reazione dello shunt di Rapoport-Luebering.[3][4] Essendo lo shunt di Rapoport-Luebering una deviazione dalla via glicolitica a monte delle reazioni che portano alla sintesi dell’ATP, la produzione di 2,3-bisfosfoglicerato comporta un costo energetico di due molecole di ATP per molecola prodotta.[5]
Il 2,3-BPG ed è uno dei regolatori dell’affinità dell’emoglobina per l’ossigeno.[6] E’ inoltre necessario all’attività catalitica della fosfoglicerato mutasi (EC 5.4.2.1), ed è un attivatore allosterico della ribosio fosfato pirofosfochinasi (EC 2.7.6.1).[2][7]
Legandosi all’emoglobina ne riduce l’affinità per l’ossigeno facilitandone il rilascio a livello dei tessuti periferici.[6]
Nel globulo rosso la concentrazione del 2,3-bisfosfoglicerato può variare in risposta a modifiche nel flusso di carbonio attraverso la via glicolitica. Tali modificazioni possono essere conseguenti a variazioni di altitudine, a malattie che compromettano l’ossigenazione del sangue, o difetti a carico di enzimi glicolitici, e si riflettono sull’affinità di legame tra emoglobina e ossigeno.[8][9][10]
Il 2,3-bisfosfoglicerato è la base coniugata dell’acido 2,3-bisfosfoglicerico. Ha un peso molecolare di 261,00 g/mol e formula molecolare C3H3O10P2-5.[1]
Secondo la nomenclatura IUPAC il suo nome è (2R)-2,3-bis(fosfonoossi)propanoato.[11]
Il pKa dell’acido 2,3-bisfosfoglicerico è pari a 0,48. Ne consegue che a pH fisiologico è presente quasi esclusivamente in forma ionizzata, il 2,3-BPG.[11]
E’ solubile in acqua, con un valore pari a 9,69 g/L.[1]
Il 2,3-bisfosfoglicerato è un isomero dell’1,3-bisfosfoglicerato.
Metabolismo
Il 2,3-bisfosfoglicerato viene prodotto a partire dall’intermedio glicolitico 1,3-bisfofoglicerato nella prima delle due tappe che compongono lo shunt di Rapoport-Luering.[3][4]
Entrambe le reazioni dello shunt sono catalizzate dalla bisfosfoglicerato mutasi (EC 5.4.2.4), enzima multifunzionale in grado di svolgere tre attività principali, potendo agire come 2,3-BPG sintasi, l’attività principale, 2,3-BPG fosfatasi, e 2,3-BPG mutasi.[12]
Nella prima reazione dello shunt di Rapoport-Luebering l’enzima agisce come sintasi e catalizza l’isomerizzazione dell’1,3-bisfosfoglicerato a 2,3-bisfosfoglicerato. L’enzima catalizza il trasferimento intermolecolare di un gruppo fosforico dal C-1 dell’1,3-bisfosfoglicerato al C-2 del 3-fosfoglicerato, che quindi deve essere presente nel sito attivo. La reazione comporta la conversione del 3-fosfoglicerato a 2,3-bisfosfoglicerato mentre l’1,3-BPG diviene il nuovo 3-fosfoglicerato.[6]
Nella seconda reazione dello shunt, il 2,3-bisfosfoglicerato viene defosforilato a 3-fosfoglicerato. La reazione è catalizzata dall’attività fosfatasica della bisfosfoglicerato mutasi. Il 3-fosfoglicerato rientra quindi nella via glicolitica a livello dell’ottava tappa, catalizzata dalla fosfoglicerato mutasi (EC 5.4.2.1).[5]
Costo energetico della sintesi del 2,3-bisfosfoglicerato
La sintesi del 2,3-bisfosfoglicerato comporta un costo energetico per il globulo rosso pari a due molecole di ATP per molecola prodotta.[5]
L’1,3-BPG è il substrato della settima reazione della glicolisi, il trasferimento catalizzato dalla fosfoglicerato chinasi (EC 2.7.2.3) del gruppo fosforico ad alta energia legato al C-1 all’ADP, con formazione di 3-fosfoglicerato e ATP. La fosfoglicerato chinasi catalizza infatti la prima delle due fosforilazioni a livello del substrato che, lungo la via glicolitica, portano alla conservazione di parte dell’energia chimica presente nel glucosio in forma di ATP, la moneta di scambio energetico della cellula.[6]
Dato che, in condizioni fisiologiche, lo shunt di Rapoport-Luebering intercetta circa il 20% del flusso glicolitico, il costo energetico pagato dal globulo rosso per la sintesi del 2,3-bisfosfoglicerato implica l’esistenza di un equilibrio finemente regolato tra il fabbisogno energetico della cellula e la necessità di mantenere l’emoglobina in un adeguato stato di deossigenazione e ossigenazione.[13][14]
Ruolo del 2,3-bisfosfoglicerato
Il 2,3-bisfosfoglicerato svolge almeno tre funzioni.
E’ necessario per l’attività catalitica della fosfoglicerato mutasi. Ne consegue che la molecola ha anche un ruolo nel controllo del livello degli intermedi glicolitici.[7]
E’ uno degli attivatori allosterici della ribosio fosfato pirofosfochinasi o PRPP sintetasi. L’enzima catalizza l’attivazione del ribosio-5-fosfato, uno dei due prodotti della via del pentoso fosfato, a 5-fosforibosil-1-pirofosfato (PRPP), intermedio nella sintesi de novo delle purine.[2]
Il 2,3-bisfosfoglicerato è coinvolto, assieme ad altri fattori, nella regolazione allosterica dell’affinità dell’emoglobina per l’ossigeno, concorrendo a regolarne il rilascio a livello dei tessuti periferici.[15] Nell’uomo, nella maggior parte dei primati e in molti altri mammiferi il 2,3-bisfosfoglicerato riduce l’affinità dell’emoglobina per l’ossigeno. In questo modo la molecola favorisce la dissociazione dell’ossigeno dall’emoglobina, facilitandone la cessione ai tessuti.[6] L’azione del 2,3-BPG è un esempio di regolazione allosterica eterotropica, ossia un regolazione allosterica in cui l’effettore è diverso dal normale ligando della proteina.[5]
Emoglobina
L’emoglobina è presente nei globuli rossi, e, in quasi tutti i vertebrati, ha il compito di trasportare l’ossigeno molecolare ai tessuti.
La proteina ha una struttura quaternaria formata da quattro subunità e contiene quattro gruppi prostetici eme, uno per subunità, ognuno dei quali in grado di legare, reversibilmente, una molecola di ossigeno.[13]
In condizioni fisiologiche nell’uomo si distinguono due tipi di emoglobina: quella presente nell’adulto, indicata come emoglobina A, e quella fetale, l’emoglobina F.
L’emoglobina A è formata da due catene α, di 141 residui amminoacidici, e due catene β, di 146 residui amminoacidici. Dunque è un tetramero α2β2.
Nell’emoglobina F le catene β sono sostituite da catene γ, molto simili, ma non identiche alle β. Ne risulta un tetramero α2γ2.[13] Questa differenza ha una grande influenza sull’affinità della proteina per l’ossigeno.[5]
L’emoglobina può esistere in due conformazioni indicate come stato T, teso, e stato R, rilassato.
Sebbene l’ossigeno si leghi all’emoglobina sia nello stato T che in quello R, ha un’affinità maggiore per lo stato R, che viene stabilizzato dal legame. Il legame dell’ossigeno da parte dell’emoglobina è modulato da diversi fattori, alcuni con azione a breve termine e altri, come il 2,3-bisfosfoglicerato, che agiscono più a lungo termine.[6]
Cooperatività del legame emoglobina-ossigeno
L’emoglobina nello stato deossigenato si trova in conformazione T.
Il legame dell’ossigeno all’emoglobina nello stato T può avvenire solo sul gruppo eme di una delle subunità α, essendo i gruppi eme delle subunità β nello stato T virtualmente inaccessibili.[6]
Il legame della prima molecola di ossigeno innesca una variazione conformazionale nella subunità α che la converte nello stato R. Tale modifica è trasmessa alle subunità adiacenti mediante interazioni subunità-subunità, modifica che determina il passaggio della seconda subunità α allo stato R. Ciò innesca cambiamenti conformazionali sequenziali che portano al passaggio della struttura quaternaria dell’emoglobina che lega due molecole di ossigeno, Hb:(O2)2, dallo stato T a quello R.
A questo punto anche le subunità β possono legare l’ossigeno.[6]
Questo tipo di legame è detto cooperativo ed è alla base dell’andamento sigmoidale della curva di saturazione dell’emoglobina.[16]
Affinità dell’emoglobina per l’ossigeno
Nei capillari polmonari l’ossigeno si lega all’emoglobina, e, a mezzo della circolazione sanguigna, raggiunge i tessuti periferici cui viene rilasciato.
L’affinità di legame tra emoglobina e ossigeno è determinata principalmente dalla struttura dell’emoglobina. Tuttavia risulta modulata da vari fattori quali:
Le modifiche nell’affinità del legame Hb-O2 che si verificano nel sistema circolatorio ad opera dei suddetti fattori sono tali da ottimizzare il carico di ossigeno a livello polmonare e la sua cessione nei tessuti periferici.[10]
Graficamente, le modifiche dell’affinità di legame indotte dagli effettori allosterici e dalla temperatura, si traducono in spostamenti della curva di saturazione dell’emoglobina verso destra, nel caso di una diminuzione dell’affinità di legame, o verso sinistra, quando l’affinità di legame aumenta.[5]
Temperatura, pH, e CO2 operano un regolazione a breve termine.[10] Se ad esempio si considera il muscolo scheletrico durante l’esercizio fisico, vi si osserva un aumento della temperatura, della concentrazione degli ioni idrogeno e della pressione parziale della CO2. Alte concentrazioni di H⁺ e CO2 riducono l’affinità dell’emoglobina per l’ossigeno, un effetto noto come effetto Bohr. Anche l’aumento della temperatura diminuisce l’affinità. Tutto ciò favorisce il rilascio di ossigeno al tessuto muscolare.[17]
Anche gli ioni cloro e il 2,3-bisfosfoglicerato riducono l’affinità dell’emoglobina per l’ossigeno, ma si associano a una regolazione più a lungo termine.[10]
2,3-Bisfosfoglicerato ed emoglobina A
Il 2,3-bisfosfoglicerato si va a legare all’emoglobina A a mezzo di legami ionici che si stabiliscono con residui delle catene β carichi positivamente.
I legami tra il 2,3-bisfosfoglicerato e l’emoglobina si stabiliscono con le catene laterali della lisina 82, istidina143, e il gruppo ammino-terminale di ciascuna della due catene β. Questi residui vanno a formare una tasca elettrostatica complementare sia alla conformazione che alla distribuzione di carica del 2,3-BPG.[5]
Il 2,3-BPG si lega all’emoglobina deossigenata e stabilizza lo stato T. Anche gli ioni idrogeno, cloro e l’anidride carbonica stabilizzano lo stato T.[10]
Nel passaggio dallo stato T a quello R, tra le varie modifiche conformazionali che occorrono nella struttura quaternaria dell’emoglobina, si verifica anche il restringimento della tasca di legame per 2,3-BPG tra le subunità β, impedendone quindi il legame. Degno di nota è il fatto che mentre l’emoglobina ha quattro siti di legame per l’ossigeno molecolare, ne ha solo uno per il 2,3-BPG.[6] Inoltre, il sito di legame per il 2,3-BPG è lontano da quello per l’ossigeno.[5]
2,3-Bisfosfoglicerato ed emoglobina F
Le catene γ dell’emoglobina F, analogamente alle catene β dell’emoglobina A, vanno a formare la tasca di legame per il 2,3-BPG.[5]
Tuttavia, nella tasca di legame sono presenti due cariche positive in meno rispetto all’emoglobina A. Infatti, in posizione 143 delle catene γ l’istidina è sostituita da una serina.[2]
Questa piccola differenza nella struttura primaria fa si che il 2,3-bisfosfoglicerato si leghi con minor affinità, e di conseguenza che l’emoglobina F abbia un’affinità per l’ossigeno maggiore rispetto all’emoglobina materna. Ciò concorre ad assicurare il trasferimento dell’ossigeno dall’emoglobina A a quella F, quindi dalla circolazione materna a quella fetale.[6]
Variazioni della concentrazione del 2,3-bisfosfoglicerato
La concentrazione del 2,3-bisfosfoglicerato nel globulo rosso può essere influenzata da diversi fattori, sia fisiologici che patologici.[2]
Tra le condizioni fisiologiche c’è l’adattamento all’alta montagna, mentre tra le condizioni patologiche si hanno difetti genetici a carico di enzimi glicolitici sia a valle che a monte della reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12), o malattie che causano ipossia.[18][19]
In tutti i casi le variazioni sono conseguenti a modifiche nella sua velocità di sintesi, e si riflettono sull’affinità dell’emoglobina per l’ossigeno.
Adattamento all’alta montagna
L’adattamento all’alta montagna è un processo piuttosto complesso che comporta anche un aumento del numero dei globuli rossi e del loro contenuto in emoglobina. Queste modifiche richiedono, per completarsi, settimane. Tuttavia, già dopo un giorno di permanenza in alta quota viene percepito un certo grado di adattamento. Questo effetto è conseguenza dell’aumento della concentrazione eritrocitaria del 2,3-bisfosfoglicerato.[2]
Ciò che accade è che la bassa pressione parziale dell’ossigeno attiva la glicolisi. A sua volta questo comporta un aumento del flusso di carbonio attraverso lo shunt di Rapoport-Luebering con conseguente aumento della sintesi del 2,3-bisfosfoglicerato, la cui concentrazione che può arrivare sino a 8 mM.
Tale aumento determina una riduzione dell’affinità dell’emoglobina per l’ossigeno, che ne facilita il rilascio ai tessuti periferici, concorrendo così all’adattamento all’alta montagna.[5]
Anemia
Variazioni della concentrazione eritrocitaria del 2,3-bisfosfoglicerato si hanno anche in due malattie a trasmissione autosomica recessiva: l’anemia emolitica non sferocitica da deficit di esochinasi e l’anemia emolitica da deficit di piruvato chinasi negli eritrociti.[18][19]
Nell’anemia emolitica dovuta a un deficit di piruvato chinasi negli eritrociti (EC 2.7.1.40), la carenza dell’enzima causa un incremento della sintesi di 2,3-BPG e, di conseguenza, una riduzione dell’affinità dell’emoglobina per l’ossigeno.[8] Un incremento della concentrazione del 2,3-BPG si verifica anche nei soggetti affetti da condizioni che limitano l’ossigenazione del sangue, che causano cioè ipossia. Un esempio è l’insufficienza cardiopolmonare.[2]
Nell’anemia emolitica non sferocitica causata da un deficit di esochinasi (EC 2.7.1.1), il deficit enzimatico eritrocitario causa una riduzione della concentrazione del 2,3-bisfosfoglicerato, portando così a un aumento dell’affinità dell’emoglobina per l’ossigeno.[9]
^ abcdefg Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
^ ab Rapoport S., Luebering J. Glycerate-2,3-diphosphatase. J Biol Chem 1951;189(2):683-694.
^ ab Rapoport S., Luebering J. The formation of 2,3-diphosphoglycerate in rabbit erythrocytes: the existence of a diphosphoglycerate mutase. J Biol Chem 1950;183(2):507-516.
^ abcdefghi Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012.
^ ab Oslund R.C., Su X., Haugbro M., Kee J-M., Esposito M., David Y., Wang B., Ge E., Perlman D.H., Kang Y., Muir T.W., Rabinowitz J.D. Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat Chem Biol 2017;13:1081-1087. doi:10.1038/nchembio.2453
^ ab Enegela O.A., Anjum F. Pyruvate Kinase Deficiency. [Updated 2023 Apr 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560581/
^ ab Paglia D.E., Shende A., Lanzkowsky P., Valentine W.N.. Hexokinase “New Hyde Park”: a low activity erythrocyte isozyme in a Chinese kindred. Am J Hematol 1981;10(2):107-17. doi:10.1002/ajh.2830100202
^ abcde Webb K.L., Dominelli P.B., Baker S.E., Klassen S.A., Joyner M.J., Senefeld J.W., Wiggins C.C. Influence of high hemoglobin-oxygen affinity on humans during hypoxia. Front Physiol 2022;12:763933. doi:10.3389/fphys.2021.763933
^ Aljahdali A.S., Musayev F.N., Burgner J.W. 2nd, Ghatge M.S., Shekar V., Zhang Y., Omar A.M., Safo M.K. Molecular insight into 2-phosphoglycolate activation of the phosphatase activity of bisphosphoglycerate mutase. Acta Crystallogr D Struct Biol 2022;78(Pt 4):472-482. doi:10.1107/S2059798322001802
^ abc Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ Hodgkins S.R. 6 – Erythrocyte metabolism and membrane structure and function. Editor(s): Keohane E.M., Otto C.N., Walenga J.M. Rodak’s Hematology (Sixth Edition), Elsevier. 2020:78-90. doi:10.1016/B978-0-323-53045-3.00015-5
^ ab Mairbäurl H., Oelz O., Bärtsch P. Interactions between Hb, Mg, DPG, ATP, and Cl determine the change in Hb-O2 affinity at high altitude. J Appl Physiol 1993;74(1):40-8. doi:10.1152/jappl.1993.74.1.40
^ Henry E.R., Bettati S., Hofrichter J., Eaton W.A. A tertiary two-state allosteric model for hemoglobin. Biophys Chem 2002;98(1-2):149-64. doi:10.1016/s0301-4622(02)00091-1
^ Böning D., Schwiegart U., Tibes U., Hemmer B. Influences of exercise and endurance training on the oxygen dissociation curve of blood under in vivo and in vitro conditions. Eur J Appl Physiol Occup Physiol 1975;34(1):1-10. doi:10.1007/BF00999910
Lo shunt di Rapoport-Luebering è una via metabolica presente prevalentemente nei globuli rossi ed è una deviazione dalla via glicolitica, da cui si diparte a livello dell’1,3-bisfosfoglicerato.[1][2]
Le due reazioni dello shunt sono catalizzate da un enzima multifunzionale, la bisfosfoglicerato mutasi (EC 5.4.2.4). Tuttavia di recente è stato identificato un secondo enzima che partecipa allo shunt, la 2,3-bisfosfoglicerato 3-fosfatasi (EC 3.1.3.80).[3]
Il prodotto più importante è il 2,3-bisfosfoglicerato (2,3-BPG), un effettore allosterico dell’emoglobina di cui riduce l’affinità per l’ossigeno molecolare, concorrendo così a regolarne il rilascio a livello tissutale.[4][5]
Essendo una deviazione dalla via glicolitica a monte delle reazioni che portano alla sintesi di ATP, lo shunt di Rapoport-Luebering comporta un costo energetico per la cellula.[3]
Difetti a carico degli enzimi glicolitici dei globuli rossi determinano anomalie nella sintesi del 2,3-BPG, e di conseguenza nell’affinità dell’emoglobina per l’ossigeno molecolare.[6]
La bisfosfoglicerato mutasi è un enzima centrale nello shunt di Rapoport-Luebering, e si ritrova nei globuli rossi.[4]
E’ un enzima multifunzionale essendo una 2,3-bisfosfoglicerato sintasi e una 2,3-bisfosfoglicerato fosfatasi. Inoltre è anche una 2,3-bisfosfoglicerato mutasi, attività analoga a quella della fosfoglicerato mutasi glicolitica (EC 5.4.2.1), ma che non partecipa allo shunt. Le tre attività catalitiche avvengono nello stesso sito attivo, con l’attività di sintasi come attività principale, che è inibita dal 2,3-bisfosfoglicerato libero.[7][8]
L’attività di fosfatasi è bassa, circa 1000 volte inferiore all’attività di sintasi, ed è stimolata da diversi anioni, come cloruro, fosfato, solfito e, in modo più marcato, dal 2-fosfoglicolato.[9]
Sembra infine che l’attività di mutasi sia fisiologicamente non significativa, catalizzando circa il 5% della conversione reversibile del 3-fosfoglicerato a 2-fosfoglicerato.[10][11]
La bisfosfoglicerato mutasi è una delle tre isoforme con cui nei mammiferi è presente la fosfoglicerato mutasi. Le altre due isoforme sono la fosfoglicerato mutasi 1 o tipo M, presente nel muscolo, e la fosfoglicerato mutasi 2 o tipo B presente in tutti gli altri tessuti. Anche queste due isoforme sono enzimi trifunzionali.[4]
Reazioni
Lo shunt di Rapoport-Luebering si compone di due reazioni.
Nella prima la bisfosfoglicerato mutasi, agendo come sintasi, catalizza l’isomerizzazione dell’1,3-bisfosfoglicerato (1,3-BPG) a 2,3- bisfosfoglicerato (2,3-BPG). L’enzima necessita della presenza del 3-fosfoglicerato in quanto catalizza il trasferimento intermolecolare di un gruppo fosforico dal C-1 dell’1,3-bisfosfoglicerato al C-2 del 3-fosfoglicerato. Nella reazione quindi il 3-fosfoglicerato di partenza diviene il 2,3-BPG, mentre l’1,3-BPG diviene il nuovo 3-fosfoglicerato.[12]
Nel secondo passaggio l’attività fosfatasica della bisfosfoglicerato mutasi catalizza la sintesi del 3-fosfoglicerato, un intermedio glicolitico, a seguito dell’idrolisi del gruppo fosforico in posizione C-2 del 2,3-BPG.Il 3-fosfoglicerato prodotto può quindi rientrare nella via glicolitica a livello della reazione catalizzata dalla fosfoglicerato mutasi, ossia l’ottava tappa della via.[5]
2,3-Bisfosfoglicerato 3-fosfatasi
E’ stato identificato un secondo enzima coinvolto nello shunt Rapoport-Luebering.
Oltre alla bisfosfoglicerato mutasi, è stata scoperta un’attività di 2,3-bisfosfoglicerato 3-fosfatasi catalizzata da una inositolo polifosfato fosfatasi multipla evolutivamente conservata, nota come MIPP1, che è stata trovata nelle amebe del genere Dictyostelium, negli uccelli e nei mammiferi.
Questa scoperta rivela quindi che la formazione di 3-fosfoglicerato, un precursore per la biosintesi della serina e un attivatore della proteina chinasi attivata da AMP, può essere bypassata. Manipolando geneticamente l’espressione di MIPP1 in Dictyostelium, è stato dimostrato il ruolo fisiologicamente rilevante dell’enzima nella regolazione del contenuto cellulare di 2,3-BPG.[3]
Ruolo dello shunt di Rapoport-Luebering
Lo shunt di Rapoport-Luebering porta alla formazione del 2,3-bisfosfoglicerato, che con l’anidride carbonica e gli ioni idrogeno, è uno degli effettori allosterici dell’emoglobina.[5]
Il 2,3-bisfosfoglicerato è presente in elevate concentrazioni nel globulo rosso dell’uomo, mentre nelle altre cellule è presente in tracce.[6] Nella maggior parte dei primati e molti altri mammiferi riduce l’affinità dell’emoglobina per l’ossigeno molecolare, facilitandone così il rilascio ai tessuti.[3]
Il 2,3-BPG si lega alla deossiemoglobina a mezzo di legami ionici con le catene β nello stato T, stato nel quale queste subunità formano una tasca elettrostatica complementare sia alla conformazione che alla distribuzione di carica della molecola. Il legame stabilizza lo stato T, quindi la deossiemoglobina, ed è principalmente responsabile della natura cooperativa del legame dell’ossigeno molecolare da parte dell’emoglobina.[12]
In tracce, il 2,3-bisfosfoglicerato è anche necessario all’attività catalitica della fosfoglicerato mutasi. Ne consegue che un’altra funzione della bisfosfoglicerato mutasi, e quindi dello shunt di Rapoport-Luebering, è quella di controllare il livello degli intermedi glicolitici.[13]
Costo energetico dello shunt di Rapoport-Luebering
L’1,3-bisfoglicerato è un intermedio della via glicolitica. Infatti, nella settima tappa della glicolisi, la fosfoglicerato chinasi (EC 2.7.2.3) catalizzata il trasferimento del gruppo fosforico ad alta energia presente sul C-1 dell’1,3-bisfosfoglicerato all’ADP, con formazione di ATP e 3-fosfoglicerato. La reazione, una fosforilazione a livello del substrato, è la prima delle due reazioni della via glicolitica in cui parte dell’energia chimica contenuta nel glucosio viene conservata in forma di ATP.[5]
Pertanto, l’ingresso dell’1,3-bisfosfoglicerato nel ciclo di Rapoport-Leubering comporta un costo energetico pari due molecole di ATP per molecola di 1,3-bisfosfoglicerato che devia dalla glicolisi ed entra nello shunt.[3] Ne consegue che esiste un delicato equilibrio tra il fabbisogno energetico del globulo rosso e la necessità di mantenere l’emoglobina in un adeguato stato di ossigenazione e deossigenazione.[14]
Rapporto tra glicolisi e shunt di Rapoport-Luebering
In condizioni fisiologiche, nei globuli rossi lo shunt di Rapoport-Luebering intercetta circa il 20% del flusso glicolitico.[8] Pertanto difetti a carico di enzimi glicolitici eritrocitari influenzano lo shunt, e quindi l’affinità dell’emoglobina per l’ossigeno molecolare.[6]
Due condizioni che evidenziano tale connessione sono l’anemia emolitica non sferocitica da deficit di esochinasi e l’anemia emolitica da deficit di piruvato chinasi negli eritrociti, malattie a trasmissione autosomica recessiva.[15][16]
Nell’anemia emolitica non sferocitica da deficit di esochinasi c’è una carenza di esochinasi eritrocitaria (EC 2.7.1.1).[17] L’enzima catalizza la fosforilazione del glucosio a glucosio-6-fosfato, e un suo deficit comporta una diminuzione della concentrazione eritrocitaria di 2,3-BPG, e quindi un aumento dell’affinità dell’emoglobina per l’ossigeno molecolare.[6]
Di contro, nell’anemia emolitica da deficit di piruvato chinasi negli eritrociti c’è una carenza della piruvato chinasi (EC 2.7.1.40), che ha come conseguenze un aumento della sintesi del 2,3-BPG e una diminuzione dell’affinità dell’emoglobina per l’ossigeno molecolare.[18]
Bibliografia
^ Rapoport S., Luebering J. Glycerate-2,3-diphosphatase. J Biol Chem 1951;189(2):683-694.
^ Rapoport S., Luebering J. The formation of 2,3-diphosphoglycerate in rabbit erythrocytes: the existence of a diphosphoglycerate mutase. J Biol Chem 1950;183(2):507-516.
^ abcde Cho J., King J.S., Qian X., Harwood A.J., Shears S.B. Dephosphorylation of 2,3-bisphosphoglycerate by MIPP expands the regulatory capacity of the Rapoport-Luebering glycolytic shunt. Proc Natl Acad Sci U S A 2008;105(16):5998-6003. doi:10.1073/pnas.0710980105
^ abc DiMauro S., Miranda A.F., Khan S., Gitlin K., Friedman R. Human muscle phosphoglycerate mutase deficiency: newly discovered metabolic myopathy. Science 1981;212(4500):1277-9. doi:10.1126/science.6262916
^ abcd Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012.
^ abcd Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
^ Aljahdali A.S., Musayev F.N., Burgner J.W. 2nd, Ghatge M.S., Shekar V., Zhang Y., Omar A.M., Safo M.K. Molecular insight into 2-phosphoglycolate activation of the phosphatase activity of bisphosphoglycerate mutase. Acta Crystallogr D Struct Biol 2022;78(Pt 4):472-482. doi:10.1107/S2059798322001802
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ Reynolds C.H. Activation of human erythrocyte 2,3-bisphosphoglycerate phosphatase at physiological concentrations of substrate. Arch Biochem Biophys 1986;250(1):106-11. doi:10.1016/0003-9861(86)90706-x
^ Lemarchandel V., Joulin V., Valentin C., Rosa R., Galactéros F., Rosa J., Cohen-Solal M. Compound heterozygosity in a complete erythrocyte bisphosphoglycerate mutase deficiency. Blood 1992;80(10):2643-9. doi:10.1182/blood.V80.10.2643.2643
^ Sasaki R., Chiba H. Role and induction of 2,3-bisphosphoglycerate synthase. Mol Cell Biochem 1983;53-54(1-2):247-56. doi:10.1007/BF00225257
^ Oslund R.C., Su X., Haugbro M., Kee J-M., Esposito M., David Y., Wang B., Ge E., Perlman D.H., Kang Y., Muir T.W., Rabinowitz J.D. Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat Chem Biol 2017;13:1081-1087. doi:10.1038/nchembio.2453
^ Hodgkins S.R. 6 – Erythrocyte metabolism and membrane structure and function. Editor(s): Keohane E.M., Otto C.N., Walenga J.M. Rodak’s Hematology (Sixth Edition), Elsevier. 2020:78-90. doi:10.1016/B978-0-323-53045-3.00015-5
^ Paglia D.E., Shende A., Lanzkowsky P., Valentine W.N. Hexokinase “New Hyde Park”: a low activity erythrocyte isozyme in a Chinese kindred. Am J Hematol 1981;10(2):107-17. doi:10.1002/ajh.2830100202
^ Enegela O.A., Anjum F. Pyruvate Kinase Deficiency. [Updated 2023 Apr 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560581/
L’acido piruvico, un alfa-chetoacido, è una molecola con un ruolo centrale nel metabolismo cellulare.[1][2]
Può essere prodotto attraverso diverse vie metaboliche, per la maggior parte citosoliche, tra le quali glicolisi è di solito la più importante, mentre il suo destino dipende dal tipo di cellula e dalla disponibilità di ossigeno, potendo essere utilizzato sia a fini energetici, biosintetici e anaplerotici.[3]
Dato il suo ruolo centrale nel metabolismo cellulare, mutazioni a carico dei geni che codifica per le proteine coinvolte nel suo metabolismo sono causa, nell’uomo, di malattie da lievi a gravi.[4]
L’acido piruvico o, secondo la nomenclatura IUPAC, l’acido 2-ossopropanoico, ha formula molecolare C3H4O3, formula condensata CH3COCOOH, e peso molecolare molecolare di 88,06 g/mol.
E’ classificato tra gli alfa-chetoacidi, ossia chetoacidi dove il gruppo carbonilico è adiacente al carbonio carbossilico. E, tra gli alfa-chetoacidi è quello con la struttura chimica più semplice.[1]
In forma purificata si presenta come un liquido incolore, con un odore che ricorda quello dell’acido acetico.
La sua costante di dissociazione o pKa, a 25 °C, è pari a 2,45. Si tratta dunque di un acido molto forte. Pertanto a pH fisiologico, sia nelle cellule che nei fluidi extracellulari, è presente quasi totalmente nella sua forma anionica, il piruvato.
Il suo punto di fusione è pari a 13,8 °C (56,84 °F; 286,95 K), mentre il punto di ebollizione è pari a 163,5 °C (329 °F; 438 K).[1]
Metabolismo del piruvato
Le vie metaboliche che portano alla sintesi dell’acido piruvico, nonché il suo successivo utilizzo, dipendono dal tipo di cellula e/o dalla disponibilità di ossigeno.[3]
Nelle cellule dotate di mitocondri, il metabolismo del piruvato si compone di una fase citosolica e una mitocondriale.
Nel citosol diverse vie metaboliche portano alla sua formazione. Tra queste, la glicolisi, l’ossidazione del lattato, la reazione catalizzata dall’enzima malico citosolico, e il catabolismo di almeno sei aminoacidi, tra cui l’alanina è il più importante. L’acido piruvico può essere prodotto anche nella matrice mitocondriale dall’alanina e dal lattato.[5] L’acido piruvico prodotto nel citosol, a mezzo di trasportatori specifici, entra nel mitocondrio dove potrà essere utilizzato a fini energetici, entrando nel ciclo dell’acido citrico, e/o biosintetici/anaplerotici, sulla base delle necessità della cellula.[6]
Se invece si considerano le cellule prive di mitocondri, come i globuli rossi, e, in condizioni di ipossia, anche le cellule dotate di mitocondri, il piruvato è ridotto a lattato e/o lascia la cellula come tale per essere quindi metabolizzato in altri tessuti, ad esempio il muscolo cardiaco.[7]
Glicolisi
In condizioni fisiologiche nella maggior parte delle cellule l’acido piruvico deriva principalmente dalla glicolisi, di cui ne rappresenta uno dei tre prodotti, con l’ATP e il NADH.
Nell’ultima reazione della via glicolitica la piruvato chinasi (EC 2.7.1.40) catalizza una fosforilazione a livello del substrato che porta al trasferimento del gruppo fosforico dal fosfoenolpiruvato all’ADP. La reazione, essenzialmente irreversibile, porta dalla formazione di una molecola di ATP e una di piruvato.
Fosfoenolpiruvato + ADP + H+ → Piruvato + ATP
Dunque, a mezzo della via glicolitica, da una molecola di glucosio sono prodotte due molecole di piruvato, oltre che di ATP e NADH.[2][8]
Lattato deidrogenasi
Nel citosol l’acido piruvico può essere prodotto dall’acido lattico.
L’enzima lattato deidrogenasi o LDH (EC 1.1.1.27) catalizza la conversione, reversibile, del piruvato a lattato e del NADH a NAD+.
Piruvato + NADH + H+ ⇄ Lattato + NAD+
La direzione della reazione dipende dagli isozimi della lattato deidrogenasi presenti e dal rapporto NADH/NAD+ nel citosol.[3][5]
Gli isoenzimi in cui predomina la subunità H, come LDH-1, predominano nel muscolo cardiaco, un tessuto completamente aerobico, dove catalizzano l’ossidazione del lattato, proveniente da altri tessuti, a piruvato, che sarà quindi utilizzato a fini energetici.
L’ossidazione del lattato prodotto ad esempio dai globuli rossi o dal muscolo scheletrico che lavora in condizioni ipossiche, può avvenire anche negli epatociti, favorita dal basso rapporto NADH/NAD+ nel citosol, sebbene l’isozima prevalente sia LDH-5. Nell’epatocita l’acido piruvico prodotto potrà essere utilizzato nella gluconeogenesi, che in questo caso è parte del più ampio ciclo di Cori, o essere ossidato a fini energetici.[9]
Di contro, nella fibra muscolare scheletrica che lavora in condizioni ipossiche, essendo inibito il complesso della piruvato deidrogenasi o PDC e bloccata fosforilazione ossidativa, perché la glicolisi possa procedere, il piruvato sarà ridotta a lattato con la concomitante ossidazione del NADH a NAD+.[10] Si noti che la conversione del glucosio in acido lattico è definita fermentazione lattica. Tale riduzione è favorita dal fatto che nella fibra muscolare scheletrica predominano isoenzimi con prevalenza della subunità M, quali LDH-4 e LDH-5.[11]
Alanina aminotransferasi
Un’altra fonte di acido piruvico è rappresentata dalla alanina, un aminoacido particolarmente abbondante nelle proteine muscolari.
L’utilizzazione degli scheletri carboniosi degli aminoacidi a fini energetici e anabolici implica la rimozione del gruppo amminico, che si verifica in reazioni catalizzate da enzimi chiamati transaminasi (EC 2.6.1-), e il successivo smaltimento dell’azoto in forma non tossica, attraverso il ciclo dell’urea.[2][8]
La rimozione del gruppo amminico dell’alanina è catalizzata dalla alanina aminotransferasi o ALT (EC 2.6.1.2). La reazione, reversibile, porta alla formazione di piruvato e glutammato.
Sono state identificate due forme della ALT, una a localizzazione citosolica, ALT1, e l’altra presente nella matrice mitocondriale, indicata come ALT2.[4] Ne risulta che l’acido piruvico può essere prodotto per transaminazione dell’alanina anche nella matrice mitocondriale.
L’alanina è uno dei principali precursori gluconeogenetici, e, a mezzo del ciclo glucosio-alanina, rappresenta un collegamento tra il metabolismo degli aminoacidi e quello dei carboidrati.[12]
Nel citosol gli scheletri carboniosi di altri cinque aminoacidi, ossia cisteina, glicina, serina, treonina e triptofano, possono essere convertiti in toto o in parte in piruvato.[2]
Enzima malico
Altra fonte piruvato è il malato.
Nella reazione, catalizzata dall’enzima malico citosolico, una decarbossilazione ossidativa (EC 1.1.1.40), il malato subisce una decarbossilazione ossidativa a dare piruvato.[3]
Malato + NADP+ → Piruvato + CO2 + NADPH + H+
L’enzima svolge un ruolo importante nel trasporto di intermedi del ciclo dell’acido citrico, quali, oltre al malato, l’ossalacetato e il citrato, tra il citosol e la matrice mitocondriale.[7]
Trasportatori mitocondriali del piruvato
Nelle cellule dotate di mitocondri, la maggior parte del piruvato prodotto nel citoplasma passa nella matrice mitocondriale, superando prima la membrana mitocondriale esterna e quindi quella interna.
Una volta nella matrice mitocondriale il piruvato può essere utilizzato sia per fini anabolici che catabolici.
Il passaggio attraverso la membrana mitocondriale esterna avviene per libera diffusione mediata dalle porine o VDAC, dall’inglese voltage dependent anion-selective canne, canali anionici voltaggio-dipendenti relativamente non specifici, nonché le più proteine più abbondanti della membrana mitocondriale esterna, la cui funzione è quella di mediare lo scambio di ioni e piccole molecole, tra cui oltre al piruvato anche l’ATP e il NADH, tra il citosol e lo spazio intermembrana dei mitocondri.[13][14]
La membrana mitocondriale interna è invece impermeabile alle molecole dotate di carica, il che permette il mantenimento del gradiente protonico necessario alla fosforilazione ossidativa. Ne consegue che il passaggio del piruvato necessita di specifici trasportatori, indicati come MPC, dall’inglese mitochondrial pyruvate carriers, un complesso etero-oligomerico di due subunità indicate come MPC1 e MPC2, che operano un simporto di protoni. Questi trasportatori legano quindi il metabolismo citosolico e mitocondriale del piruvato.[15][16]
Complesso della piruvato deidrogenasi
Una volta nella matrice mitocondriale, l’acido piruvico è per la maggior parte ossidato ad anidride carbonica al fine di supportare la produzione di ATP.
Il primo passaggio di questo processo di ossidazione vede l’intervento del complesso della piruvato deidrogenasi, uno tra i più importanti complessi multienzimatici presenti nelle cellule. Il complesso catalizza la decarbossilazione ossidativa del piruvato, con formazione di acetil-CoA e di una molecola di anidride carbonica, con il rilascio di due elettroni, che sono trasportati dal NADH.
Piruvato + CoA + NAD+ → Acetil-CoA + NADH + H+ + CO2
Il gruppo acetilico può ora entrare nel ciclo dell’acido citrico per essere completamente ossidato ad anidride carbonica, con produzione di una molecola di GTP, 3 molecole di NADH, una di FADH2. I due coenzimi ridotti, a mezzo della fosforilazione ossidativa, permetteranno la formazione di ulteriori molecole di ATP.[2]
In alternativa, il gruppo acetilico derivante dal piruvato può essere utilizzato a fini anabolici, tra cui la sintesi di alcuni lipidi quali gli acidi grassi, i fosfolipidi e il colesterolo, o essere utilizzato a fini regolatori a mezzo dell’acetilazione degli istoni.
Poiché la biosintesi dei lipidi si verifica nel citosol e l’acetilazione degli istoni nel nucleo, l’acetil-CoA dovrà lasciare la matrice mitocondriale, processo che necessita della formazione di citrato, a mezzo della condensazione tra il gruppo acetilico e il carbonile dell’ossalacetato, reazione catalizzata dalla citrato sintasi (EC 2.3.3.1). Una volta raggiunto il citosol, a mezzo di un trasportatore del citrato, una proteina integrale della membrana mitocondriale interna, il citrato viene scisso ad acetil-CoA e ossalacetato nella reazione catalizzata dalla citrato liasi (EC 4.1.3.6).[17]
Piruvato carbossilasi
Nella matrice mitocondriale l’acido piruvico può essere carbossilato a ossalacetato in una reazione, irreversibile catalizzata dalla piruvato carbossilasi o PC (EC 6.4.1.1).[8]
Piruvato + HCO3– + ATP ⇄ Ossalacetato + ADP + Pi
Molti intermedi del ciclo dell’acido citrico sono anche precursori per la sintesi di diverse molecole. Ne consegue che ognuno di questi intermedi rimossi dal ciclo dell’acido citrico dovrà essere reintegrato perché il ciclo possa proseguire. Le reazioni che reintegrano il ciclo sono definite anaplerotiche. In quest’ottica, la reazione catalizzata dalla piruvato carbossilasi svolge una funzione anaplerotica catalizzando la formazione di ossalacetato dal piruvato.[6]
^ abcde Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ abcd Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002
^ ab Gray L.R., Tompkins S.C., Taylor E.B. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-604. doi:10.1007/s00018-013-1539-2
^ ab Markert C.L., Shaklee J.B., Whitt G.S. Evolution of a gene. Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science 1975;189(4197):102-14. doi:10.1126/science.1138367
^ ab Owen O.E., Kalhan S.C., Hanson R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 2002;277(34):30409-12. doi:10.1074/jbc.R200006200
^ ab McCommis K.S. and Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J 2015;466(3):443-454. doi:10.1042/BJ20141171
^ abc Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
^ Gleeson T.T. Post-exercise lactate metabolism: a comparative review of sites, pathways, and regulation. Annu Rev Physiol 1996;58:565-81. doi:10.1146/annurev.ph.58.030196.003025
^ Li X., Yang Y., Zhang B., Lin X., Fu X., An Y., Zou Y., Wang J.X., Wang Z., Yu T. Lactate metabolism in human health and disease. Signal Transduct Target Ther 2022;7(1):305. doi:10.1038/s41392-022-01151-3
^ Farhana A., Lappin S.L. Biochemistry, lactate dehydrogenase. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557536/
^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
^ Colombini M. The VDAC channel: molecular basis for selectivity. Biochim Biophys Acta 2016;1863(10):2498-502. doi:10.1016/j.bbamcr.2016.01.019
^ Young M.J., Bay D.C., Hausner G., Court D.A. The evolutionary history of mitochondrial porins. BMC Evol Biol 2007;7:31. doi:10.1186/1471-2148-7-31
^ Bricker D.K., Taylor E.B., Schell J.C., Orsak T., Boutron A., Chen Y.C., Cox J.E., Cardon C.M., Van Vranken J.G., Dephoure N., Redin C., Boudina S., Gygi S.P., Brivet M., Thummel C.S., Rutter J. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 2012;337(6090):96-100. doi:10.1126/science.1218099
^ Herzig S., Raemy E., Montessuit S., Veuthey J.L., Zamboni N., Westermann B., Kunji E.R., Martinou J.C. Identification and functional expression of the mitochondrial pyruvate carrier. Science 2012;337(6090):93-6. doi:10.1126/science.1218530
^ Zara V., Assalve G., Ferramosca A. Multiple roles played by the mitochondrial citrate carrier in cellular metabolism and physiology. Cell Mol Life Sci 2022;79(8):428. doi:10.1007/s00018-022-04466-0
La lattato deidrogenasi o LDH (EC 1.1.1.27) è una famiglia di ossidoreduttasi che catalizza la conversione reversibile del piruvato a lattato, con la concomitante interconversione di NADH e NAD+, i quali agiscono da cofattori.
Sono enzimi a struttura tetramerica, dove ogni subunità è dotata di attività catalitica. Le subunità, codificate da geni distinti, si possono combinare in proporzioni differenti a dare isoenzimi con proprietà cinetiche e regolatorie specifiche.[1]
La lattato deidrogenasi è presente in quasi tutti i tessuti animali e nelle piante, ma anche nei microorganismi. Sebbene sia presente per la maggior parte nel citosol, la sua presenza è stata dimostrata anche nei mitocondri, dove catalizza l’ossidazione del lattato a piruvato, e nei perossisomi.[2][3]
Nell’uomo in tessuti differenti prevalgono isoenzimi differenti, sulla base dello specifico fenotipo metabolico del tessuto.
La lattato deidrogenasi è un enzima cruciale nel metabolismo cellulare, essendo coinvolta nella produzione di energia dai carboidrati in condizioni anaerobiche, nella sintesi del glucosio dal lattato e nell’utilizzo dello scheletro carbonioso del lattato a fini energetici in condizioni aerobiche.[1][4]
Nei mammiferi esistono diversi geni che codificano per le subunità della lattato deidrogenasi, indicati come LDHA, LDHB, LDHC, LDHx e LDHD.
I primi quattro codificano enzimi che riconoscono come substrato gli L-isomeri dell’acido lattico, la principale forma enantiomerica della molecola presente nei vertebrati, e hanno come cofattore il NAD, mentre LDHD codifica per un enzima che riconosce come substrato l’isomero D dell’acido lattico e ha come cofattore il FAD.[5][6]
LDHA, presente sul cromosoma 11p15.4, codifica per subunità M, dall’inglese muscle, mentre LDHB, presente sul cromosoma 12p12.2-p12.1, codifica per la subunità H, dall’inglese heart.[2][7]
LDHC, presente sul cromosoma 11p15.5-p15.3 e probabilmente derivante dalla duplicazione del gene LDHA, codifica per una subunità indicata come C.[8]
Infine, LDHx codifica per la subunità specifica dei perossisomi.[2] LDHx è la forma readthrough del gene LDHB; infatti quello che succede è che il ribosoma, nel momento in cui arriva al codone di stop dell’mRNA per la subunità H inserisce un aminoacido anziché terminare la traduzione, che procede aggiungendo altri sette aminoacidi che costituiscono il targeting perossisomiale di quella che sarebbe una normale subunità H.[3]
Struttura
Gli enzimi appartenenti alla famiglia della lattato deidrogenasi hanno struttura oligomerica, nello specifico sono tetrameri derivanti dall’assemblaggio di subunità, di circa 35 kD, che possono essere di uno stesso tipo o due tipi differenti. Ognuna subunità è dotata di attività catalitica, quindi il tetramero presenta quattro siti attivi. Tuttavia le subunità, prese singolarmente, risultano cataliticamente inattive.[7][9]
Considerando le subunità H e M, le loro strutture primarie sono per circa il 75% identiche, e di conseguenza anche la loro struttura tridimensionale risulta molto simile, ma con piccole differenze a livello del sito di legame per il substrato che portano a grosse differenze nelle proprietà cinetiche delle proteine.[1][10] Altra conseguenza delle differenze nella struttura primaria riguarda la carica netta di superficie che risulta essere di -6 per la subunità M e +1 per la H.[11]
La struttura secondaria è costituita per circa un 40% da alfa-eliche e un 23% da foglietti beta, a formare strutture beta-alfa-beta, con i foglietti beta paralleli come componente principale.[12]
Nell’uomo gli isoenzimi prevalenti sono quelli formati dalle subunità H e M. L’assemblaggio, in proporzioni differenti, delle due subunità porta alla formazione di cinque isoenzimi, indicati come:
La subunità C va a formare una sesta isoforma, omotetramerica, indicata come LDH-6 o C4.[8]
Una settima isoforma deriva dall’assemblaggio della subunità LDHx.[3][8]
Reazione
La lattato deidrogenasi catalizza l’interconversione del piruvato e lattato, che le forme ionizzate rispettivamente dell’acido piruvico e dell’acido lattico, e del NADH e NAD+. In entrambe i casi si verifica la rimozione, da quello che è l’agente riducente, di due atomi di idrogeno, e il successivo trasferimento di uno ione idruro, ossia un protone e due elettroni, all’agente ossidante, mentre il protone rimanente viene rilasciato nel mezzo acquoso. L’enzima è in grado di aumentare la velocità di reazione di 14 volte.[5]
Quando la reazione procede da piruvato a lattato, il primo passaggio è il legame del NADH, l’agente riducente, all’enzima, legame che è seguito da cambiamenti conformazionali che facilitano la formazione di legami idrogeno tra specifici residui delimitanti il sito attivo e il carbonile del piruvato, ossia il C2, e la successiva interazione tra NADH e il piruvato.[13] A questo punto si verifica il trasferimento di uno ione idruro dall’anello nicotinamidico del NADH al C2 del piruvato, che viene quindi ridotto a lattato. Ciò provoca l’ossidazione del coenzima e la neutralizzazione della carica positiva dell’azoto dell’anello nicotinamidico, e la riduzione del piruvato a lattato. Pertanto, in questo caso il C2 del piruvato viene ridotto da chetone ad alcol.
Quando la reazione procede in direzione opposta lo ione idruro viene trasferito dal C2 del lattato, che in questo caso è l’agente riducente, al C-4 dell’anello nicotinamidico. In questo caso il C2 del lattato è ossidato da alcool a chetone.[14][15]
Sito attivo
Il sito attivo delle subunità H e M ha una struttura ben conservata nelle differenti specie, con gli stessi aminoacidi che partecipano alla reazione.[16] Tra questi, non solo per la lattato deidrogenasi umana, ma anche per quella di molte altre specie, c’è His-193, aminoacido che si trova vicino al sito di legame per il coenzima.
Tuttavia, le piccole differenze nella struttura primaria delle due subunità, determinano differenze nelle proprietà biochimiche. Tra le differenze, una in particolare risulta cruciale per le proprietà catalitiche: una alanina della subunità M è sostituita, nella subunità H, da una glutammina, ossia un aminoacido non polare è sostituito da uno polare.[11] Di seguito una breve rassegna delle differenza catalitiche tra le due subunità.
La subunità H lega il substrato più velocemente della subunità M, ma ha, rispetto alla M un’attività catalitica circa cinque volte inferiore.
La subunità M ha una maggiore affinità per il piruvato, favorendo quindi la sintesi di lattato e NAD+, e non è inibita da alte concentrazioni di piruvato. Di contro, la subunità H ha un’affinità maggiore per il lattato, il che favorisce la sintesi di piruvato e NADH, mentre è inibita da elevate concentrazioni di piruvato.[17][18]
Da tutto ciò consegue che le proprietà cinetiche/catalitiche dei differenti isoenzimi dipendono dalla prevalenza di una delle due subunità.[19]
Regolazione ad opera del piruvato e lattato
Gli isoenzimi della lattato deidrogenasi sono soggetti a inibizione da parte del piruvato e lattato.
Gli isoenzimi dove predomina la subunità H sono inibiti da concentrazioni di piruvato più basse rispetto a quelli dove predomina la subunità M. Ad esempio, H4 è inibito da concentrazioni di piruvato pari a circa 0,2 mM, mentre M4 è debolmente inibito dal piruvato fino a concentrazioni di 5 mM.
Di contro, H4 è inibito dal lattato in concentrazione superiori a 20-40 mM mentre M4 è inibito in misura minore da elevate concentrazioni di lattato.[20]
Distribuzione tissutale
Nell’uomo i differenti isoenzimi della lattato deidrogenasi hanno localizzazione tissutali preferenziali, che in genere riflettono il fenotipo metabolico del tessuto.
Infatti, tessuti con un fenotipo metabolico prevalentemente o esclusivamente aerobico, come il cuore, producono in misura maggiore isoenzimi a prevalenza della subunità H.[21]
Al contrario, nei tessuti dove il metabolismo anaerobico è importante, come fibre muscolari scheletriche nel corso di un esercizio inteso, o alcune zone scarsamente ossigenate nei tumori solidi, sono prodotti isoenzimi con prevalenza della subunità M.[5]
Esistono tuttavia eccezioni, come ad esempio il fegato, organo con fenotipo metabolico aerobico dove predomina LDH-5, ma dove l’ossidazione del lattato a piruvato, che può essere considerata come parte del ramo epatico del ciclo di Cori, è favorita dal basso rapporto NADH/NAD+ presente nel citosolico dell’epatocita.[13]
Di seguito una breve panoramica.
LDH-1 o H4: muscolo cardiaco, rene e globuli rossi;
LDH-2 o M1H3 ha una distribuzione simile a LDH-1;
LDH-3 o M2H2: milza, cervello, globuli bianchi, rene e polmone;
LDH-4 o M3H1: milza, polmone, muscolo scheletrico, globuli rossi e rene;
LDH-5 o M4: fegato, muscolo scheletrico e polmone.
LDH-6: sperma e testicolo
Nei singoli organi, LDH-1 e LDH-2 predominano nel cuore, nei reni e nei globuli rossi, LDH-3 nei polmoni, LDH-4 e LDH-5 nel muscolo scheletrico e LDH-5 nel fegato.
Gli isoenzimi presenti nel siero, e il cui dosaggio viene utilizzato a fini diagnostici, sono sempre di derivazione tissutale.[5]
Infine, in uno stesso tessuto/organo, i differenti isoenzimi possono essere presenti in quantità significative in differenti tipi cellulari, come ad esempio accade nel muscolo scheletrico, nel cervello, ma anche nei tumori solidi.
Ruolo
La lattato deidrogenasi è un enzima essenziale sia per la produzione che l’utilizzazione/rimozione del lattato.[4]
In condizioni ipossiche la cellula ricava l’ATP dall’ossidazione anaerobica del glucosio. A mezzo della via glicolitica il monosaccaride viene ossidato a due molecole di piruvato, ricavando da ciò 2 molecole di ATP e due di NADH. Tuttavia, affinché la glicolisi possa procedere, il NADH prodotto dovrà essere riossidato a NAD+, essendo il coenzima ossidato necessario alla reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12). Poiché in condizioni anaerobiche il complesso della piruvato deidrogenasi inibito e la fosforilazione ossidativa bloccata, l’ossidazione del NADH è portata a termine dalla reazione catalizzata dalla lattato deidrogenasi, che quindi consente alla glicolisi di procedere. Questa via metabolica porta quindi alla produzione di lattato dal glucosio, ed è nota come fermentazione lattica.[13][22]
L’acido lattico è spesso considerato un prodotto finale del metabolismo dei glucosio, e il suo accumulo nel corpo risulta essere pericoloso, potendo essere causa di acidosi lattica.[22] Ne consegue che la molecola deve essere rapidamente rimossa dai tessuti e dal circolo. Considerando ad esempio il lattosio prodotto dai globuli rossi, o quello prodotto dalle fibre muscolari che lavorano in condizioni di scarso apporto di ossigeno, come durante un esercizio intenso, questi può raggiungere, trasportato dal circolo ematico, tra gli altri, il fegato e il cuore. Nel fegato, sebbene LDH-5 favorisca la riduzione del lattato a piruvato, il basso rapporto NADH/NAD+ citosolico sposta l’equilibrio della reazione verso la formazione del piruvato, che potrà essere utilizzato per la produzione di energia, o entrare nella gluconeogenesi.[23] Nel cardiomiocita invece, l’isoenzima prevalente, LDH-1, ossida il lattato a piruvato che sarà utilizzato a fini energetici.
Bibliografia
^ abc Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002
^ abc Markert C.L., Shaklee J.B., Whitt G.S. Evolution of a gene. Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science 1975;189(4197):102-14. doi:10.1126/science.1138367
^ abc Schueren F., Lingner T., George R., Hofhuis J., Dickel C., Gärtner J., Thoms S. Peroxisomal lactate dehydrogenase is generated by translational readthrough in mammals. Elife 2014;3:e03640. doi:10.7554/eLife.03640
^ abc Laughton J.D., Charnay Y., Belloir B., Pellerin L., Magistretti P.J., Bouras C. Differential messenger RNA distribution of lactate dehydrogenase LDH-1 and LDH-5 isoforms in the rat brain. Neuroscience 2000;96(3):619-25. doi:10.1016/s0306-4522(99)00580-1
^ abcd Farhana A., Lappin S.L. Biochemistry, lactate dehydrogenase. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557536/
^ Jin S., Chen X., Yang J., Ding J. Lactate dehydrogenase D is a general dehydrogenase for D-2-hydroxyacids and is associated with D-lactic acidosis. Nat Commun 2023;14(1):6638. doi:10.1038/s41467-023-42456-3
^ ab Krieg A.F., Rosenblum L.J., Henry J.B. Lactate dehydrogenase isozymes a comparison of pyruvate-to-lactate and lactate-to-pyruvate assays. Clin Chem 1967;13(3):196-203. doi:10.1093/clinchem/13.3.196
^ abc Zinkham W.H., Blanco A., Clowry L.J. Jr. An unusual isozyme of lactic dehydrogenase in mature testes: localization, ontogeny, and kinetic properties. Ann N Y Acad Sci 1964;121:571-88. doi:10.1111/j.1749-6632.1964.tb14227.x
^ Bellamacina C.R. The nicotinamide dinucleotide binding motif: a comparison of nucleotide binding proteins. FASEB J 1996;10(11):1257-69. doi:10.1096/fasebj.10.11.8836039
^ ab Read J.A., Winter V.J., Eszes C.M., Sessions R.B., Brady R.L. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins 2001;43(2):175-85.
^ Auerbach G., Ostendorp R., Prade L., Korndörfer I., Dams T., Huber R., Jaenicke R. Lactate dehydrogenase from the hyperthermophilic bacterium thermotoga maritima: the crystal structure at 2.1 A resolution reveals strategies for intrinsic protein stabilization. Structure 1998;6(6):769-81. doi:10.1016/s0969-2126(98)00078-1
^ abc Forkasiewicz A., Dorociak M., Stach K., Szelachowski P., Tabola R., Augoff K. The usefulness of lactate dehydrogenase measurements in current oncological practice. Cell Mol Biol Lett 2020;25:35. doi:10.1186/s11658-020-00228-7
^ Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
^ Nelson D.L., M. M. Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ Holmes R.S., Goldberg E. Computational analyses of mammalian lactate dehydrogenases: human, mouse, opossum and platypus LDHs. Comput Biol Chem 2009;33(5):379-85. doi:10.1016/j.compbiolchem.2009.07.006
^ Feng Y., Xiong Y., Qiao T., Li X., Jia L., Han Y. Lactate dehydrogenase A: a key player in carcinogenesis and potential target in cancer therapy. Cancer Med 2018;7(12):6124-6136. doi:10.1002/cam4.1820
^ Gray L.R., Tompkins S.C., Taylor E.B. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-604. doi:10.1007/s00018-013-1539-2
^ Rogatzki M.J., Ferguson B.S., Goodwin M.L., Gladden L.B. Lactate is always the end product of glycolysis. Front Neurosci 2015;9:22. doi:10.3389/fnins.2015.00022
^ Stambaugh R., Post D. Substrate and product inhibition of rabbit muscle lactic dehydrogenase heart (H4) and muscle (M4) isozymes. J Biol Chem 1966;241(7):1462-7. doi:10.1016/S0021-9258(18)96733-5
^ Wroblewski F, Gregory KF. Lactic dehydrogenase isozymes and their distribution in normal tissues and plasma and in disease states. Ann N Y Acad Sci 1961;94:912-32. doi:10.1111/j.1749-6632.1961.tb35584.x
^ ab Li X., Yang Y., Zhang B., Lin X., Fu X., An Y., Zou Y., Wang J.X., Wang Z., Yu T. Lactate metabolism in human health and disease. Signal Transduct Target Ther 2022;7(1):305. doi:10.1038/s41392-022-01151-3
^ Gleeson T.T. Post-exercise lactate metabolism: a comparative review of sites, pathways, and regulation. Annu Rev Physiol 1996;58:565-81. doi:10.1146/annurev.ph.58.030196.003025
I chetoacidi sono composti organici contenenti due gruppi funzionali: un gruppo carbossilico (−COOH) e uno carbonilico (˂C=O).
In base alla posizione del gruppo carbonilico rispetto a gruppo carbossilico, cui le regole della nomenclatura IUPAC assegnano la priorità più alta, sono suddivisi in alfa-chetoacidi, beta-chetoacidi, e gamma-chetoacidi.[1]
I chetoacidi, e in particolare gli alfa-chetoacidi, sono molto importanti in biochimica, essendo coinvolti in molte vie metaboliche.
Alfa-chetoacidi
Negli alfa-chetoacidi il gruppo carbonilico è adiacente al carbonio carbossilico. Molti di questi composti, in forma delle rispettive basi coniugate, svolgono importanti funzioni biologiche. Di seguito alcuni esempi.[2]
L’acido piruvico, il più semplice alfa-chetoacido, è il prodotto finale della glicolisi.
L’acido ossalacetico e l’acido alfa-chetoglutarico sono intermedi del ciclo dell’acido citrico.
Gli alfa-chetoacidi possono derivare da reazioni di transaminazione e di deaminazione ossidativa di aminoacidi. Nelle reazioni di transaminazione il gruppo amminico in alfa dell’amminoacido viene trasferito a un alfa-chetoacido, di solito l’alfa-chetoglutarato, con formazione di un nuovo amminoacido e di un alfa-chetoacido. Queste reazioni sono catalizzate da enzimi detti transaminasi o aminotransferasi (EC 6.1.-).
alfa-Chetoacido + Aminoacido ⇄ Nuovo aminoacido + Nuovo alfa-chetoacido
Nelle reazioni di deaminazione ossidativa gli amminoacidi sono convertiti nei corrispettivi alfa-chetoacidi a mezzo dell’eliminazione del gruppo amminico, che è convertito in ammoniaca e sostituito da un gruppo carbonilico. Essendo le reazione reversibile gli alfa-chetoacidi sono anche precursori degli aminoacidi.
Nota: l’ammoniaca è un composto tossico e, nel fegato, viene convertita in urea attraverso il ciclo dell’urea.[2]
Piruvato, ossalacetato e alfa-chetoglutarato, quest’ultimo via ossalacetato, sono i punti di ingresso nella gluconeogenesi degli scheletri carboniosi di molti amminoacidi glucogenici.[3]
E’ stato inoltre osservato che, in vitro, linee cellulari tumorali murine e umane rilasciano nel microambiente tumorale 2-chetoacidi quali α-chetoisocaproato, l’α-cheto-β-metilvalerato e l’α-chetoisovalerato, molecole in grado di influenzare l’attività antitumore dei macrofagi.[4]
Beta-chetoacidi
Nei beta-chetoacidi il gruppo carbonilico è sul secondo carbonio dopo il carbonio carbossilico.
Esempi sono l’acido acetoacetico, il più semplice 3-chetoacido, e l’acido β-idrossibutirrico, i quali sono due dei tre corpi chetonici, assieme all’acetone, prodotti dall’epatocita quando l’acetil-CoA è prodotto in eccesso rispetto alla capacità della citrato sintasi (EC 2.3.3.1 ), quindi del ciclo dell’acido citrico, di ossidarlo, situazione che si presenta durante il digiuno prolungato o diete molto povere in carboidrati.
Da notare che acetoacetil-CoA e beta-idrossibutirril-CoA, ossia le due forme attivate dei rispettivi beta-chetoacidi, sono intermedi della via di sintesi dell’acido butirrico seguita dalla maggior parte dei batteri del microbiota intestinale produttori dell’acido grasso.[5][6][7]
Gamma-chetoacidi
Nei gamma-chetoacidi il gruppo carbonilico è sul terzo carbonio dopo il carbonio carbossilico. Un esempio è l’acido levulinico, il più semplice gamma-chetoacido, che deriva dal catabolismo della cellulosa.
^ ab Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ D’Andrea G. Classifying amino acids as gluco(glyco)genic, ketogenic, or both. Biochem Educ 2000;28(1):27-28. doi:10.1016/s0307-4412(98)00271-4
^ Cai Z., Li W., Brenner M., Bahiraii S., Heiss E.H., Weckwerth W. Branched-chain ketoacids derived from cancer cells modulate macrophage polarization and metabolic reprogramming. Front Immunol 2022;13:966158. doi:10.3389/fimmu.2022.966158
^ Miller T.L., Wolin M.J. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl Environ Microbiol 1996;62(5):1589-92. doi:10.1128/aem.62.5.1589-1592
^ Portincasa P., Bonfrate L.,Vacca M., De Angelis M., Farella I., Lanza E., Khalil M.,Wang D.Q.-H., Sperandio M., Di Ciaula A. Gut microbiota and short chain fatty acids: implications in glucose homeostasis. Int J Mol Sci 2022;23:1105. doi:10.3390/ijms23031105
^ Pryde S.E., Duncan S.H., Hold G.L., Stewart C.S., Flint H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett 2002;217(2):133-9. doi:10.1111/j.1574-6968.2002.tb11467.x
Un ciclo futile, o ciclo del substrato, si instaura quando due reazioni opposte, non all’equilibrio e catalizzate da enzimi differenti, o due vie metaboliche opposte avvengono simultaneamente senza altro effetto complessivo se non un dispendio di energia.[1]
Il nome di ciclo futile fu coniato in quanto all’apparenza questi cicli sembravano non portare alcun beneficio alla cellula, una sorta di imperfezione metabolica.[2]
Tuttavia, recentemente, sono stati riconosciuti come importanti per la produzione di calore, l’amplificazione dei segnali metabolici, la redistribuzione di energia, in forma di trigliceridi, tra tessuto adiposo e fegato, e per le modificazioni a carico degli acidi grassi immagazzinati nei trigliceridi del tessuto adiposo.[3][4][5]
Per evitare un dispendio incontrollato di energia, i cicli futili sono strettamente controllati.
Esempi di cicli futili sono la glicolisi e la gluconeogenesi quando procedono simultaneamente a elevata velocità nella stessa cellula, il ciclo di Cori, e il ciclo trigliceridi/acidi grassi.[6]
Consideriamo una via ciclica che da A porta a B e viceversa. Supponiamo che il tasso di conversione di A in B sia 100, e quella di B in A 90. Ne risulta un flusso netto di A verso B di 10. Si immagini ora un effettore che aumenti la conversione di A in B del 30%, portandola a 130, e contemporaneamente riduca del 30% la conversione di B in A, portandola a 63. Il flusso netto risultante è pari a 130-63 = 67. Ne risulta un aumento del flusso netto pari a 570% a seguito di una variazione del 30% nella velocità delle reazioni opposte. Modifiche di questo tipo potrebbero, almeno in parte, spiegare l’aumento anche di 1000 volte del flusso di carbonio lungo la glicolisi nella fase iniziale di un esercizio intenso.[2]
Regolazione
Nel corso dell’evoluzione, la selezione di enzimi differenti per la catalisi di reazioni irreversibili e contrapposte ha permesso di evitare l’instaurarsi di cicli futili o di metterli sotto stretto controllo. In che modo? La selezione di un enzima che catalizza la conversione di A in B, e di un secondo enzima che catalizza la conversione di B in A, le cui attività sono regolate separatamente, permette il controllo della direzione netta del flusso metabolico.[7]
Il controlla delle attività enzimatiche può avvenire mediante:
meccanismi allosterici;
modificazioni covalenti;
modificazioni nella concentrazione degli enzimi coinvolti, conseguenti a variazioni del rapporto tra la loro sintesi e degradazione. Un meccanismo differente regola la glucochinasi epatica (EC 2.7.1.2). Durante il digiuno, l’enzima si lega in modo reversibile alla GKPR, una delle proteine specifiche del fegato, che lo ancora all’interno del nucleo, separandolo dagli altri enzimi glicolitici, e impedendo così l’instaurarsi di un ciclo futile tra glicolisi e gluconeogenesi.
Grazie a questi meccanismi di controllo è così possibile ottenere una regolazione coordinata delle due vie contrapposte, evitando un ciclo futile incontrollato. Ovviamente, una regolazione così fine non potrebbe essere ottenuta se un singolo enzima operasse in entrambe le direzioni.[8]
Glicolisi e gluconeogenesi
Considerando la glicolisi e la gluconeogenesi, la prima via porta alla conversione del glucosio in piruvato, la base coniugata dell’acido piruvico, con produzione di ATP, mentre la seconda converte il piruvato in glucosio, con consumo di ATP. Se le due vie procedessero simultaneamente ad alta velocità nella stessa cellula si avrebbe solo un consumo netto di ATP, quindi un ciclo futile. Ciò è evitato attraverso il controllo delle tappe irreversibili dei due processi, in particolare delle reazioni catalizzate dalla fosfofruttochinasi-1 o PFK-1 (EC 2.7.1.11) e dalla fruttosio-1,6-bisfosfatasi o FBPasi (EC 3.1.3.11), principalmente ad opera dall’effettore allosterico fruttosio 2,6-bifosfato.[7]
Da notare che mentre nella glicolisi il controllo allosterico coinvolge tutte e tre le reazioni irreversibili, nella gluconeogenesi i punti di regolazione cruciali sono le reazioni catalizzate dalla piruvato carbossilasi (EC 6.4.1.1) e dalla fruttosio-1,6-bisfosfatasi.[8]
Ciclo di Cori
Con il ciclo di Cori una parte dell’acido lattico prodotto dal muscolo e da altri tessuti extraepatici raggiunge il fegato, dove è convertito in glucosio che, rilasciato in circolo, torna al muscolo e ad altri tessuti extraepatici chiudendo il ciclo. Dal punto di vista energetico il ciclo di Cori può essere considerato un ciclo futile perché per ogni ciclo si ha come unico effetto apparente un consumo netto di 4 ATP, come per il ciclo glicolisi/gluconeogenesi. Tuttavia, questo ciclo permette il funzionamento di molti differenti tipi di cellule extraepatiche a spese del fegato.[6]
Ciclo trigliceridi/acidi grassi
Nel ciclo trigliceridi/acidi grassi, i trigliceridi nel tessuto adiposo vengono parzialmente o completamente idrolizzati in acidi grassi liberi e glicerolo, in un processo chiamato lipolisi; quindi, gli acidi grassi rilasciati vengono utilizzati per sintetizzare nuove molecole di trigliceridi.[3][6][7] Per ogni mole di trigliceridi che completa il ciclo vengono consumate 4 moli di ATP.
Questo ciclo futile può avvenire:
tra il tessuto adiposo, che rilascia acidi grassi, e il fegato, che li riesterifica in trigliceridi, portando a una ridistribuzione dell’energia immagazzinata;[4]
negli adipociti, dove può contribuire alla termogenesi e alla modifica degli acidi grassi immagazzinati.
Per quanto riguarda le modifiche degli acidi grassi immagazzinati, il ciclo li rende accessibili a reazioni di allungamento e desaturazione, che consentono la conversione degli acidi grassi saturi in acidi grassi insaturi. Tuttavia, sembra che l’efficienza del processo dipenda dal tipo di acidi grassi. Ad esempio, il metabolismo degli acidi grassi a catena media è più rapido della conversione dell’acido palmitico, uno degli acidi grassi a catena lunga, negli acidi palmitoleico e oleico, e poi, negli epatociti, in acido arachidonico. Si noti che la conversione degli acidi grassi a catena media e dell’acido palmitico in acidi grassi insaturi a catena lunga riduce il rischio per la salute associato al loro accumulo nei trigliceridi immagazzinati.[5]
Generazione di calore
In alcuni casi un ciclo futile ha la sola funzione di produrre calore a mezzo dell’idrolisi di ATP.
Questo si verifica ad esempio nei muscoli alari dei bombi, i quali, per volare devono mantenere una temperatura toracica di circa 30 °C, anche quando la temperatura esterna è bassa, fino anche a 10 °C. La temperatura toracica è mantenuta ai livelli ottimali per consentire il volo grazie al ciclo futile che si instaura tra le reazioni catalizzate dagli enzimi PFK-1 e FBPasi. Infatti, la FBAasi dei muscoli alari non è inibita dall’AMP, il che sembra suggerire che nel corso dell’evoluzione questa proteina sia stata selezionata appositamente per la produzione di calore.
Al contrario dei bombi, nei muscoli alari delle api mellifere non è quasi presente la FBPasi, e pertanto questi insetti non possono volare quando la temperatura esterna è bassa.[2]
Bibliografia
^ Newsholme E.A. Substrate cycles: their metabolic, energetic and thermic consequences in man. Biochem Soc Symp. 1978;43:183–205.
^ abc Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002
^ ab Reshef L., Olswang Y., Cassuto H., Blum B., Croniger C.M., Kalhan S.C., Tilghman S.M., Hanson R.W. Glyceroneogenesis and the triglyceride/fatty acid cycle. J Biol Chem 2003;278(33):30413-6. doi:10.1074/jbc.R300017200
^ ab Sharma A.K., Wolfrum C. Lipid cycling isn’t all futile. Nat Metab 2023;5(4):540-541. doi:10.1038/s42255-023-00779-x
^ ab Wunderling K., Zurkovic J., Zink F., Kuerschner L., Thiele C. Triglyceride cycling enables modification of stored fatty acids. Nat Metab 2023;5(4):699-709. doi:10.1038/s42255-023-00769-z
^ abc Brownstein A.J., Veliova M., Acin-Perez R., Liesa M., Shirihai O.S. ATP-consuming futile cycles as energy dissipating mechanisms to counteract obesity. Rev Endocr Metab Disord 2022;23(1):121-131. doi:10.1007/s11154-021-09690-w
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
Gli acidi grassi a catena corta o SCFAs sono acidi grassi saturi con catena carboniosa lineare o ramificata composta da un numero di atomi di carbonio compreso tra 2 e 5, e sono l’acido acetico, l’acido propionico, l’acido butirrico, l’acido isobutirrico, l’acido valerico, l’acido isovalerico, e l’acido 2-metilbutirrico.[1]
Nell’uomo sono, con i sali biliari secondari, i principali metaboliti prodotti dai batteri del microbiota intestinale nel cieco e nel colon, e derivano quasi interamene dalla fermentazione anerobica di carboidrati non digeribili.[11] I più abbondanti sono l’acido acetico, l’acido propionico e l’acido butirrico, che costituiscono circa il 95% del totale degli acidi grassi a catena corta prodotti.[6] La percentuale rimanente è costituita da quelli a catena ramificata.
Sono i principali anioni presenti nel colon. La loro concentrazione è maggiore nel cieco e nella parte prossimale del colon rispetto a quella distale, dove i substrati per la loro sintesi vanno esaurendosi.[1][2][4] Sono in grado di ridurre il valore del pH del lume intestinale e quindi acidificare le feci.
Circa il 90-95% viene assorbito nel cieco e nel colon, mentre il restante 5-10% viene escreto con le feci.[13]
Si ritiene forniscano circa il 70% del fabbisogno energetico necessario ai colonociti.[4]
Gli acidi grassi a catena corta sono in grado di modulare sia la fisiologia che la composizione del microbiota intestinale.[7] Inoltre, un numero crescente di ricerche suggerisce che svolgano un ruolo importante nel concorrere al mantenimento della salute dell’uomo.[4]
Al pari degli acidi grassi a catena media e degli acidi grassi a catena lunga, gli acidi grassi catena corta sono presenti nei tessuti animali e vegetali principalmente in forma di trigliceridi, sebbene in quantità molto inferiori rispetto a quelli a catena lunga.
Nell’adulto la principale fonte alimentare è costituita da latte e derivati, dove l’acido butirrico è quello presente in concentrazione maggiore. Altre fonti sono alcuni oli vegetali, come l’olio di cocco e l’olio di palmisti.
Nei neonati allattati al seno la principale fonte è il latte materno.[10]
Tuttavia, per l’uomo, e per la maggior dei mammiferi, la fonte più importante è costituita dalla fermentazione anaerobica delle fibre e dell’amido resistente, ossia di carboidrati indigeribili, ad opera dei batteri del microbiota intestinale.[6] Questa via porta alla produzione di circa 500-600 mM di SCFAs al giorno. Gli acidi acetico, propionico e butirrico sono presenti in un rapporto molare rispettivamente di circa 60:20:20, sebbene la proporzione relativa di ciascuno essi dipenda dal substrato, dalla composizione del microbiota, e dal tempo di transito intestinale.[7][11]
Proprietà
Gli acidi grassi a catena corta hanno catene carboniose composte da un numero di atomi di carbonio compreso tra 2 e 5, caratteristica che influenza fortemente le loro proprietà fisiche.[9]
L’acido acetico, l’acido butirrico, l’acido propionico e l’acido valerico sono acidi grassi a catena lineare, mentre l’acido isobutirrico, l’acido isovalerico e l’acido 2-metil butirrico sono acidi grassi a catena ramificata.
Sono molecole di piccole dimensioni, e tra i lipidi sono i più piccoli.
A temperatura ambiente sono liquidi, e sono solubili in solventi polari come l’acqua, differendo in questo dagli acidi grassi con catene carboniose più lunghe, la cui solubilità in solventi polari, considerando quelli a catena lineare, va diminuendo all’aumentare della lunghezza della catena stessa, essendo questa la parte della molecola dotata di carattere non polare, a differenza del gruppo carbossilico che è polare.[10]
Va infine notato che l’acido butirrico e l’acido isobutirrico, che hanno formula molecolare C4H8O2, sono un esempio di isomeria di catena, al pari dell’acido valerico, dell’acido isovalerico e dell’acido 2-metilbutirrico, i quali hanno formula molecolare C5H10O2.
Effetti sulla salute
Gli acidi grassi a catena corta sono ritenuti svolgere un ruolo cruciale nel mantenimento della salute dell’uomo.[4] La loro azione sembra esplicarsi attraverso effetti diretti e indiretti su processi cellulari come la proliferazione, la differenziazione e l’espressione genica, concorrendo in questo modo alla regolazione di processi quali l’omeostasi del glucosio, la funzionalità intestinale e immunitaria, e la regolazione dell’asse microbiota-intestino-cervello.[6] Tutto ciò sembra essere confermato anche da studi che mostrano come una disbiosi intestinale appaia essere implicata in patologie metaboliche, come disturbi che coinvolgono l’omeostasi del glucosio, e comportamentali e neurologiche, come l’Alzheimer, il Parkinson, e la depressione.[11]
Sintesi
Nell’uomo, il corredo enzimatico responsabile della digestione dei carboidrati è privo degli enzimi in grado di digerire le fibre e l’amido resistente, quest’ultimo così chiamato proprio perché resiste all’azione idrolitica della alfa-amilasi. Al contrario, i batteri del microbiota intestinale codificano per un gran numero di differenti glicoside idrolasi, oltre 260, grazie alle quali sono in grado di liberare monosaccaridi dalle fibre e dall’amido resistente.[7] Gli esosi e i desossiesosi entrano nella glicolisi, e i pentosi nella via del pentoso fosfato, a dare il piruvato, la base coniugata dell’acido piruvico, che è il principale precursore per la sintesi degli acidi grassi a catena corta.[2][4][5]
La sintesi degli SCFAs è influenzata da diversi fattori, alcuni dei quali sono di seguito elencati.[4][11][13]
Il contenuto di fibre della dieta. Ad esempio, una dieta ricca di fibre, come la dieta mediterranea, può influenzarne la sintesi.
La composizione del microbiota intestinale.
Il pH del lume intestinale, in quanto per valori di circa 5,5 dominano i batteri che producono acido butirrico, mentre per valori di circa 6,5 dominano i batteri che producono acido acetico e acido propionico.
La velocità di transito intestinale.
La quantità di ossigeno nel lume intestinale.
L’acido acetico e l’acido propionico sono prodotti principalmente da specie appartenenti al phylum Bacteroides, mentre l’acido butirrico, per la cui sintesi è particolarmente importante l’amido resistente, da specie batteriche appartenenti al phylum Firmicutes.[6]
Sintesi dell’acido acetico
L’acido acetico, il più abbondante tra gli SCFAs presenti nel colon, può essere sintetizzato attraverso la via di Wood-Ljungdahl nella direzione riduttiva, a mezzo della riduzione della CO2 ad acetato, o dall’acetil-CoA, la via metabolica più importante essendo responsabile della produzione di circa due terzi dell’acido grasso presente nel lume intestinale.[4]
Sintesi dell’acido propionico
L’acido propionico può essere sintetizzato attraverso tre differenti vie metaboliche: le vie dell’acrilato e del succinato, che utilizzano come precursore l’acido lattico prodotto da altri batteri, e la via del propandiolo, nella quale i precursori sono desossiesosi.[2][3][10]
Nella via dell’acrilato, l’acido lattico viene convertito in propionil-Coa via lattoil-CoA. Nel passaggio finale il propionil-Coa è idrolizzato a dare acido propionico.
Nella via del succinato, il lattato viene ridotto a piruvato, che a sua volta viene carbossilato a ossalacetato, che è convertito in propionil-CoA attraverso una via metabolica che ha come intermedi il malato, il fumarato, il succinato e il metilmalonil-CoA. Infine il propionil-CoA viene idrolizzato a coenzima A e acido propionico. La via del succinato è considerata la via dominante per la sintesi dell’acido propionico nell’intestino.
Nella via del propandiolo, alcuni desossiesosi, come il fucosio e il ramnosio, sono convertiti, via 1,2-propandiolo, in propionil-Coa e quindi in acido propionico.
Sintesi dell’acido butirrico
La sintesi dell’acido butirrico può seguire due vie.[7][10]
Nella maggior parte dei batteri produttori di acido butirrico, l’acido grasso a catena corta è prodotto attraverso una via metabolica che ha inizio con la condensazione di due molecole di acetil-CoA a dare acetoacetil-CoA. Di seguito l’acetoacetil-CoA viene convertito in beta-idrossibutirril-CoA, crotonil-CoA, e butirril-CoA. Nell’ultima tappa si ha la liberazione di acido butirrico dal butirril-CoA.[8]
In un numero ridotto di specie batteriche il butirril-CoA viene convertito in butirrilfosfato, da cui viene infine liberato acido butirrico.[2]
Sintesi dagli aminoacidi
L’acido acetico, l’acido propionico e l’acido butirrico possono essere prodotti anche a partire da aminoacidi derivanti dalla degradazione di peptidi e proteine, sebbene la quantità prodotta da queste vie sia minima.[7]
Questi processi si verificano nella parte distale del colon, spesso ad opera di batteri non commensali.[3] Dal metabolismo di aminoacidi differenti sono prodotti acidi grassi a catena corta diversi; di seguito alcuni esempi.[4]
Dall’acido glutammico sono prodotti principalmente acido acetico e acido butirrico.
Dall’acido aspartico derivano principalmente acido acetico e acido propionico.
Dagli aminoacidi basici lisina, arginina e istidina sono prodotti acido acetico e acido butirrico.
Dalla cisteina derivano gli acidi acetico, propionico e butirrico.
Dal metabolismo della metionina derivano principalmente l’acido propionico e l’acido butirrico.
Dagli aminoacidi ramificati leucina, isoleucina e valina si originano acidi grassi a catena corta ramificati.
Il pH del lume intestinale influenza il metabolismo delle proteine da parte del microbiota intestinale, a partire dalla loro degradazione negli aminoacidi costituenti, più probabile a pH neutro o debolmente alcalino.
Si noti che dal metabolismo intestinale degli aminoacidi sono prodotti anche composti potenzialmente tossici, quali ammoniaca, solfiti e fenoli.
Sintesi endogena
L’uomo, e più in generale i mammiferi, hanno il corredo enzimatico per la sintesi endogena di acidi grassi a catena corta. La sintesi, prevalentemente epatica, avviene a mezzo di cicli di reazioni di beta-ossidazione che portano alla formazione di acil-CoA con catena carboniosa via via più corta dell’acido grasso di partenza. L’acido grasso legato al coenzima A è quindi rilasciato a seguito dell’idrolisi operata da una acil-CoA tioesterasi (EC 3.1.2.20).[10][12]
Recettori di membrana
Gli acidi grassi a catena corta sono in grado di legarsi a specifici recettori presenti sulla membrana plasmatica, i recettori accoppiati alle proteine G, tra cui GPR41, GPR43 e GPR109A.[6]
Gli effetti del legame ai recettori dipendono dal tipo di cellula. Ad esempio, il legame ai recettori presenti sulle cellule L dell’intestino è associato al rilascio di GLP-1, o glucagon-like peptide-1, e del peptide YY, ormoni che hanno effetto sull’appetito e sull’assunzione di cibo. Il legame alle cellule enterocromaffini induce il rilascio di serotonina, il che potrebbe avere effetto sulla motilità intestinale. Infine il legame ai recettori presenti sulle cellule beta del pancreas aumenta il rilascio di insulina.[11]
I diversi acidi grassi a catena corta hanno capacità differenti di attivare i recettori: GPR43 è più probabile sia attivato dall’acido acetico e dall’acido propionico, GPR41 dall’acido propionico e dall’acido butirrico, mentre GPR109A dall’acido butirrico.[4]
Assorbimento
Circa il 90% degli acidi grassi a catena corta presenti nel lume intestinale è assorbito dai colonociti. Il passaggio attraverso la membrana plasmatica può avvenire per diffusione passiva o a mezzo di un trasporto attivo mediato da due tipi di trasportatori di membrana: i trasportatori per i monocarbossilati H+-dipendenti e quelli Na+-dipendenti, indicati rispettivamente come MCTs e SMCTs.[6][7]
Il trasporto passivo interessa le forme protonate degli SCFAs, quindi è influenzato dal pH presente nel colon. Una debole acidificazione del lume intestinale, che può essere conseguente all’attività metabolica dei microrganismi presenti, aumenta la prevalenza della forma protonata e quindi del trasporto passivo.[10]
Funzioni nel colonocita
Nei colonociti, gli acidi grassi a catena corta possono essere utilizzati a scopi energetici e regolatori.
Quando utilizzati a fini energetici, l’acido acetico e l’acido butirrico sono convertiti in acetil-CoA, e l’acido propionico in propionil-CoA. Attraverso la produzione di ATP contribuiscono al mantenimento dell’omeostasi cellulare, ma anche, ad esempio, al mantenimento dell’integrità delle giunzioni strette presenti agli apici cellulari, e quindi dell’integrità della barriera intestinale.[11] Dei tre principali acidi grassi a catena corta prodotti nell’intestino, l’acido butirrico è la principale fonte di energia per i colonociti, mentre gli acidi acetico e propionico sono scarsamente metabolizzati e per la maggior parte drenati dalla vena porta.[2][13]
A fini regolatori gli SCFAs sono ad esempio in grado di inibire l’attività delle istone deacetilasi (EC 3.5.1.98), enzimi che catalizza la rimozione dei gruppi acetilici da residui di lisina delle proteine istoniche, gruppi acetilici precedentemente inseriti dalla istone acetiltransferasi (EC 2.3.1.48).[7] I gruppi R delle lisine deacetilate portano cariche positive, il che fa si che le proteine istoniche avvolgano più strettamente il DNA, che porta cariche negative. Ciò rende il nucleosoma più compatto, e di conseguenza più difficile eseguire la trascrizione e l’espressione genica. I diversi acidi grassi a catena corta hanno differenti capacità di inibire l’attività delle istone deacetilasi:
fino all’80% per l’acido butirrico;
fino al 60% per l’acido propionico;
l’acido acetico ha la minor capacità inibitoria.[4]
Questa modalità d’azione sulle istone deacetilasi è stata osservata non solo nell’intestino e nel tessuto immunitario associato, ma anche nel sistema nervoso centrale e periferico.[11]
Trasporto
Gli acidi grassi a catena corta che non sono stati utilizzati nel colonocita lasciano la cellula per diffusione passiva e trasporto attivo, a livello della membrana basolaterale, per entrare nella circolazione portale, dove la concentrazione maggiore è raggiunta dall’acido acetico, circa 260 mM/L, mentre gli acidi propionico e butirrico raggiungono concentrazione di circa 30 mM/L.[2]
Nel retto, una piccola quantità di questi piccoli lipidi può passare direttamente nella circolazione sistemica, bypassando il fegato, a mezzo della vena iliaca interna.[13]
A differenza degli acidi grassi a catena lunga, quelli a catena corta e media viaggiano in circolo in forma libera, come acidi grassi non esterificati, e, legati all’albumina, raggiungono il fegato. Il successivo uptake e trasporto citosolico non richiede l’intervento delle proteine leganti gli acidi grassi, di traslocasi per gli acidi grassi nella membrana plasmatica, o di proteine leganti gli acidi grassi nel citosol. Pertanto, muovendosi liberamente, la loro ossidazione potrebbe essere assai più veloce rispetto a quella degli acidi grassi a catena lunga e ai membri più lunghi degli acidi grassi a catena media, ossia quelli con catene carboniose con più di 8 atomi.[10]
Metabolismo epatico ed extraepatico
Il fegato è un sito importante per il metabolismo degli acidi grassi a catena corta.[13] Infatti l’organo è in grado di assorbire circa il 40% dell’acido acetico e l’80% del dell’acido propionico presenti nella vena porta. L’acido propionico è per la maggior parte metabolizzato nel fegato, dove può essere utilizzato anche come substrato per la gluconeogenesi.[7]
Una piccola quantità degli SCFAs prodotti nell’intestino, circa il 36% per l’acido acetico, il 9% per l’acido propionico e solo il 2% per l’acido butirrico, raggiunge, attraverso la circolazione sistemica, i tessuti periferici. Nel muscolo, l’acido acetico può essere utilizzato per la sintesi dei lipidi o essere ossidato per la produzione di energia. Inoltre, si ritiene che le concentrazioni presenti nella circolazione sistemica, seppure piccole, siano in grado di influenzare il metabolismo e la fisiologia delle cellule dei tessuti periferici.
Bibliografia
^ ab Abdul Rahim M.B.H., Chilloux J., Martinez-Gili L., Neves A.L., Myridakis A., Gooderham N., Dumas M.-E. Diet-induced metabolic changes of the human gut microbiome: importance of short-chain fatty acids, methylamines and indoles. Acta Diabetol 2019;56:493-500. doi:10.1007/s00592-019-01312-x
^ abcde Deleu S., Machiels K., Raes J., Verbeke K., Vermeire S. Short chain fatty acids and its producing organisms: an overlooked therapy for IBD? EBioMedicine 2021;66:103293. doi:10.1016/j.ebiom.2021.103293
^ ab Koh A., De Vadder F., Kovatcheva-Datchary P., Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 2016;165(6):1332-1345. doi:10.1016/j.cell.2016.05.041
^ abcdefghij Liu X.F., Shao J.H., Liao Y.T., Wang L.N., Jia Y., Dong P.J., Liu Z.Z., He D.D., Li C., Zhang X. Regulation of short-chain fatty acids in the immune system. Front Immunol 2023;14:1186892. doi:10.3389/fimmu.2023.1186892
^ Miller T.L., Wolin M.J. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl Environ Microbiol 1996;62(5):1589-92. doi:10.1128/aem.62.5.1589-1592
^ abcdef Parada Venegas D., De la Fuente M.K., Landskron G., González M.J., Quera R., Dijkstra G., Harmsen H.J.M., Faber K.N., Hermoso M.A. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol 2019;10:1486. doi:10.3389/fimmu.2019.00277
^ abcdefgh Portincasa P., Bonfrate L., Vacca M., De Angelis M., Farella I., Lanza E., Khalil M.,Wang D.Q.-H., Sperandio M., Di Ciaula A. Gut microbiota and short chain fatty acids: implications in glucose homeostasis. Int J Mol Sci 2022;23:1105. doi:10.3390/ijms23031105
^ Pryde S.E., Duncan S.H., Hold G.L., Stewart C.S., Flint H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett 2002;217(2):133-9. doi:10.1111/j.1574-6968.2002.tb11467.x
^ abcdefg Schönfeld P., Wojtczak L. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res 2016;57(6):943-54. doi:10.1194/jlr.R067629
^ abcdefg Silva Y.P., Bernardi A., Frozza R.L. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front Endocrinol (Lausanne) 2020;11:25. doi:10.3389/fendo.2020.00025
^ abcde Xiong R.G., Zhou D.D., Wu S.X., Huang S.Y., Saimaiti A., Yang Z.J., Shang A., Zhao C.N., Gan R.Y., Li H.B. Health benefits and side effects of short-chain fatty acids. Foods 2022;11(18):2863. doi: 10.3390/foods11182863
L’amido fosforilasi o alfa-glucano fosforilasi (EC 2.4.1.1) è una proteina multimerica, dotata di attività enzimatica e regolatoria, che svolge un ruolo importante nel metabolismo dei carboidrati, sia nei procarioti che negli eucarioti.[10]
L’enzima catalizza il trasferimento di un’unità glucosidica dal glucosio-1-fosfato all’estremità non riducente di un α-(1→4)-glucano nascente, a cui viene legata per mezzo di un legame glicosidico α-(1→4).[6][8] La reazione è reversibile e la direzione dipende dal rapporto fosfato/glucosio-1-fosfato presente in vivo.[2] L’enzima, al pari delle amido sintasi (EC 2.4.1.21), che intervengono nella sintesi dell’amilosio e dell’amilopectina, i polisaccaridi che compongono i granuli di amido, della glicogeno fosforilasi (EC 2.4.1.1), enzima che interviene nella glicogenolisi, e della glicogeno sintasi (EC 2.4.1.11) che interviene nella sintesi del glicogeno, appartiene alla famiglia delle glucosiltransferasi (EC 2.4).[6]
Si noti che il substrato donatore dell’unità glucosidica nella reazione catalizzata dalla amido fosforilasi non è ne l’ADP-glucosio, utilizzato dalle amido sintasi, ne l’UDP-glucosio utilizzato dalla glicogeno sintasi, ma il glucosio-1-fosfato.[2]
L’amido fosforilasi sembra intervenire sia nella sintesi che nella degradazione dell’amilosio e amilopectina.[9]
A livello industriale l’azione fosforolitica della fosforilasi è utilizzata nella produzione del glucosio-1-fosfato e nella preparazione di carboidrati quali glucani e amidi modificati.[6]
Nelle piante sono presenti almeno due isoforme dell’amido fosforilasi, una nello stroma dei plastidi, indicata come Pho1, e una a localizzazione citosolica, denominata Pho2. Entrambe gli isoenzimi svolgono un ruolo fondamentale nella sintesi e degradazione dell’amido.[6][8]
Degradazione dell’amido
Sebbene il primo enzima a intervenire nella degradazione del polisaccaride sia l’alfa-amilasi (EC 3.2.1.1) nelle prime fasi della germinazione, e la beta-amilasi (EC 3.2.1.2) nel caso della degradazione dell’amido transitorio nei cloroplasti, anche altre attività enzimatiche sono state chiamate in causa, quali l’alfa-glucano acqua dichinasi (EC 2.7.9.4) e la fosfoglucano acqua dichinasi (EC 2.7.9.5), e l’enzima deramificante.[10]
Dei due isoenzimi dell’amido fosforilasi, Pho1 sembra avere un’azione indiretta o regolatrice in grado di influenzare l’attività degli altri enzimi che intervengono nella degradazione dell’amido, mentre Pho2 è in grado di degradare i granuli di amido e altri glucani ramificati.
Sintesi dell’amido
Nel corso della sintesi dell’amido, l’amido fosforilasi, in particolare Pho1, sembra svolgere sia un’azione enzimatica che regolatrice.
Pho1, sembra essere coinvolta nei passaggi iniziali della sintesi dell’amido, concorrendo all’allungamento della catena nascente.[1][8]
La sintesi dell’amido coinvolge un set di almeno cinque classi di enzimi, ossia la ADP-glucosio pirofosforilasi (EC 2.7.7.27), le amido sintasi, gli enzimi ramificanti l’amido (EC 2.4.1.18), gli enzimi deramificanti l’amido (EC2.1.41) e le amido fosforilasi.[5][10] A queste vanno aggiunte proteine prive di attività catalitica ma necessarie alla corretta sintesi del granulo di amido. Nell’endosperma dei cereali la formazione di complessi multienzimatici tra le amido sintasi e gli enzimi ramificanti dipendono, oltre che da specifiche fosforilazioni delle proteine stesse, anche dalla presenza di Pho1.[1][3]
L’amido fosforilasi è in grado di formare anche un complesso con l’enzima disproporzionante (EC 2.4.1.25).[1][10] Questo complesso sembra in grado di sintetizzare brevi malto-oligosaccaridi o MOS, che sono α-(1→4)-glucani con un grado di polimerizzazione compreso tra 2 e 7.[7] I MOS agiscono come primer per l’amido sintasi IV e l’amido sintasi legata ai granuli nelle fasi iniziali della sintesi rispettivamente dell’amilopectina e dell’amilosio, una funzione questa analoga a quella svolta dalla glicogenina nella della sintesi del glicogeno.[4] I MOS possono derivare anche dall’azione degli enzimi deramificanti l’amido nel corso del rifinitura di molecole di amilopectina vicine.
Bibliografia
^ abc Crofts N., Abe N., Oitome N.F., Matsushima R., Hayashi M., Tetlow I.J., Emes M.J., Nakamura Y., Fujita N. Amylopectin biosynthetic enzymes from developing rice seed form enzymatically active protein complexes. J Exp Bot 2015;66(15):4469-82. doi:10.1093/jxb/erv212
^ ab Orzechowski S. Starch metabolism in leaves. Acta Biochim Pol 2008;55(3):435-45. doi:10.18388/abp.2008_3049
^ Pfister B., Zeeman S.C., Rugen M.D., Field R.A., Ebenhöh O., Raguin A. Theoretical and experimental approaches to understand the biosynthesis of starch granules in a physiological context. Photosynth Res 2020;145:55-70. doi:10.1007/s11120-019-00704-y
^ Qu J., Xu S., Zhang Z., Chen G., Zhong Y., Liu L., Zhang R., Xue J., Guo D. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci Rep 2018;8(1):12736. doi:10.1038/s41598-018-30411-y
^ abcd Rathore R.S., Garg N., Garg S., Kumar A. Starch phosphorylase: role in starch metabolism and biotechnological applications. Crit Rev Biotechnol 2009;29(3):214-24. doi:10.1080/07388550902926063
^ Tetlow I.J., Bertoft E. A review of starch biosynthesis in relation to the building block-backbone model. Int J Mol Sci 2020;21(19):7011. doi:10.3390/ijms21197011
^ abc Tickle P., Burrell M.M., Coates S.A., Emes M.J., Tetlow I.J., Bowsher C.G. Characterization of plastidial starch phosphorylase in Triticum aestivum L. endosperm. J Plant Physiol 2009;166(14):1465-78. doi:10.1016/j.jplph.2009.05.004
^ Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011
^ abcd Yu G., Shoaib N., Xie Y., Liu L., Mughal N., Li Y., Huang H., Zhang N., Zhang J., Liu Y., Hu Y., Liu H., Huang Y. Comparative study of starch phosphorylase genes and encoded proteins in various Monocots and Dicots with emphasis on maize. Int J Mol Sci 2022;23:4518. doi:doi.org/10.3390/ijms23094518
L’amilopectina è un polisaccaride altamente ramificato formato da unità di alfa-D-glucosio. Insieme all’amilosio è uno dei due principali costituenti dei granuli di amido, il mezzo attraverso cui le piante immagazzinano energia, e la più diffusa e abbondante forma di accumulo di carboidrati presente sulla Terra.[12]
I residui glucosidici sono legati da legami glicosidici α-(1→4) a formare catene da cui si dipartono ramificazioni a mezzo di legami glicosidici α-(1→6).[2]
La sua sintesi richiede l’intervento coordinato di almeno quattro distinte classi di enzimi: le amido sintasi, gli enzimi ramificanti l’amido (EC 2.4.1.18), gli enzimi deramificanti l’amido, e l’amido fosforilasi (EC 2.4.1.1).[17]
Nel granulo di amido l’amilopectina è presente in quantità maggiore rispetto all’amilosio, e forma una matrice semicristallina all’interno della quale si ritiene venga depositato l’amilosio.
Il rapporto amilosio/amilopectina influenza fortemente le proprietà chimico-fisiche dell’amido, e di conseguenza sia i suoi usi industriali, come la produzione di additivi alimentari, che i possibili effetti sulla salute.[3]
L’amilopectina è un polisaccaride altamente ramificato con un peso molecolare compreso tra i 107 e 108 Dalton, quindi molto più grande dell’amilosio. Contiene 104-105 molecole di glucosio organizzate a formare molte catene relativamente corte, composte in media da 18-25 molecole di glucosio legate da legami glicosidici α-1,4.[17] La lunghezza delle catene glucosidiche varia a seconda della fonte dell’amido nonché delle condizioni ambientali e nutritive prevalenti nel corso della crescita della pianta e della formazione del seme.
Le catene sono legate tra di loro attraverso legami glicosidici α-1,6, a formare numerose ramificazioni che creano una struttura ad albero, con le catene vicine organizzate in strutture simili a grappoli o cluster.[1]
Nella maggior parte degli amidi i legami glicosidici α-1,6 rappresentano circa il 5% di tutti i legami glicosidici, una percentuale più bassa rispetto al 9% che si osserva nella molecola del glicogeno, dove le ramificazioni sono anche più uniformemente distribuite. La lunghezza e la posizione della ramificazioni influenzano direttamente le proprietà fisico-chimiche dell’amilopectina, quali la solubilità, la viscosità, la facilità di retrogradazione e la temperatura di gelificazione e incollaggi.[3] Ad esempio, il glicogeno è solubile in acqua mentre l’amilopectina non lo è.
Catene dell’amilopectina
Le catene glucosidiche dell’amilopectina possono essere classificate sulla base della loro lunghezza o della presenza o assenza di ramificazioni.
La classificazione basata sulla lunghezza individua due tipi principali di catene: le catene corte e lunghe. Le catene corte hanno un grado di polimerizzazione compreso tra 6 e 36, sebbene il limite superiore dipenda dalla fonte di amilopectina, mentre le catene lunghe hanno un grado di polimerizzazione maggiore o uguale a 36. La distribuzione molare delle catene corte e lunghe è dell’ordine di 6-19 nella maggior parte degli amidi, ed è di solito più bassa negli amidi B-cristallini, come quelli delle patate, rispetto agli amidi A-cristallini, come gli amidi di deposito dell’endosperma dei cereali.[17]
La classificazione basata sulle connessioni con altre catene individua tre categorie: catene A, catene B e catene C.[7]
Le catene A non hanno ramificazioni, sono corte, con un grado di polimerizzazione di circa 13, e sono le catene esterne.
Le catene B hanno almeno una ramificazione,ossia legano una catena A o B, sono più lunghe delle catene A, e si trovano nella parte più interna della molecola.
Le catene B a loro volta sono state suddivisi in catene B1, che hanno un grado di polimerizzazione di circa 22, le catene B2, con un grado di polimerizzazione di circa 42, le B3, con un grado di polimerizzazione di circa 69, B4 e così via.
La catene C è la catena B che porta l’estremità libera riducente.
Le catene A e le catene B1 partecipano alla formazione dei cluster, mentre si ritiene che le catene B2, B3, e B4 si estendano rispettivamente in due, tre e quattro cluster.[7][11]
Polimorfi di tipo A, B e C
All’interno dei cluster, i segmenti di catena lineari vicini formano doppie eliche, parallele tra di loro, con un periodo di 2,1 nm, e dove ogni giro conta sei unità glucosidiche per catena.[10] Le doppie eliche si dispongono a formare due tipi di strutture cristalline indicate come polimorfo di tipo A, più denso e tipico dell’amido dei cereali, e polimorfo di tipo B, a struttura esagonale, meno denso, più idratato e tipico degli amidi dei tuberi e degli amidi ad alto contenuto di amilosio. In alcuni tipi di amido, come quello presente nelle radici, legumi e alcuni frutti, sono presenti polimorfi miscele dei due precedenti, chiamati polimorfi di tipo C.[2]
Questo tipo di organizzazione è alla base della natura semicristallina dei granuli di amido.
Anelli di crescita
Sebbene i granuli di amido abbiamo forme molto differenti, la loro architettura interna è notevolmente conservata tra le differenti specie. Infatti, quando osservati al microscopio, la maggior parte degli amidi presenta uno schema regolare di strutture ad anello più scure e più chiare, strutture note come anelli di crescita, così chiamati in quanto ricordano gli anelli di crescita delle piante.[10]
Gli anelli di crescita circondano l’ilo del granulo, ossia, il centro del granulo, la cui struttura non è nota, sebbene sembri essere formata da una struttura relativamente disordinata di alfa-glucani. Gli anelli hanno uno spessore di 200-400 nm, e sono formati da regioni amorfe e semicristalline che si alternano, meno dense le prime e più dense le seconde.[10]
Secondo il modello a cluster le regioni semicristalline sono formate dall’alternanza di lamelle cristalline e lamelle amorfe, impilate con una periodicità di circa 9-10 nm.[11] Le lamelle cristalline sono formate dalle catene lineari dell’amilopectina disposte a dare i polimorfi di tipo A, B o C, e si estendono per circa 6-7 nm, mentre le lamelle amorfe presentano la maggior parte dei punti di ramificazione e si estendono per circa 3 nm.[5]
Sintesi dell’amilopectina
La sintesi dell’amilopectina si ritiene avvenga a partire dall’ilo.[22]
La sintesi richiede l’intervento coordinato di almeno quattro distinte classi di enzimi: le amido sintasi, l’amido fosforilasi, gli enzimi ramificanti l’amido e gli enzimi deramificanti l’amido.[12] Ciascuna classe si compone di diverse isoforme con proprietà biochimiche distinte.
Al pari della sintesi dell’amilosio, anche per la sintesi dell’amilopectina sono necessari corti malto-oligosaccaridi o MOS, α-(1→4)-glucani lunghi da 2 a 7 residui glucosidici, che agiscono come primer e sono allungati dalle amido sintasi, una funzione analoga a quella svolta dalla proteina glicogenina nelle fasi iniziali della sintesi del glicogeno.[16]
I MOS sembra possano avere diverse origini, che chiamano tutte in causa enzimi coinvolti nella sintesi del granulo di amido, ossia:
amido sintasi III e amido fosforilasi, quest’ultima in combinazione con l’enzima disproporzionate (EC 2.4.1.25), che utilizzano come substrati rispettivamente l’ADP-glucosio e il glucosio-1-fosfato;
gli enzimi deramificanti l’amido, nel corso del rifinitura di altre molecole di amilopectina.[16]
Grazie alla loro scarsa solubilità in ambiente acquoso, i MOS sembrano in grado di eludere l’azione idrolitica delle alfa-amilasi (EC 3.2.1.1) e delle beta-amilasi (EC 3.2.1.2).
La coordinazione, sia spaziale che temporale, degli enzimi coinvolti, che in molti casi interagiscono anche fisicamente a formare complessi multienzimatici, è essenziale per permettere la conversione dei prodotti della fotosintesi nella struttura organizzata e insolubile del polisaccaride. E, come nel caso della sintesi dell’amilosio e del glicogeno, la polimerizzazione del glucosio in amilopectina, e più in generale di monosaccaridi osmoticamente attivi in polisaccaridi osmoticamente inerti, permette il deposito di grandi quantità di monosaccaridi all’interno della cellula senza alcun aumento sostanziale della pressione osmotica.
Amido sintasi
Sono note sei isoforme dell’amido sintasi, tutte strutturalmente correlate, di cui cinque sono coinvolte nella sintesi dell’amilopectina, isoforme indicate come amido sintasi I, II, III, IV e V o, rispettivamente, SSI, SSII, SSIII, SSIV e SSV (EC 2.4.1.21), presenti nello stroma dei plastidi o suddivise tra stroma e granuli, mentre la sesta, la amido sintasi legata ai granuli o GBSS (EC 2.4.1.242), è quasi esclusivamente legata ai granuli ed è coinvolta nella sintesi dell’amilosio.[10]
Le prime quattro isoforme sono dotate di attività catalitica e appartengono, come GBSS, la glicogeno fosforilasi (EC 2.4.1.1) e la glicogeno sintasi (EC 2.4.1.11), enzimi che intervengono rispettivamente nella glicogenolisi e nella sintesi del glicogeno, alla famiglia delle glicosiltransferasi (EC 2.4). La amido sintasi V è invece priva di attività catalitica.
Nel corso della sintesi dell’amilopectina, l’amido sintasi catalizza il trasferimento di un’unità di glucosio all’estremità non riducente di un α-(1→4)-glucano, cui il monosaccaride è legato per mezzo di un legame glicosidico α-(1→4).[5]
Si noti che, a differenza della glicogeno sintasi, le amido sintasi utilizzano come donatore dell’unità glucosidica l’ADP-glucosio e non l’UDP glucosio.
La modalità d’azione dell’amido sintasi I, II, III, e IV è differente rispetto a quella della GBSS in quanto sono in grado di catalizzare l’aggiunta di una sola unità di glucosio per incontro con il substrato, modalità d’azione definita distributiva, mentre l’amido sintasi legata ai granuli è in grado di catalizzare l’aggiunta di più di un’unità glucosidica per incontro con l’α-(1→4)-glucano nascente, modalità d’azione definita processuale.[17]
Ruolo delle amido sintasi
Le fasi iniziali della sintesi dell’amilopectina, nonché la formazione di un granulo di amido normale, richiedono la presenza di SSIV, sebbene anche SSIII sembra svolgere un ruolo, sovrapponendo la sua azione a quella di SSIV.[16]
Al pari della GBSS, SSIV richiede la presenza di PTST2, una proteina della famiglia PTST. PTST2 è priva di attività catalitica, ma è in grado, grazie alla presenza di uno specifico dominio che lega i carboidrati, di facilitare il legame dell’enzima agli alfa-glucani. SSIV è anche capace di formare dimeri, una caratteristica importante sia per l’attività catalitica che per la capacità di interagire con altre proteine.
Secondo un modello d’azione di PTST2, la proteina, a mezzo del dominio che lega i carboidrati, riconosce e forma un complesso con MOS dotati di una specifica forma tridimensionale elicoidale. In complesso proteina-MOS a sua volta interagisce con un dimero SSIV, che è ora in grado di catalizzare l’allungamento dell’alfa-glucano, mentre PTST2 viene rilasciata così da permetterle di legare un altro malto-oligosaccaride e facilitarne una successiva interazione con un altro dimero di SSIV.[15]
L’azione di SSIV è seguita da quella delle altre isoforme dell’amido sintasi. SSI catalizza l’allungamento di malto-oligosaccaridi lunghi da 6 a 7 residui glucosidici, a dare oligosaccaridi con un grado di polimerizzazione compreso tra 8 e 12, che a loro volta sono ottimi substrati per SSII, che porta il grado di polimerizzazione tra 12 e 30. Questi alfa-glucani sono ulteriormente allungati dall’azione catalitica di SSIII, a dare catene lineari con un grado di polimerizzazione superiore a 30.[12] Quindi SSIII sembra agire non solo nelle fasi iniziali della sintesi del granulo di amido, ma anche nelle fasi successive.
SSIV e SSV sembrano necessarie per la sintesi di un numero regolare di granuli di amido di normale morfologia.[1][16]
Enzimi ramificanti l’amido
Gli enzimi ramificanti l’amido catalizzano la formazione di legami glicosidici α-(1→6), creando quindi punti di ramificazione nella catena lineare di alfa-glucani, di cui i principali sono il glicogeno e l’amilopectina.[20] La loro azione aumenta il numero delle estremità non riducenti, che possono servire come accettori di unità glucosidiche nelle reazioni di allungamento.[14]
SBE catalizza dapprima la scissione idrolitica di un legame α-(1→4) all’interno di una catena di alfa-glucano, liberando un oligosaccaride la cui estremità riducente viene legata al gruppo idrossilico in posizione C6 di un’unità glucosidica di un alfa-glucano. Quindi le due catene sono legate a mezzo di un legame glicosidico α-(1→6).
La catena cui l’oligosaccaride viene legato può essere la stessa da cui è stato staccato, e in questo caso di parla di trasferimento intracatena, o una catena differente, e in questo caso si parla di trasferimento intercatena.
Tra i fattori determinanti il tipo di trasferimento sembra esserci la concentrazione relativa della catene lineari α-(1→4). In particolare sembra che le catena strettamente associate, come nelle doppie eliche che si formano nei cluster, promuovano il trasferimento inter-catena.[19] Infine sembra che l’interazione tra l’amido sintasi I e l’enzima ramificante sia cruciale nel determinare la distribuzione bimodale della lunghezza delle catene osservata negli amidi delle piante.
Isoforme degli enzimi ramificanti l’amido
Nelle piante sono presenti due isoforme dell’enzima ramificante l’amido, indicate come SBEI e SBEII.
Codificate da geni distinti, hanno proprietà biochimiche distinte, il che suggerisce che svolgano ruoli differenti nella determinazione della struttura dell’amilopectina e dell’amilosio.[17]
SBEI sembra essere espresso maggiormente nei tessuti di deposito, il che suggerisce un suo ruolo importante nel determinare le proprietà strutturali degli amidi di deposito, mostra una preferenza di substrato per l’amilosio, e in grado di trasferire oligosaccaridi con un grado di polimerizzazione superiore ai 30, sebbene la maggior parte sia tra 10 e 13.[6] Sembra inoltre intervenire nella formazione delle catene superlunghe ed extralunghe dell’amilopectina, mentre sembrano meno importanti altri suoi contributi alla struttura dell’amilopectina. Non tutte le piante esprimono SBEI; ad esempio Arabidopsis e Canola (Brassica napus L.), che immagazzinano olio, e dove l’amido è presente solo nei tessuti fotosintetici, hanno solo SBEII.
SBEII è maggiormente espresso nelle graminacee, nei cereali, e in molte altre piante. La sua perdita determina importanti alterazioni nell’architettura dell’amilopectina e influenza anche la quantità totale di amido, riducendone il contenuto. L’enzima mostra una preferenza di substrato per l’amilopectina e trasferisce oligosaccaridi relativamente corti, formati da 6-14 unità di glucosio. Nei cereali e nelle graminacee SBEII è presente con due isoforme tessuto-specifiche e codificate da geni distinti, indicate come SBEIIa, presente principalmente nelle foglie, e SBEIIb, presente prevalentemente nell’endosperma.[17]
Enzimi deramificanti l’amido
Gli enzimi deramificanti l’amido catalizzano l’idrolisi dei legami glicosidici α-(1→6), e appartengono alla superfamiglia delle alfa-amilasi.
Nelle piante sono presenti due tipi di enzimi deramificanti: le isoamilasi (EC 3.2.1.68) e le pullulanasi (EC 3.2.1.41).[8] Le isoamilasi deramificano l’amilopectina e altri poliglucani, mentre le pullulanasi l’amilopectina e il pullulano, un polisaccaride fungino. Isoamilasi e pullulanasi differiscono anche riguardo alla specificità di substrato in quanto le isoamilasi richiedono che le ramificazioni siano formate da almeno tre residui glucosidici, mentre le pullulanasi sono in grado di agire su ramificazioni con non meno di due residui glucosidici.[18]
Nel corso della formazione del granulo di amido gli enzimi deramificanti svolgono un ruolo cruciale nel determinare le proprietà insolubili e la struttura fine dell’amilopectina. Si ritiene infatti che le idrolisi operate permettano il raggruppamento delle ramificazioni rimanenti, promuovendo in questo modo le interazioni tra catene adiacenti e la formazione delle alfa-eliche, che a loro volta sono importanti nella formazione delle strutture semicristalline insolubili in acqua dell’amilopectina, e quindi dell’amido.[17] Nella struttura semicristallina le ramificazioni sono presumibilmente inaccessibili all’azione della alfa-amilasi e beta-amilasi e dell’enzima deramificante stesso.
Gli enzimi deramificanti l’amido sono utilizzati industrialmente nella preparazione dell’amido resistente e delle ciclodestrine, che sono oligosaccaridi ciclici.
Amido fosforilasi
L’amido fosforilasi appartiene, al pari delle amido sintasi, alla famiglia delle glicosiltransferasi, ed è una fosforilasi simile alla glicogeno fosforilasi. Nelle piante è presente con almeno due forme isoenzimatiche, Pho1, che si ritrova nello stroma dei plastidi, ed è considerata la vera amido fosforilasi che si ritiene agisca nella sintesi dell’amido, e Pho2, isoforma a localizzazione citosolica.[4]
L’amido fosforilasi sembra essere coinvolta nei passaggi iniziali della sintesi dell’amido, catalizzando il trasferimento, reversibile, di unità glucosidiche a un alfa-glucano, unità che sono legate attraverso un legame glicosidico α-(1→4).[3] A differenza delle amido sintasi, il substrato donatore dell’unità glucosidica è il glucosio-1-fosfato e non l’ADP-glucosio.[4]
Fosforilazione dell’amilopectina
L’amilopectina, analogamente al glicogeno, lega covalentemente gruppi fosfato in quantità variabili a seconda dell’origine botanica dell’amido. Ad esempio, l’amido delle patate ha un contenuto di gruppi fosfato relativamente alto, con un grado di sostituzione di circa lo 0,1-0,3%, mentre negli amidi di deposito dell’endosperma dei cereali è generalmente inferiore allo 0,01%.[9]
La fosforilazione dell’amilopectina è catalizzata da due dichinasi presenti nei plastidi: la alfa-glucano acqua dichinasi (EC 2.7.9.4) e la fosfoglucano acqua dichinasi (EC 2.7.9.5). I due enzimi catalizzano il trasferimento del gruppo fosfato beta dell’ATP a un residuo glucosidico di una di catena di alfa-glucano, mentre il gruppo fosfato gamma è trasferito all’acqua. Nello specifico, la alfa-glucano acqua dichinasi catalizza la fosforilazione del gruppo ossidrilico in posizione C6, mentre la fosfoglucano acqua dichinasi del gruppo ossidrilico in posizione C3, di solito di un alfa-glucano prefosforilato.[13] La posizione C6 raccoglie circa i due terzi dei gruppi fosfato, mentre la posizione C3 circa il 20-30%. Gruppi fosfato si ritrovano anche in posizione C2, sebbene in percentuale molto ridotta rispetto alle altre posizioni. Non è noto quale enzima catalizzi questa fosforilazione.
Riguardo alla specificità di substrato sembra che le fosforilazioni si accumulino più facilmente sulle catene più lunghe. Inoltre sembra esistere anche una correlazione inversa tra il contenuto totale di fosfato e la frequenza delle ramificazioni dell’amilopectina.
Le cariche negative portate dai gruppi fosfato determinano modificazioni localizzate della struttura del polisaccaride, in quanto tendono a far allontanare le une dalle altre le catene fosforilate adiacenti. Queste repulsioni sembra permettano l’apertura e l’idratazione delle catene, influenzando in questo modo l’attività degli enzimi della biosintesi e rendendo anche più suscettibili le catene stesse all’attacco delle amilasi.[21]
Rapporto amilosio/amilopectina
I granuli di amido sono costituiti per la maggior da amilosio e amilopectina.[12] I due polisaccaridi sono presenti in percentuali variabili, con l’amilosio che non costituisce più del 35% del peso secco del granulo.[5] Esistono tuttavia piante nelle quali i granuli di amido sono costituiti per la maggior parte, o quasi interamente, da amilopectina, amidi definiti cerosi, e altre dove, al contrario, l’amilosio costituisce la maggior parte, e in alcuni casi, la quasi totalità della componente polisaccaridica del granulo.[19]
Il rapporto amilosio/amilopectina influenza le proprietà fisico-chimiche dell’amido, quali la capacità di assorbire acqua, la gelatinizzazione e la retrogradazione, o la resistenza all’idrolisi enzimatica, quest’ultima importante nel determinare la velocità con cui, nel corso della digestione dei carboidrati, amilosio e amilopectina sono idrolizzati a maltosio e maltotriosio ad opera della alfa-amilasi.[3] Pertanto, il rapporto amilosio/amilopectina influenza gli effetti dei differenti tipi di amido sulla salute nonché i possibili usi industriali.
Bibliografia
^ ab Abt M.R., Pfister B., Sharma M., Eicke S., Bürgy L., Neale I., Seung D., Zeeman S.C. STARCH SYNTHASE5, a noncanonical starch synthase-like protein, promotes starch granule initiation in Arabidopsis. Plant Cell 2020;32(8):2543-2565. doi:10.1105/tpc.19.00946
^ ab Cornejo-Ramírez Y.I., Martínez-Cruz O., Del Toro-Sánchez C.L., Wong-Corral F.J., Borboa-Flores J. & Cinco-Moroyoqui F.J. The structural characteristics of starches and their functional properties. CYTA J Food 2018;16(1):1003-1017. doi:10.1080/19476337.2018.1518343
^ abcd Crofts N., Abe N., Oitome N.F., Matsushima R., Hayashi M., Tetlow I.J., Emes M.J., Nakamura Y., Fujita N. Amylopectin biosynthetic enzymes from developing rice seed form enzymatically active protein complexes. J Exp Bot 2015;66(15):4469-82. doi:10.1093/jxb/erv212
^ ab Cuesta-Seijo J.A., Ruzanski C., Krucewicz K., Meier S., Hägglund P., Svensson B., Palcic M.M. Functional and structural characterization of plastidic starch phosphorylase during barley endosperm development. PLoS One 2017;12(4):e0175488. doi:10.1371/journal.pone
^ abc Gous P.W., Fox G.P. Review: Amylopectin synthesis and hydrolysis – Understanding isoamylase and limit dextrinase and their impact on starch structure on barley (Hordeum vulgare) quality. Trends Food Sci Technol 2017;62:23-32. doi:10.1016/j.tifs.2016.11.013
^ Guan H.P., Preiss J. Differentiation of the properties of the branching isozymes from maize (Zea mays). Plant Physiol 1993;102(4):1269-1273. doi:10.1104/pp.102.4.1269
^ ab Li G., Yacine Y., Zhu F. Relationships between supramolecular organization and amylopectin fine structure of quinoa starch. Food Hydrocoll 2021;117:106685. doi:10.1016/j.foodhyd.2021.106685
^ Møller M.S., Henriksen A., Svensson B. Structure and function of α-glucan debranching enzymes. Cell Mol Life Sci. 2016;73(14):2619-41. doi:10.1007/s00018-016-2241-y
^ Nitschke F., Wang P., Schmieder P., Girard J.M., Awrey D.E., Wang T., Israelian J., Zhao X., Turnbull J., Heydenreich M., Kleinpeter E., Steup M., Minassian B.A. Hyperphosphorylation of glucosyl C6 carbons and altered structure of glycogen in the neurodegenerative epilepsy Lafora disease. Cell Metab 2013;17(5):756-67. doi:10.1016/j.cmet.2013.04.006
^ abcd Pfister B., Zeeman S.C. Formation of starch in plant cells. Cell Mol Life Sci 2016;73(14):2781-807. doi:10.1007/s00018-016-2250-x
^ ab Pfister B., Zeeman S.C., Rugen M.D., Field R.A., Ebenhöh O., Raguin A. Theoretical and experimental approaches to understand the biosynthesis of starch granules in a physiological context. Photosynth Res 2020;145:55-70. doi:10.1007/s11120-019-00704-y
^ abcd Qu J., Xu S., Zhang Z., Chen G., Zhong Y., Liu L., Zhang R., Xue J., Guo D. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci Rep 2018;8(1):12736. doi:10.1038/s41598-018-30411-y
^ Ritte G., Heydenreich M., Mahlow S., Haebel S., Kötting O., Steup M. Phosphorylation of C6- and C3-positions of glucosyl residues in starch is catalysed by distinct dikinases. FEBS Lett 2006;580(20):4872-6. doi:10.1016/j.febslet.2006.07.085
^ Sawada T., Nakamura Y., Ohdan T., Saitoh A., Francisco P.B. Jr, Suzuki E., Fujita N., Shimonaga T., Fujiwara S., Tsuzuki M., Colleoni C., Ball S. Diversity of reaction characteristics of glucan branching enzymes and the fine structure of α-glucan from various sources. Arch Biochem Biophys 2014;562:9-21. doi:10.1016/j.abb.2014.07.032
^ Seung D., Boudet J., Monroe J., Schreier T.B., David L.C., Abt M., Lu K.J., Zanella M., Zeeman S.C. Homologs of PROTEIN TARGETING TO STARCH control starch granule initiation in Arabidopsis leaves. Plant Cell 2017;29(7):1657-1677. doi:10.1105/tpc.17.00222
^ abcd Szydlowski N., Ragel P., Raynaud S., Lucas M.M., Roldán I., Montero M., Muñoz F.J., Ovecka M., Bahaji A., Planchot V., Pozueta-Romero J., D’Hulst C., Mérida A. Starch granule initiation in Arabidopsis requires the presence of either class IV or class III starch synthases. Plant Cell 2009;21(8):2443-57. doi:10.1105/tpc.109.066522
^ abcdefg Tetlow I.J., Bertoft E. A review of starch biosynthesis in relation to the building block-backbone model. Int J Mol Sci 2020;21(19):7011. doi:10.3390/ijms21197011
^ Xia W., Zhang K., Su L., Wu J. Microbial starch debranching enzymes: developments and applications. Biotechnol Adv 2021;50(3):107786. doi:10.1016/j.biotechadv.2021.107786
^ ab Wang J., Hu P., Lin L., Chen Z., Liu Q., Wei C. Gradually decreasing starch branching enzyme expression is responsible for the formation of heterogeneous starch granules. Plant Physiol 2018;176(1):582-595. doi:10.1104/pp.17.01013
^ Wilkens C., Svensson B., Møller M.S. Functional roles of starch binding domains and surface binding sites in enzymes involved in starch biosynthesis. Front Plant Sci 2018;9:1652. doi:10.3389/fpls.2018.01652
^ Zhou W., He S., Naconsie M., Ma Q., Zeeman S.C., Gruissem W. & Zhang P. Alpha-glucan, water dikinase 1 affects starch metabolism and storage root growth in Cassava (Manihot esculenta Crantz). Sci Rep 2017;7:9863 doi:10.1038/s41598-017-10594-6
^ Ziegler G.R., Creek J.A., Runt J. Spherulitic crystallization in starch as a model for starch granule initiation. Biomacromolecules 2005;6(3):1547-54. doi:10.1021/bm049214p
L’amido sintasi (EC 2.4.1.21) è un enzima che catalizza il trasferimento di molecole di glucosio dall’ADP-glucosio all’estremità non riducente di un α-(1→4)-glucano preesistente, cui i monosaccaridi sono legati attraverso un legame glicosidico α-(1→4).[4]
L’enzima è coinvolto nella sintesi dell’amilosio e dell’amilopectina, i due polisaccaridi che costituiscono la quasi totalità del granulo di amido, la più abbondante forma di accumulo di carboidrati ed energia presente nelle piante.[7]
L’amido sintasi appartiene alla famiglia delle glicosiltransferasi (EC 2.4), al pari dell’amido fosforilasi (EC 2.4.1.1), un altro enzima coinvolto nel metabolismo dell’amido, e della glicogeno sintasi (EC 2.4.1.11) e glicogeno fosforilasi (EC 2.4.1.1), enzimi coinvolti rispettivamente nella sintesi del glicogeno e nella degradazione del glicogeno o glicogenolisi.[6] Tuttavia, mentre la glicogeno sintasi utilizza come donatore dell’unità glucosidica l’UDP-glucosio, e l’amido fosforilasi il glucosio-1-fosfato, l’amido sintasi utilizza l’ADP-glucosio.[2][3]
Nelle piante sono note sei isoforme di amido sintasi. Sono proteine strutturalmente correlate, cinque coinvolte nella sintesi dell’amilopectina, indicate come amido sintasi I, II, III, IV e V o, rispettivamente, SSI, SSII, SSIII, SSIV e SSV, e una coinvolta nella sintesi dell’amilosio, l’amido sintasi legata ai granuli o GBSS (EC 2.4.1.242).[6] GBSS, SSI, SSII, SSIII, e SSIV sono dotate di attività catalitica, mentre la SSV ne è priva.[1]
GBSS è quasi esclusivamente legata ai granuli, sembra per la maggior parte posizionata al suo interno, come evidenziato dal trattamento della superficie dei granuli con proteasi. Le altre isoforme dell’amido sintasi sono presenti o solamente nello stroma dei plastidi o suddivise tra stroma e granuli di amido, e sono dette solubili.[8]
Le amido sintasi I, II, III, e IV catalizzano l’aggiunta di una sola unità di glucosio per incontro con l’α-(1→4)-glucano nascente, modalità d’azione definita distributiva, mentre GBSS è in grado di catalizzare l’aggiunta di più unità glucosidiche per incontro con il substrato, modalità d’azione definita processuale.[11]
Amido sintasi e MOS
Le amido sintasi coinvolte nelle prime fasi della sintesi di amilosio e amilopectina necessitano della presenza di corti malto-oligosaccaridi o MOS per iniziare la sintesi de novo dei due polisaccaridi.[11]
Questi piccoli oligosaccaridi, ossia α-(1→4)-glucani lunghi da 2 a 7 residui glucosidici, fungono da primer e sono allungati, funzione analoga a quella svolta dalla glicogenina nella sintesi del glicogeno.
I MOS possono derivare dall’azione dell’amido sintasi III, dell’amido fosforilasi e degli enzimi deramificanti l’amido, in quest’ultimo caso nel corso del rifinitura di molecole di amilopectina.
Essendo scarsamente solubili, i MOS sembrano in grado di evitare l’azione idrolitica delle alfa-amilasi (EC 3.2.1.1) e delle beta-amilasi.[8]
Amido sintasi e sintesi dell’amilopectina
La sintesi dell’amilopectina richiede l’azione temporalmente coordinata di almeno quattro classi di enzimi, ossia gli isoenzimi dell’amido sintasi, l’amido fosforilasi, gli enzimi ramificanti l’amido (EC 2.4.1.18), e gli enzimi deramificanti l’amido.[2][7] Si ritiene inoltre che, in molti casi, questi enzimi interagiscono fisicamente tra loro a dare complessi multienzimatici, strutture in grado di aumentare l’efficienza di una via metabolica.[11]
E’ generalmente accettato che la crescita del granulo di amido avvenga a partire da una zona centrale detta ilo, la cui esatta struttura non è nota, sebbene sembra sia formata da una struttura relativamente disordinata di α-glucani.[13] L’iniziazione dell’ilo, la formazione di un granulo di amido strutturalmente normale, e il grado di accumulo dell’amido richiedono la presenza di SSIV, sebbene sia stato suggerito che anche SSIII e SSV possano svolgere un ruolo, sovrapponendo la loro azione con quella della SSIV.[10]
GBSS e sintesi dell’amilosio
La sintasi legata ai granuli è coinvolta nella sintesi dell’amilosio.
Questo enzima fu scoperto nei primi anni sessanta del secolo scorso dal gruppo di Luis Federico Leloir, lo stesso ricercatore che nel 1948 aveva scoperto la principale via per il metabolismo del galattosio, la via di Leloir.[5]
La sua azione catalitica non è del tutto contemporanea a quella delle altre amido sintasi in quanto necessita della presenza di un’impalcatura di amilopectina per essere indirizzata verso i granuli nascenti.[6]
Nelle Graminacee si conoscono due isoforme dell’enzima, codificate da geni distinti, e indicate come GBSSI e GBSSII.[12]
GBSS richiede, per la sua azione catalitica, la presenza di una proteina della famiglia PTST, la PTST1 che permette il suo legame al granulo di amido, e la cui azione sembra essere più importante nei cloroplasti rispetto agli amiloplasti.[9]
La proteina sembra si associ, nello stoma del plastidio, a GBSS. Il complesso formato si lega al granulo nascente, la proteina si dissocia dall’enzima che inizia a catalizzare l’allungamento dei malto-oligosaccaridi. PTST1 torna quindi nello stroma dove recluta un altro enzima.
Bibliografia
^ Abt M.R., Pfister B., Sharma M., Eicke S., Bürgy L., Neale I., Seung D., Zeeman S.C. STARCH SYNTHASE5, a noncanonical starch synthase-like protein, promotes starch granule initiation in Arabidopsis. Plant Cell 2020;32(8):2543-2565. doi:10.1105/tpc.19.00946
^ ab Crofts N., Abe N., Oitome N.F., Matsushima R., Hayashi M., Tetlow I.J., Emes M.J., Nakamura Y., Fujita N. Amylopectin biosynthetic enzymes from developing rice seed form enzymatically active protein complexes. J Exp Bot 2015;66(15):4469-82. doi:10.1093/jxb/erv212
^ Cuesta-Seijo J.A., Ruzanski C., Krucewicz K., Meier S., Hägglund P., Svensson B., Palcic M.M. Functional and structural characterization of plastidic starch phosphorylase during barley endosperm development. PLoS One 2017;12(4):e0175488. doi:10.1371/journal.pone
^ Gous P.W., Fox G.P. Review: Amylopectin synthesis and hydrolysis – Understanding isoamylase and limit dextrinase and their impact on starch structure on barley (Hordeum vulgare) quality. Trends Food Sci Technol 2017;62:23-32. doi:10.1016/j.tifs.2016.11.013
^ Leloir L.F., de Fekete M.A., Cardini C.E. Starch and oligosaccharide synthesis from uridine diphosphate glucose. J Biol Chem 1961;236:636-41. doi:10.1016/S0021-9258(18)64280-2
^ abc Pfister B., Zeeman S.C. Formation of starch in plant cells. Cell Mol Life Sci 2016;73(14):2781-807. doi: 10.1007/s00018-016-2250-x
^ ab Qu J., Xu S., Zhang Z., Chen G., Zhong Y., Liu L., Zhang R., Xue J., Guo D. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci Rep 2018;8(1):12736. doi:10.1038/s41598-018-30411-y
^ ab Seung D., Boudet J., Monroe J., Schreier T.B., David L.C., Abt M., Lu K.J., Zanella M., Zeeman S.C. Homologs of PROTEIN TARGETING TO STARCH control starch granule initiation in Arabidopsis leaves. Plant Cell 2017;29(7):1657-1677. doi:10.1105/tpc.17.00222
^ Seung D., Soyk S., Coiro M., Maier B.A., Eicke S., and Zeeman S.C. PROTEIN TARGETING TO STARCH is required for localising GRANULE-BOUND STARCH SYNTHASE to starch granules and for normal amylose synthesis in Arabidopsis. PLoS Biol 2015;13:e1002080. doi:10.1371/journal.pbio.1002080
^ Szydlowski N., Ragel P., Raynaud S., Lucas M.M., Roldán I., Montero M., Muñoz F.J., Ovecka M., Bahaji A., Planchot V., Pozueta-Romero J., D’Hulst C., Mérida A. Starch granule initiation in Arabidopsis requires the presence of either class IV or class III starch synthases. Plant Cell 2009;21(8):2443-57. doi:10.1105/tpc.109.066522
^ abc Tetlow I.J., Bertoft E. A review of starch biosynthesis in relation to the building block-backbone model. Int J Mol Sci 2020;21(19):7011. doi:10.3390/ijms21197011
^ Vrinten P.L., Nakamura T. Wheat granule-bound starch synthase I and II are encoded by separate genes that are expressed in different tissues. Plant Physiol 2000;122(1):255-64. doi:10.1104/pp.122.1.255
^ Ziegler G.R., Creek J.A., Runt J. Spherulitic crystallization in starch as a model for starch granule initiation. Biomacromolecules 2005;6(3):1547-54. doi:10.1021/bm049214p
L’amilosio è un polisaccaride formato da unità di α-D-glucosio legate da legami glicosidici α-(1→4), con poche ramificazioni connesse alla catena principale da legami glicosidici α-(1→6).[17]
Insieme all’amilopectina è uno dei due principali costituenti dell’amido, la più diffusa e abbondante forma di accumulo di energia e carboidrati presente nella Biosfera.[12]
La sua sintesi è catalizzata dall’enzima amido sintasi legata ai granuli o GBSS (EC 2.4.1.242), e necessita della presenza di una seconda proteina, la PTST1, che è priva di attività catalitica.[15]
Nel granulo di amido l’amilosio viene depositato all’interno della matrice semicristallina formata dall’amilopectina.[13] Sebbene a differenza dell’amilopectina l’amilosio non sia necessario per la formazione dei granuli semicristallini di amido, dove è presente in quantità inferiore rispetto all’amilopectina, l’amilosio ha una grande influenza sulle proprietà fisico-chimiche dell’amido.[7]
Sono state selezionate piante dove il granulo di amido può contenere quantità molto piccole di amilosio, e altre dove invece i granuli sono formati per la quasi totalità dal polisaccaride. Questi fenotipi possono avere vantaggi sia dal punto di vista della salute che industriale.[4][20]
Le molecole di amilosio hanno un peso molecolare di circa 106 daltons, sono per la maggior parte lineari e formate da residui di α-D-glucosio, di seguito indicato come glucosio, legati a mezzo di legami glicosidici α-(1→4), ossia legami in cui ciascuna unità glucosidica è legata alla successiva a mezzo di un legame covalente tra il C-1 di una unità e il gruppo ossidrilico sul C-4 dell’unità successiva.[1] Le catene lineari sono costituite da un numero di monosaccaridi variabile da poche centinaia a diverse migliaia, risultando quindi molto più lunghe di quelle dell’amilopectina.[17]
Le poche ramificazioni sono connesse alla catena lineare da legami glicosidici α-(1→6), al pari di quanto accade con l’amilopectina e il glicogeno, la forma di accumulo dei carboidrati presente negli animali. Nel caso dei legami glicosidici α-(1→6), le due unità di glucosio sono unite da un legame covalente tra il C-1 di un’unità e il gruppo ossidrilico sul C-6 dell’altra unità. Il numero di ramificazioni è compreso tra 5 e 20, a seconda dell’origine botanica dell’amido, ramificazioni che, rispetto all’amilopectina, non sono raggruppate.[6] Studi sulla lunghezza delle ramificazioni dell’amilosio hanno evidenziato una loro distribuzione bimodale, con le due frazioni indicate come:
AM1, che raggruppa le catene più corte, con un grado di polimerizzazione compreso tra i 100 e i 700 daltons;
AM2, dove si ritrovano le catene più lunghe, con un grado di polimerizzazione compreso tra i 700 e i 40.000 daltons.[19]
Analoga distribuzione bimodale della lunghezza delle ramificazioni si osserva nell’amilopectina, le cui frazioni sono indicate come AP1, più corte e abbondanti, e AP2.
La variazione intraspecie della distribuzione delle frazioni AM1 e AM2 è relativamente piccola, mentre è grande tra specie differenti, variazioni che hanno una base genetica.[19]
Localizzazione dell’amilosio nel granulo di amido
L’esatta localizzazione dell’amilosio all’interno del granulo di amido non è nota, sebbene si ritenga che la maggior parte si trovi nelle regioni amorfe. Tuttavia alcuni studi hanno suggerito che la sua localizzazione non sia limitata alle regioni amorfe, ma sia anche presente tra le catene di amilopectina e sulla superficie dei granuli.[13] Ne consegue che l’amilosio potrebbe avere diverse localizzazione all’interno del granulo di amido.
Sintesi
Nelle piante la sintesi dell’amilosio è catalizzata dalla GBSS, una selle sei isoforme dell’amido sintasi.[11] L’enzima, la cui azione è la principale determinante del contenuto in amilosio del granulo di amido, necessita della presenza della proteina PTST1.[15] L’inserzione delle ramificazioni sembra essere catalizzata dall’enzima ramificante l’amido o SBE (EC 2.4.1.18).
Come nel caso della sintesi dell’amilopectina, si ritiene che gli enzimi coinvolti interagiscano fisicamente a formare complessi multienzimatici, strutture in grado di ottimizzare l’efficienza dell’intero processo.[17]
Poiché la sintesi dell’amilosio necessita della presenza di un’impalcatura di amilopectina per poter indirizzare l’amido sintasi legata ai granuli verso i granuli di amido, la sintesi dei due polisaccaridi non è del tutto contemporanea.[11]
Nelle fasi iniziali, l’amido sintasi legata ai granuli, al pari delle altre amido sintasi, necessita della presenza di corti malto-oligosaccaridi o MOS, α-(1→4)-glucani lunghi da 2 a 7 residui glucosidici, che fungono da primer e sono allungati.[17] I MOS possono originare da diverse fonti, tra cui il processo di rifinitura di molecole di amilopectina nascenti per opera dell’enzima deramificante, o dall’attività dell’amido fosforilasi (EC 2.4.1.1), un altro enzima coinvolto nel metabolismo dell’amido.[16] Essendo scarsamente solubili in acqua, i MOS sembrano in grado di eludere l’azione idrolitica delle alfa-amilasi (EC 3.2.1.1) e delle beta-amilasi (EC 3.2.1.2), e diffondono all’interno della matrice dei granuli di amido in formazione dove sono allungati per azione della GBSS.[14] Una volta superati i sette residui glucosidici non riescono a diffondere facilmente al di fuori del granulo e sono ulteriormente allungati.
La fase iniziale della sintesi dell’amilosio e dell’amilopectina, richiedendo la presenza di un primer, somiglia a quella del glicogeno. Tuttavia, il primer richiesto nel corso della sintesi del glicogeno non è un oligosaccaride ma una proteina auto-glicosilante, la glicogenina.
Da notare che la polimerizzazione del glucosio in amilopectina, amilosio, o glicogeno, e più in generale di monosaccaridi osmoticamente attivi in polisaccaridi osmoticamente inerti, permette il deposito di grandi quantità di molecole osmoticamente attive all’interno della cellula senza che si verifichi un aumento della pressione osmotica.
Amido sintasi legata ai granuli
GBSS e le altre isoforme dell’amido sintasi appartengono alla famiglia delle glicosiltransferasi (EC 2.4), al pari dell’amido fosforilasi.[11]
L’enzima, scoperto dal gruppo di Louis Federico Leloir, che in precedenza aveva scoperto la principale via per il metabolismo del galattosio, la via di Leloir, è la più abbondante tra le proteine associate ai granuli di amido.[9] E’ presente quasi esclusivamente legata ai granuli, a differenza delle altre isoforme della amido sintasi che sono per la maggior parte presenti nello stroma del plastidio, o suddivise tra stroma e granuli. Inoltre, il trattamento della superficie dei granuli con proteasi ha evidenziato che la maggior parte di GBSS è presente all’interno del granulo piuttosto che sulla sua superficie, posizionamento coerente con la sintesi dell’amilosio all’interno dei granuli nascenti.[15]
Nelle graminacee la sintasi è presente in due isoforme, codificate da geni distinti, indicate come GBSSI e GBSSII, presenti rispettivamente negli amiloplasti dei tessuti di deposito, quindi non fotosintetici, e nei cloroplasti, quindi nei tessuti fotosintetici dove interviene nella sintesi dell’amido transiente.[18]
GBSS catalizza il trasferimento di una molecola di glucosio dall’ADP-glucosio all’estremità non riducente di un α-(1→4)-glucano, cui l’unità glucosidica viene legata attraverso un legame glicosidico α-(1→4).[5]
Si noti che, mentre le amido sintasi utilizzano come donatore dell’unità glucosidica l’ADP-glucosio, la glicogeno sintasi, che interviene nella sintesi del glicogeno, utilizza l’UDP-glucosio.[3]
L’amido sintasi legata ai granuli è in grado di aggiungere più di un’unità di glucosio per incontro con il substrato, modalità d’azione definita processiva, a differenza delle altre isoforme di amido sintasi che aggiungono una sola unità di glucosio per incontro con la catena di glucano nascente, azione definita distributiva. L’azione processiva permette la sintesi di lunghe catene lineari, e sembra essere fortemente incrementata dalla presenza di amilopectina.[17]
PTST1
La sintesi dell’amilosio richiede la presenza di una proteina della famiglia PTST, ossia PTST1, scoperta cinquanta anni dopo GBSS.[15]
PTST1 non ha attività catalitica, ma permette il legame di GBSS al granulo di amido, azione che sembra essere più importante nei cloroplasti, e quindi per la sintesi dell’amido transiente, che negli amiloplasti. Secondo un modello di azione di PTST1, la proteina si associa a GBSS nello stroma del plastidio, il complesso si lega al granulo di amido nascente, PTST1 si dissocia dall’enzima che inizia la sintesi dell’amilosio, mentre la proteina torna nello stroma per reclutare un’altra molecola di enzima.
L’importanza di PTST1 è sottolineata dal fatto che è conservata in tutto il regno vegetale e la sua perdita causa il distacco dell’enzima dal granulo di amido nascente.
Enzima ramificante l’amido
Non è noto l’enzima che catalizza l’inserzione delle poche ramificazioni presenti nelle molecole di amilosio, sebbene sembra possa essere coinvolta un’isoforma dell’enzima ramificante l’amido, la SBEI.[13]
Presente per la maggior parte nello stroma dei plastidi, e in piccola parte legato al granulo, è principalmente espressa nei tessuti di deposito. La bassa frequenza con cui sono presenti le ramificazioni nell’amilosio potrebbe essere il risultato della sintesi del polisaccaride all’interno dei granuli, dove la molecola nascente sarebbe protetta dall’azione dell’enzima ramificante l’amido, ivi scarsamente presente.[8]
Rapporto amilosio/amilopectina
Nei granuli di amido delle piante terrestri l’amilosio è quasi sempre presente, ma in percentuali variabili, in genere comprese tra il 5 e il 35%.[5] La variabilità si osserva non solo tra specie differenti, ma anche all’interno di una stessa specie in base all’organo o tessuto considerato, e, nei tuberi e semi, in base alla fase di sviluppo, essendo in genere il suo contenuto basso nelle prime fasi, per poi aumentare fino al raggiungimento del valore finale, una modalità coerente con la sua sintesi all’interno di una matrice di amilopectina.[13]
Esistono tuttavia piante il cui contenuto di amilosio è molto basso, o addirittura assente. I loro amidi sono detti cerosi, per l’aspetto simile a cera dell’endosperma dei chicchi crudi. Al contrario, ci sono piante i cui granuli di amido sono formati per la maggior parte, o in alcuni casi per la quasi totalità, da amilosio.[20]
Sebbene il suo ruolo biologico non sia ancora stato chiarito, la sua quasi costante presenza sembra indicare che questo polisaccaride svolga un ruolo strutturale importante nel granulo di amido, e conferisca alla pianta un qualche vantaggio nella fase di crescita e sviluppo.[13]
Il rapporto amilosio/amilopectina influenza fortemente le proprietà fisico-chimiche dell’amido, quali la sua capacità di assorbire acqua, che a sua volta influenza processi come la gelatinizzazione e la retrogradazione dell’amido, o la resistenza all’idrolisi enzimatica, quest’ultima importante ad esempio nel determinare la velocità con cui il maltosio e maltotriosio sono rilasciati per azione della alfa-amilasi e delle beta-amilasi.[10] Tutto ciò è in grado di influenzare gli utilizzi industriali dell’amido nonché i suoi effetti sulla salute.
Amidi ricchi di amilosio
I cerali ad alto contenuto di amilosio sono ottenuti attraverso l’aumento dell’espressione del gene per GBSSI, e dalla soppressione o eliminazione dei geni codificanti l’enzima ramificante l’amido, SSIIa, o altri enzimi e proteine coinvolti nella sintesi dell’amilopectina. Tuttavia, almeno per l’endosperma dei cereali, la metodica più efficace è risultata essere la soppressione o l’eliminazione di una o più isoforme dell’enzima ramificante l’amido.[4][20]
Gli amidi ad alto contenuto di amilosio hanno proprietà chimico-fisiche peculiari, tra cui una elevata forza gelificante, una maggior facilità di retrogradazione, e un’ottima capacità di formare film, caratteristiche che li rendono idonei ad applicazioni industriali come la produzione di plastica biodegradabile, di carta e di adesivi.[7][10]
Gli amidi ricchi in amilosio sono ricchi di amido resistente, un tipo di amido che resiste alla digestione intestinale ad opera della alfa-amilasi, una delle idrolasi coinvolte nella digestione dei carboidrati. Studi condotti utilizzando cibi arricchiti con amido resistente hanno evidenziato un miglioramento delle risposte insuliniche e glicemiche, nonché una riduzione del rischio di sviluppare malattie cardiovascolari, obesità e diabete mellito di tipo II.[2]
Quale potrebbe essere la loro modalità d’azione?
L’amido resistente, essendo in grado di eludere l’azione dell’alfa-amilasi, potrebbe ridurre l’indice glicemico degli alimenti in cui è presente, contribuendo così a una migliore regolazione della glicemia. Inoltre, una volta raggiunto il colon, l’amido resistente può essere fermentato dai batteri del microbiota intestinale, che è parte del più ampio microbiota umano, con produzione di acidi grassi a catena corta, in particolari l’acido butirrico, l’acido acetico e l’acido propionico, acidi grassi essenziali per la salute intestinale.
^ Birt D.F., Boylston T., Hendrich S., Jane J.L., Hollis J., Li L., McClelland J., Moore S., Phillips G.J., Rowling M., Schalinske K., Scott M.P., Whitley E.M. Resistant starch: promise for improving human health. Adv Nutr 2013;4(6):587-601. doi:10.3945/an.113.004325
^ Crofts N., Abe N., Oitome N.F., Matsushima R., Hayashi M., Tetlow I.J., Emes M.J., Nakamura Y., Fujita N. Amylopectin biosynthetic enzymes from developing rice seed form enzymatically active protein complexes. J Exp Bot 2015;66(15):4469-82. doi:10.1093/jxb/erv212
^ ab Funnell-Harris D.L., Sattler S.E., O’Neill P.M., Eskridge K.M., and Pedersen J.F. Effect of waxy (low amylose) on fungal infection of Sorghum grain. Phytopathology 2015;105(6):716-846. doi:10.1094/PHYTO-09-14-0255-R
^ ab Gous P.W., Fox G.P. Review: amylopectin synthesis and hydrolysis – Understanding isoamylase and limit dextrinase and their impact on starch structure on barley (Hordeum vulgare) quality. Trends Food Sci Technol 2016;62:23-32. doi:10.1016/j.tifs.2016.11.013
^ Hizukuri S., Takeda Y., Yasuda M., Suzuki A. Multi-branched nature of amylose and the action of debranching enzymes. Carbohydr Res 1981;94:205-213. doi:10.1016/S0008-6215(00)80718-1
^ ab Jobling S. Improving starch for food and industrial applications. Curr Opin Plant Biol 2004;7(2):210-8. doi:10.1016/j.pbi.2003.12.001
^ Kram A.M., Oostergetel G.T., Van Bruggen E. Localization of branching enzyme in potato tuber cells with the use of immunoelectron microscopy. Plant Physiol 1993;101(1):237-243. doi:10.1104/pp.101.1.237
^ Leloir L.F., de Fekete M.A., Cardini C.E. Starch and oligosaccharide synthesis from uridine diphosphate glucose. J Biol Chem 1961;236:636-41. doi:10.1016/S0021-9258(18)64280-29
^ ab Magallanes-Cruz P.A., Flores-Silva P.C., Bello-Perez L.A. Starch structure influences its digestibility: a review. J Food Sci 2017;82(9):2016-2023. doi:10.1111/1750-3841.13809
^ abc Pfister B., Zeeman S.C. Formation of starch in plant cells. Cell Mol Life Sci 2016;73(14):2781-807. doi:10.1007/s00018-016-2250-x
^ Qu J., Xu S., Zhang Z., Chen G., Zhong Y., Liu L., Zhang R., Xue J., Guo D. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci Rep 2018;8(1):12736. doi:10.1038/s41598-018-30411-y
^ abcde Seung D. Amylose in starch: towards an understanding of biosynthesis, structure and function. New Phytol 2020;228:1490-1504. doi:10.1111/nph.16858
^ Seung D., Boudet J., Monroe J., Schreier T.B., David L.C., Abt M., Lu K.J., Zanella M., Zeeman S.C. Homologs of PROTEIN TARGETING TO STARCH control starch granule initiation in Arabidopsis leaves. Plant Cell 2017;29(7):1657-1677. doi:10.1105/tpc.17.00222
^ abcd Seung D., Soyk S., Coiro M., Maier B.A., Eicke S., Zeeman S.C. PROTEIN TARGETING TO STARCH is required for localising GRANULE-BOUND STARCH SYNTHASE to starch granules and for normal amylose synthesis in Arabidopsis. PLOS Biol 2015;13(2):e1002080. doi:10.1371/journal.pbio.1002080
^ Szydlowski N., Ragel P., Raynaud S., Lucas M.M., Roldán I., Montero M., Muñoz F.J., Ovecka M., Bahaji A., Planchot V., Pozueta-Romero J., D’Hulst C., Mérida A. Starch granule initiation in Arabidopsis requires the presence of either class IV or class III starch synthases. Plant Cell 2009;21(8):2443-57. doi:10.1105/tpc.109.066522
^ abcde Tetlow I.J., Bertoft E. A review of starch biosynthesis in relation to the building block-backbone model. Int J Mol Sci 2020;21(19):7011. doi:10.3390/ijms21197011
^ Vrinten P.L., Nakamura T. Wheat granule-bound starch synthase I and II are encoded by separate genes that are expressed in different tissues. Plant Physiol 2000;122(1):255-64. doi:10.1104/pp.122.1.255
^ ab Wang K., Hasjim J., Wu A.C., Henry R.J., Gilbert R.G. Variation in amylose fine structure of starches from different botanical sources. J Agric Food Chem 2014;62(19):4443-53. doi:10.1021/jf5011676
^ abc Wang J., Hu P., Chen Z., Liu Q., Wei C. Progress in high-amylose cereal crops through inactivation of starch branching enzymes. Front Plant Sci 2017;8:469. doi:10.3389/fpls.2017.00469
La via di Leloir è la principale via per il metabolismo del galattosio.
Descritta per la prima volta da Leloir L.F. e colleghi nel 1948, porta alla conversione del galattosio in glucosio-1-fosfato, dunque all’inversione della configurazione del gruppo ossidrilico sul C4 del galattosio, carbonio che è uno dei centri di chiralità della molecola.[9]
Gli intermedi metabolici che portano alla isomerizzazione del galattosio a glucosio sono precursori utilizzati in vie metaboliche differenti, quali le reazioni di glicosilazione dei lipidi e proteine o la sintesi del glicogeno, a seconda della fase dello sviluppo, del tipo di tessuto e delle condizioni metaboliche presenti.[6]
Con l’esclusione della prima reazione, le altre reazioni della via di Leloir possono fluire in entrambe le direzioni, a seconda del livello dei substrati e delle richieste metaboliche del tessuto, il che permette l’interconversione di galattosio e glucosio.[4]
L’importanza della via di Leloir, e quindi del galattosio, è sottolineata dal fatto che è estremamente conservata in natura, essendo presente dai batteri fino alle piante e animali, e, nell’uomo, dalla gravità delle conseguenze dovute a mutazioni a carico di dei geni che codificano per gli enzimi che ne catalizzano le reazioni, mutazioni che provocano la malattia metabolica congenita nota come galattosemia.[2][8]
Il galattosio, con il glucosio e fruttosio, è uno dei monosaccaridi che può essere assorbito a livello intestinale. La principale fonte di galattosio è rappresentata dal lattosio che, con maltosio, trealosio e saccarosio, è uno dei disaccaridi presenti negli alimenti.
Essendo assenti trasportatori intestinali per i disaccaridi, nell’ultima tappa della digestione dei carboidrati i loro legami glicosidici sono idrolizzati con liberazione dei monosaccaridi costituenti, che per il lattosio sono glucosio e galattosio. Segue l’assorbimento dei monosaccaridi i quali, tramite il sistema portale, raggiungono il fegato, che è il sito principale per il metabolismo del galattosio e ne assorbe, attraverso diffusione passiva mediata dal trasportatore GLUT2, la maggior parte, circa l’88%.[3] La quantità residua circolante raggiunge altri organi e tessuti, come la ghiandola mammaria che, in fase di allattamento, lo utilizza per la produzione del lattosio e la glicosilazione delle proteine e dei lipidi del latte.[4]
Tappe della via di Leloir
La via di Leloir si compone di quattro reazioni catalizzate da altrettanti enzimi che sono la galattosio mutarotasi o aldoso-1-epimerasi (EC 5.1.3.3), la galattochinasi (EC 2.7.1.6), galattosio-1-fosfato uridiltransferasi (EC 2.7.7.12) e la galattosio 4-epimerasi (EC 5.1.3.2).[8]
La Via di Leloir
Reazione 1: galattosio mutarotasi
L’idrolisi del legame glicosidico β-(1→4) del lattosio porta alla liberazione di glucosio e beta-galattosio. Tuttavia il secondo enzima della via di Leloir, la galattochinasi, è specifica per l’alfa-galattosio. La conversione nell’anomero alfa è catalizzata dalla galattosio mutarotasi. L’enzima è in grado d’interconvertire le configurazioni anomeriche anche del glucosio, dello xilulosio, del maltosio e del lattosio, sebbene con efficienza variabile.[12]
Reazione 2: galattochinasi
Nel secondo passaggio l’alfa-galattosio viene fosforilato a galattosio-1-fosfato nella reazione catalizzata dalla galattochinasi.[8] La fosforilazione del galattosio è importante in quanto:
blocca il monosaccaride all’interno della cellula poiché è caricato negativamente e sono assenti sulla membrana plasmatica trasportatori per gli zuccheri fosforilati;
ne aumenta il contenuto in energia iniziandone la destabilizzazione il che faciliterà il successivo metabolismo;
ne mantiene bassa la concentrazione intracellulare, favorendone l’ulteriore passaggio all’interno della cellula.
La reazione catalizzata dalla galattochinasi è l’unica reazione irreversibile della via di Leloir.[4] Inoltre, a differenza della esochinasi e della glucochinasi (EC 2.7.1.1), che catalizzano la fosforilazione del gruppo ossidrilico sul C6 del glucosio, la galattochinasi e la fruttochinasi (EC 2.7.1.4) catalizzano la fosforilazione dei gruppi ossidrilici legati rispettivamente al C1 di galattosio e fruttosio.[11]
La conversione del galattosio-1-fosfato a glucosio-1-fosfato richiede due reazioni, rispettivamente la terza e la quarta della via di Leloir.
Nel terzo passaggio la galattosio-1-fosfato uridiltransferasi catalizza il trasferimento del gruppo UMD dall’UDP-glucosio al galattosio-1-fosfato, con formazione di UDP-galattosio e glucosio-1-fosfato. La reazione procede con un meccanismo a ping-pong con formazione di un intermedio covalente tra l’enzima e l’UMP.[7]
Reazione 4: UDP-galattosio 4-epimerasi
Nell’ultimo passaggio l’UDP-galattosio viene convertito in UDP-glucosio nella reazione catalizzata dalla UDP-galattosio 4-epimerasi.
L’enzima catalizza l’inversione della configurazione del gruppo ossidrilico sul C4 ed è il punto di interconversione tra UDP-galattosio e UDP-glucosio.[8]
L’enzima richiede come cofattore il NAD+ e la reazione procede attraverso la formazione di un intermedio chetonico sul C4, con contemporanea riduzione del NAD+ a NADH. Successivamente l’intermedio chetonico ruota presentando la faccia opposta dello zucchero al NADH. A questo punto uno ione idruro viene ritrasferito dal NADH al C4, ma in configurazione opposta.[13] Poiché il NAD nel corso della reazione viene prima ridotto e poi ossidato, non subisce alcuna ossidoriduzione netta per cui non compare nell’equazione di reazione. Sembra che la UDP-galattosio 4-epimerasi sia l’enzima limitante la via di Leloir.
L’UDP-glucosio prodotto viene riciclato nella reazione catalizzata dalla UDP-glucosio pirofosforilasi, con rilascio di glucosio-1-fosfato.[6]
Nei mammiferi la galattosio 4-epimerasi catalizza anche la interconversione tra UDP-N-acetilgalattosammina e UDP-N-acetilglucosammina.[3]
Si noti che UDP-galattosio e UDP-glucosio, o più in generale galattosio e glucosio, hanno la stessa formula molecolare. Sono quindi isomeri e differiscono solo per la configurazione del quarto atomo di carbonio. Pertanto sono un esempio di isomeria ottica.
Quali sono le funzioni della via di Leloir?
La via di Leloir permette alla cellula di utilizzare il galattosio o il glucosio derivato in diverse vie metaboliche, sia anaboliche che cataboliche, a seconda delle condizioni metaboliche della cellula o del tessuto. Inoltre, poiché la reazione catalizzata dalla UDP-galattosio 4-epimerasi è reversibile, è possibile la conversione del glucosio a galattosio e derivati nucleotidici.[4][6]
L’UDP-galattosio può essere utilizzato:
nelle reazioni di glicosilazioni di proteine e lipidi, come i galattocerebrosidi che sono i principali glicolipidi della mielina, ed è per questo che il galattosio fu inizialmente chiamato cerebrosio;
nella ghiandola mammaria in allattamento per la produzione del lattosio nella reazione catalizzata dal complesso della lattosio sintasi. Inoltre, essendo la reazione catalizzata dalla UDP-galattosio 4-epimerasi reversibile sarà anche possibile la conversione del glucosio, previa attivazione a UDP-glucosio nella reazione catalizzata dalla UDP-glucosio pirofosforilasi (EC 2.7.7.9), in UDP-galattosio per la sintesi del lattosio.
Nel fegato e nel muscolo scheletrico l’UDP-glucosio derivante dell’UDP-galattosio, può essere utilizzato:[4]
nelle reazioni di glicosilazione dei lipidi e delle proteine;
nella sintesi del glicogeno, quando la richiesta energetica della cellula è bassa; e, rispetto al glucosio e fruttosio, il galattosio è preferenzialmente incorporato nel glicogeno epatico piuttosto che essere indirizzato verso il metabolismo ossidativo;
a seguito della conversione in glucosio-1-fosfato, nella reazione catalizzata dalla UDP-glucosio pirofosforilasi, e isomerizzazione a glucosio-6-fosfato, nella reazione catalizzata dalla fosfoglucomutasi, può entrare in vie metaboliche differenti quali la glicolisi, la via del pentoso fosfato o la gluconeogenesi.[3]
Nota: l’UDP-galattosio è stato il primo zucchero nucleotidico a essere scoperto, scoperta avvenuta proprio nel corso degli studi sulla via di Leloir.[1]
Via di Leloir e galattosemia
Le glicosilazioni sono modificazioni post-traduzionali che hanno un ruolo importante nel rendere possibili e regolare differenti processi biologici. Difetti a loro carico sono stati correlati a molte condizioni patologiche come cancro, diabete ed errori congeniti del metabolismo quali i disturbi congeniti della glicosilazione, principalmente disordini monogenici autosomici recessivi.[5] Tra questi ultimi si annovera la galattosemia, descritta per la prima volta nel 1908 da von Reuss A.[14] La galattosemia è conseguente a mutazioni a carico di uno dei geni che codificano per i quattro enzimi della via di Leloir, e ne sono stati individuati quattro tipi:
tipo I, da carenza di galattosio-1-fosfato uridiltransferasi, la forma più comune, anche detta galattosemia classica;
A oggi, lo standard terapeutico per la cura della galattosemia è una dieta a ridotto contenuto di galattosio.
Galattosemia e cataratta
L’accumulo di galattosio ha quale effetto quello di attivare vie metaboliche alternative quali la sintesi del galattitolo e del galattonato.
Tra i sintomi della galattosemia si ritrova la comparsa precoce di cataratta, in genere entro i primi due anni di vita, e nei casi più severi danni al cervello, reni e fegato.[12]
Sembra che uno dei fattori che determinano l’insorgenza della cataratta sia la riduzione del galattosio, accumulatosi nella lente dell’occhio, a galattitolo, nella reazione catalizzata dalla aldoso reduttasi (EC 1.1.1.21).[10] Il galattitolo, un poliolo scarsamente metabolizzato, non diffonde attraverso la membrana plasmatica a causa della sua scarsa lipofilicità, ed essendo un composto osmoticamente molto attivo determina un aumento della pressione osmotica intracellulare, con conseguente richiamo d’acqua nella lente. Inoltre la sua sintesi, riducendo i livelli intracellulari di NADPH, può determinare una riduzione dell’attività della glutatione reduttasi (EC 1.8.1.7) causando un accumulo di radicali liberi.[3] L’effetto osmotico e l’accumulo di radicali liberi possono danneggiare l’integrità della cellula e causarne la morte. Inoltre, è stato riportato che il galattitolo è un inibitore della galattosio mutarotasi, per cui il suo accumulo potrebbe causare un ulteriore accumulo di galattosio non metabolizzato.[12]
Bibliografia
^ Cardini C.E., Paladini A.C., Caputto R., Leloir L.F. Uridine diphosphate glucose: the coenzyme of the galactose-glucose phosphate isomerization. Nature 1950;165:191-192. doi:10.1038/165191a0
^ Coelho A.I., Berry G.T., Rubio-Gozalbo M.E. Galactose metabolism and health. Curr Opin Clin Nutr Metab Care 2015;18(4):422-427. doi:10.1097/MCO.0000000000000189
^ abcd Coelho A.I., Rubio-Gozalbo M.E., Vicente J.B., Rivera I. Sweet and sour: an update on classic galactosemia. J Inherit Metab Dis 2017;40(3):325-342. doi:10.1007/s10545-017-0029-3
^ abcde Conte F., van Buuringen N., Voermans N.C., Lefeber D.J. Galactose in human metabolism, glycosylation and congenital metabolic diseases: time for a closer look. Biochim Biophys Acta Gen Subj 2021;1865(8):129898. doi:10.1016/j.bbagen.2021.129898
^ abc Frey P.A. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J 1996;10(4):461-70. doi:10.1096/fasebj.10.4.8647345
^ abcd Holden H.M., Rayment I., Thoden J.B. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem 2003;278(45):43885-43888. doi:10.1074/jbc.R300025200
^ Leloir L.F., de Fekete M.A., Cardini C.E. Starch and oligosaccharide synthesis from uridine diphosphate glucose. J Biol Chem 1961;236:636-41. doi:10.1016/S0021-9258(18)64280-2
^ Pintor J. Sugars, the crystalline lens and the development of cataracts. Biochem Pharmacol 2012;1:4. doi:10.4172/2167-0501.1000e1190
^ Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009
^ abc Thoden J.B., Timson D.J., Reece R.J., and M. Holden H.M. Molecular structure of human galactose mutarotase. J Biol Chem 2004;279(22):23431-23437. doi:10.1074/jbc.M402347200
^ Thoden J.B., Wohlers T.M., Fridovich-Keil J.L., Holden H.M. Human UDP-galactose 4-epimerase. Accommodation of UDP-N-acetylglucosamine within the active site. J Biol Chem 2001;276(18):15131-6. doi:10.1074/jbc.M100220200
^ Wada Y., Kikuchi A., Arai-Ichinoi N., Sakamoto O., Takezawa Y., Iwasawa S., Niihori T., Nyuzuki H., Nakajima Y., Ogawa E., Ishige M., Hirai H., Sasai H., Fujiki R., Shirota M., Funayama R., Yamamoto M., Ito T., Ohara O., Nakayama K., Aoki Y., Koshiba S., Fukao T., Kure S. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet Med 2019;21(6):1286-1294. doi:10.1038/s41436-018-0340-x
In una soluzione, le molecole di solvente tendono a spostarsi dalla regione dove la loro concentrazione è maggiore verso quella a concentrazione minore. Se si considerano due soluzioni separate da una membrana semipermeabile, ossia una membrana che consenta il passaggio solo a certi ioni o molecole, in questo caso le molecole di solvente, si verrà a creare attraverso la membrana un flusso netto di molecole di solvente dalla soluzione a concentrazione maggiore di solvente verso quella a concentrazione minore. Questo porta allo sviluppo di una pressione detta pressione osmotica, indicata con Π, che può essere definita come la forza che deve essere applicata per impedire lo spostamento delle molecole di solvente attraverso una membrana semipermeabile.
Assieme all’innalzamento del punto di ebollizione, all’abbassamento del punto di congelamento e alla tensione di vapore, la pressione osmotica è una delle quattro proprietà colligative delle soluzioni, proprietà che dipendono solo dal numero di particelle di soluto presenti in soluzione, ioni, molecole o strutture sopramolecolari che siano, e non dalla natura delle particelle stesse o dalla loro massa.
Per soluzioni con n soluti, l’equazione che descrive la pressione osmotica è la somma dei contributi di ciascun soluto:
Π = RT(i1c1 + i2c2 + … + incn)
L’equazione è conosciuta come equazione di van ‘t Hoff, dove:
T è la temperatura assoluta, che è espressa in Kelvin;
Il fattore di van ‘t Hoff è una misura del grado di dissociazione del soluto in soluzione, ed è descritto dall’equazione:
i = 1 + α(n-1)
dove:
α è il grado di dissociazione delle molecole di soluto, pari al rapporto tra le moli delle molecole di soluto che hanno subito dissociazione e il numero delle moli iniziali, e può assumere valori compresi tra 0, per sostanze che non si ionizzano o dissociano, e 1, per sostanze che si ionizzano o dissociano completamente in soluzione;
n è il numero di ioni ottenuti dalla dissociazione completa della molecola di soluto.
Se si considerano specie non ionizzabili, come il glucosio, il glicogeno o l’amido, n = 1 e i = 1, mentre, per specie che in soluzione diluite si dissociano completamente, come acidi e basi forti o sali, il fattore di van ‘t Hoff è un numero intero maggiore di uno, essendo α = 1 e n pari ad almeno 2. Se ad esempio si considera il cloruro di sodio, NaCl, il cloruro di potassio, KCl, o il cloruro di calcio, CaCl2, si ha:
Quindi nei primi due casi i = 2, mentre con il cloruro di calcio è pari a 3.
Infine, per sostanze che non si ionizzano completamente, come acidi e basi deboli, i non è un numero intero.
Il termine ic, il prodotto del fattore di van ‘t Hoff e la concentrazione molare del soluto, è l’osmolarità della soluzione, ossia la concentrazione delle particelle di soluto osmoticamente attive per litro di soluzione.
Pressione osmotica, osmosi e membrane cellulari
L’osmosi può essere definita come il movimento o flusso netto di un solvente attraverso una membrana semipermeabile, sotto la spinta delle differenze di pressione osmotica tra i due lati della membrana, al fine di cercare di uguagliare la concentrazione del soluto ai due lati della membrana stessa.
Nei sistemi biologici l’acqua è il solvente e le membrane plasmatiche sono le membrane semipermeabili.
Le membrane plasmatiche consentono il passaggio alle molecole d’acqua, grazie alla presenza di canali proteici, le acquaporine, nonché a piccole molecole non polari che possono diffondere rapidamente attraverso esse, mentre sono stanzialmente impermeabili a ioni e macromolecole.
La presenza all’interno della cellula di macromolecole come acidi nucleici, proteine, glicogeno e aggregati sopramolecolari, ad esempio i complessi multienzimatici, ma anche ioni in concentrazione maggiore rispetto a quella dell’ambiente extracellulare, fa si che la pressione osmotica guidi l’acqua dall’esterno all’interno della cellula. Se questo flusso netto di acqua verso l’interno della cellula non fosse controbilanciato si avrebbe in breve una distensione della membrana plasmatica tale da comportarne la rottura, ossia la cellula scoppierebbe a causa di un eccesso di acqua al suo interno, si verificherebbe cioè una lisi osmotica. In condizioni fisiologiche questo non avviene in quanto nel corso dell’evoluzione si sono sviluppati diversi meccanismi che si oppongono, e in alcuni casi addirittura sfruttano, queste forze osmotiche.
Due di questi sono le pompe ioniche energia dipendenti e, nei batteri, nei fungi e nelle cellule vegetali, la parete cellulare.
Pompe ioniche energia dipendenti
Le pompe ioniche riducono, con spesa di ATP, le concentrazioni intracellulari di determinati ioni rispetto alle loro concentrazioni nell’ambiente extracellulare, creando quindi una ineguale distribuzione degli ioni stessi sui due lati della membrana plasmatica, ossia creando un gradiente ionico. In questo modo la cellula controbilancia le forze osmotiche dovute agli ioni e macromolecole intrappolate al suo interno. Un esempio di pompa ionica energia dipendente è la Na+/K+ ATPasi, che riduce la concentrazione intracellulare di Na+ rispetto all’esterno della cellula.
Parete cellulare
Le cellule vegetali sono circondate da una matrice extracellulare, la parete cellulare, che, essendo non espandibile e posizionata in prossimità della membrana plasmatica, permette alla cellula di resistere alle forze osmotiche che potrebbero causarne il rigonfiamento e infine la lisi.
In che modo?
Nelle cellule vegetali mature i vacuoli sono gli organelli di dimensioni maggiori, arrivando a occupare circa l’80% del volume cellulare. Al loro interno vengono accumulate grandi quantità di soluti, per la maggior parte acidi organici e inorganici, i quali osmoticamente richiamano acqua, il che determina il rigonfiamento del vacuolo stesso. A sua volta questo fa si che il tonoplasto, la membrana che circonda l’organello, spinga la membrana plasmatica contro la parete cellulare, la quale, opponendosi meccanicamente a queste forze, permette di evitare la lisi osmotica. Questa pressione osmotica è chiamata pressione di turgore e può raggiungere le 20 atmosfere, 2 MPa, un valore circa 10 volte maggiore rispetto alla pressione dei pneumatici. La pressione di turgore è responsabile della rigidità delle parti non legnose delle piante, è coinvolta nella crescita della pianta, nonchè:
nell’avvizzimento della verdura, a seguito di una sua riduzione;
nei movimenti delle piante, quali:
i movimenti circadiani delle foglie;
i movimenti delle foglie della Dionaea muscipula, una pianta carnivora, o delle foglie delle Mimosa pudica.
Anche nei batteri e funghi la membrana plasmatica è circondata da una parete cellulare sufficientemente rigida e non espandibile da impedire la lisi osmotica della cellula.
Soluzioni isotoniche, ipotoniche e ipertoniche
Dal confronto tra la pressione osmotica di sue soluzioni separate da una membrana semipermeabile è possibile definire tre tipi di soluzioni, di seguito brevemente descritte.
Due soluzioni che abbiano la stessa pressione osmotica sono definite isotoniche.
Se due soluzioni hanno differenti pressione osmotica, quella a pressione maggiore viene definita ipertonica rispetto all’altra.
Se due soluzioni hanno differenti pressioni osmotiche, quella a pressione minore viene definita ipotonica rispetto all’altra.
Nei sistemi biologici la soluzione di riferimento è rappresentata dal citosol; quindi, ponendo una cellula in una soluzione:
isotonica, non si avrà trasferimento netto di acqua tra l’interno e l’esterno della cellula stessa;
ipertonica, sia avrà un trasferimento netto di acqua dalla cellula verso l’esterno, la cellula perde acqua e si raggrinzisce;
ipotonica, si verifica un trasferimento netto di acqua al suo interno, la cellula si rigonfia e può arrivare a scoppiare, ossia si può verificare una lisi osmotica.
In aggiunta alle pompe ioniche e alla parete cellulare, nel corso dell’evoluzione gli organismi pluricellulari hanno sviluppato un’altra soluzione per opporsi alle forze osmotiche: circondare le cellule con soluzioni isotoniche o prossime all’isotonicità che impediscano o comunque limitino un influsso o un efflusso netto di acqua. Un esempio è il plasma, ossia il sangue privato della componente cellulare, che, grazie alla presenza di sali e proteine, nell’uomo principalmente l’albumina, ha un’osmolarità simile a quella presente nel citosol.
Pressione osmotica, amido e glicogeno
Gli organismi immagazzinano glucosio non in forma libera ma come polimeri, glicogeno gli animali, i funghi e i batteri, amido le piante, in questo modo evitando che la pressione osmotica esercitata dalle riserve di carboidrati diventi troppo grande. Infatti, dato che la pressione osmotica, al pari delle altre proprietà colligative, dipende solo dal numero delle molecole di soluto, immagazzinare milioni di molecole di glucosio in forma di un numero notevolmente inferiore di polisaccaridi permette di evitarne un aumento abnorme. Di seguito alcuni esempi.
Se si considera un polisaccaride, come il glicogeno o amido, formato da 1000 unità di glucosio e del peso di un grammo, questi ha un effetto sulla pressione osmotica inferiore a quello di un milligrammo di glucosio libero.
Considerando un epatocita, se il glucosio immagazzinato in forma di glicogeno fosse presente come glucosio libero, la sua concentrazione sarebbe di circa 0,4 M, contro una concentrazione del glicogeno di circa 0,04 μM. Questo causerebbe un flusso netto di acqua verso l’interno della cellula tale da portare a lisi osmotica.
Inoltre, se anche si riuscisse a evitare la lisi osmotica, si avrebbero problemi riguardo al trasporto del glucosio all’interno della cellula. Nell’uomo, in condizioni fisiologiche, il sistema preposto alla regolazione della glicemia agisce in modo da mantenere i livelli glucosio nel sangue compresi tra 3,33 e 5,56 mmol/L o 60-100 mg/dL. Se il glucosio fosse immagazzinato in forma libera la sua concentrazione intracellulare sarebbe da 120 a 72 volte maggiore di quella sanguigna, e il suo trasporto nell’epatocita comporterebbe un grande dispendio energetico.
Diarrea osmotica
In presenza di malattie che causano l’accumulo di soluti non assorbiti e osmoticamente attivi nella porzione distale dell’intestino tenue e nel colon, si verifica una condizione nota come diarrea osmotica.
Tra le cause fisiopatologiche si annoverano, ad esempio, infezioni batteriche, malattie del pancreas, la celiachia, un’enteropatia autoimmune indotta dall’assunzione di glutine in soggetti geneticamente predisposti, o un deficit congenito di una delle disaccaridasi dell’orletto a spazzola degli enterociti, come nel caso dell’intolleranza al lattosio. In queste condizioni la digestione dei carboidrati può essere incompleta a causa di un deficit a carico della alfa-amilasi e/o di una o più disaccaridasi. Inoltre, i soluti osmoticamente attivi non assorbiti passano nel colon dove possono essere fermentati dai batteri del microbiota intestinale, che fa parte del microbiota umano, con conseguente eccessiva produzione di gas, come idrogeno, anidride carbonica e metano, e acidi grassi catena corta, in particolare l’acido butirrico, l’acido propionico e l’acido acetico. Ciò provoca una condizione nota come diarrea osmotico-fermentativa.
La diarrea osmotica può anche essere consente all’uso di lassativi osmotici come polietilenglicole o PEG e il solfato di magnesio.
L’accumulo di soluti osmoticamente attivi derivanti dalla digestione incompleta al pari dei lassativi osmotici portano a un aumento della pressione osmotica intraluminale e inibiscono il normale assorbimento di acqua ed elettroliti, provocando una riduzione della consistenza delle feci e un aumento della motilità intestinale.
Pressione osmotica, galattosemia e cataratta
Il galattosio, con il fruttosio e il glucosio, è uno dei tre monosaccaridi che possono essere assorbiti a livello intestinale.
Metabolizzato per la maggior parte nel fegato, e in misura minore da altri organi e tessuti, previa conversione in UDP-galattosio e UDP-glucosio attraverso la via di Leloir, il galattosio può essere utilizzato a scopi anabolici o catabolici. Mutazioni a carico di uno dei geni che codificano per gli enzimi della via di Leloir ne provocano l’accumulo e causano la galattosemia, una malattia metabolica congenita la cui unica cura è una dieta a ridotto contenuto di galattosio.
L’accumulo del monosaccaride determina l’attivazione di vie metaboliche alternative alla via di Leloir che portano alla sua conversione in galattitolo e galattonato.
Tra i sintomi della galattosemia si ritrova la comparsa precoce di cataratta e tra i fattori scatenanti sembra esserci la sintesi di galattitolo nella lente dell’occhio. Il galattitolo è un poliolo scarsamente metabolizzato e, data la sua scarsa lipofilicità, non diffonde attraverso la membrana plasmatica, accumulandosi nella cellula. Essendo osmoticamente attivo determina un aumento della pressione osmotica intracellulare e di conseguenza un richiamo d’acqua. L’effetto osmotico sembra essere uno dei meccanismi attraverso cui il galattitolo concorre allo sviluppo della cataratta in caso di galattosemia.
Bibliografia
Beauzamy L., Nakayama N., and Boudaoud A. Flowers under pressure: ins and outs of turgor regulation in development. Ann Bot 2014;114(7):1517-1533. doi:10.1093/aob/mcu187
Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002
Coelho A.I., Berry G.T., Rubio-Gozalbo M.E. Galactose metabolism and health. Curr Opin Clin Nutr Metab Care 2015;18(4):422-427. doi:10.1097/MCO.0000000000000189
Coelho A.I., Rubio-Gozalbo M.E., Vicente J.B., Rivera I. Sweet and sour: an update on classic galactosemia. J Inherit Metab Dis 2017;40(3):325-342. doi:10.1007/s10545-017-0029-3
Conte F., van Buuringen N., Voermans N.C., Lefeber D.J. Galactose in human metabolism, glycosylation and congenital metabolic diseases: time for a closer look. Biochim Biophys Acta Gen Subj 2021;1865(8):129898. doi:10.1016/j.bbagen.2021.129898
Gli enzimi multifunzionali sono proteine in cui due o più siti attivi che catalizzano reazioni consecutive di una via metabolica sono presenti in una catena polipeptidica. Sembra probabile che derivino da eventi di fusione genica rappresentando, al pari dei complessi multienzimatici, una soluzione adottata dall’evoluzione per massimizzare l’efficienza catalitica, assicurando vantaggi che non si avrebbero nel caso in cui le singole attività enzimatiche fossero presenti su proteine distinte e libere.
Quali vantaggi offrono gli enzimi multifunzionali?
Gli organismi viventi combattono con l’inevitabile processo di decadimento che, se non contrastato, conduce a un crescente aumento del disordine, sino alla morte.
A livello molecolare il mantenimento della vita è reso possibile dalla grande efficienza raggiunta dagli enzimi nell’accelerare le reazioni chimiche e nell’evitare reazioni collaterali. Un’idea della velocità con cui procede il metabolismo cellulare è fornita dalla velocità del turn over dell’ATP in una cellula di mammifero: ogni 1-2 minuti l’intero pool del trifosfato è idrolizzato e risintetizzato. Tradotto in numeri questo corrisponde al turn over di circa 107 molecole di ATP al secondo, e, per il corpo umano, a circa 1 grammo di ATP al minuto. Alcuni enzimi hanno addirittura raggiunto la perfezione catalitica, ossia sono talmente efficienti che quasi ogni collisione con il proprio substrato porta alla catalisi.
E uno dei fattori limitanti la velocità di una reazione enzimatica è la proprio frequenza con cui enzimi e substrati collidono. Il modo più semplice per aumentare la frequenza delle collisioni sarebbe quello di aumentare la concentrazione di substrati ed enzimi. Tuttavia, dato l’elevatissimo numero di differenti reazioni che avvengono all’interno della cellula, questa strada non è praticabile. Esiste cioè un limite alle concentrazioni che substrati ed enzimi possono raggiungere, concentrazioni che sono dell’ordine delle micromoli per i substrati, e anche più basse per gli enzimi. Fanno eccezione gli enzimi della glicolisi nelle cellule muscolari e negli eritrociti, presenti in concentrazioni dell’ordine delle 0.1 mM e anche maggiori.
Incanalamento dei substrati
Una delle strade percorse dall’evoluzione per aumentare la velocità con cui procedono le reazioni enzimatiche è stata quella di selezionare strutture molecolari, quali gli enzimi multifunzionali e i complessi multienzimatici, che permettano, attraverso l’ottimizzazione dell’organizzazione spaziale degli enzimi di una via metabolica, di minimizzare la distanza che il prodotto della reazione A deve percorrere per raggiungere il sito attivo che catalizza la reazione successiva B, e così via, ottenendo cioè l’incanalamento dei substrati della via stessa. Per alcuni enzimi multifunzionali e complessi multienzimatici l’incanalamento è ottenuto grazie alla presenza di veri e propri tunnel intramolecolari. L’incanalamento dei substrati migliora l’efficienza catalitica, e quindi la velocità di reazione, in vari modi, di seguito brevemente descritti.
Minimizza la diffusione nel mezzo circostante del reagente, quindi la sua diluizione, permettendo così di raggiungere concentrazioni locali elevate, anche quando la sua concentrazione nella cellula è bassa, aumentando quindi la frequenza delle collisioni enzima-substrato.
Riduce il tempo di transito dei substrati da un sito attivo al successivo.
Minimizza la probabilità che si verifichino reazioni collaterali.
Minimizza la probabilità che intermedi chimicamente labili siano degradati.
Gli enzimi multifunzionali offrono vantaggi anche dal punto di vista della regolazione della loro sintesi, essendo possibile coordinare la sintesi di tutte le attività enzimatiche interessate grazie al fatto che la struttura è codificata da un singolo gene.
Infine, analogamente ai complessi multienzimatici, è possibile regolare in modo coordinato le attività enzimatiche presenti. Se a questo si aggiunge che spesso l’enzima che catalizza la tappa di comando della sequenza è quello che catalizza la prima reazione, è possibile sia evitare la sintesi di molecole non necessarie, che sarebbero invece prodotte se la tappa di comando fosse a valle della prima reazione, come pure uno spreco di energia e la sottrazione di metaboliti ad altre vie metaboliche.
Esempi di enzimi multifunzionali
Al pari dei complessi multienzimatici, anche gli enzimi multifunzionali sono molto comuni e coinvolti in vie metaboliche sia anaboliche che cataboliche.
Di seguito alcuni esempi.
Acetil-CoA carbossilasi
L’acetil-CoA carbossilasi o ACC (EC 6.4.1.2), una carbossilasi biotina-dipendente, è formata da due enzimi, la biotina carbossilasi (EC 6.3.4.14) e una transcarbossilasi, più la proteina trasportatrice della biotina o BCCP, acronimo dell’inglese biotin carboxyl-carrier protein. ACC catalizza la sintesi del malonil-CoA via carbossilazione dell’acetil-CoA. La reazione, che rappresenta la tappa di comando della sintesi degli acidi grassi, procede in due tappe.
Nella prima la biotina carbossilasi catalizza il trasferimento di una molecola di anidride carbonica (CO2) dallo ione bicarbonato a un atomo di azoto dell’anello della biotina, che funge da trasportatore temporaneo di CO2. La reazione comporta il consumo di una molecola di ATP.
Nella seconda tappa la transcarbossilasi catalizza il trasferimento del gruppo carbossilico dalla carbossi-biotina all’acetil-CoA a dare malonil-CoA.
Il malonil-CoA verrà di seguito utilizzato come donatore di unità bicarboniose dalla acido grasso sintasi (EC 2.3.1.85) durante l’allungamento degli acidi grassi.
Nei mammiferi e negli uccelli l’acetil-CoA carbossilasi è un enzima multifunzionale essendo le due attività enzimatiche, e BCCP, presenti su un’unica catena polipeptidica. Nei batteri invece ACC è un complesso multienzimatico formato dall’aggregazione di tre catene polipeptidiche, ossia i due enzimi più BCCP.
Nelle piante superiori sono presenti entrambe le forme.
Acido grasso sintasi di tipo I
L’acido grasso sintasi o FAS, acronimo dell’inglese fatty acid synthase, catalizza la sintesi dell’acido palmitico utilizzando il malonil-CoA, che è il prodotto della reazione catalizzata dalla acetil-CoA carbossilasi, come donatore di unità bicarboniose.
Esistono due tipi di acido grasso sintasi.
Negli animali e nei funghi è un enzima multifunzionale, ed è detto di tipo I. Negli animali è un omodimero, e in ogni catena polipeptidica sono presenti le sette le attività enzimatiche e la proteina trasportatrice di acili o ACP, acronimo dell’inglese acyl carrier protein. Nei lieviti e nei funghi FAS è formata da due subunità multifunzionali, dette α and β, disposte a formare una struttura eterododecamerica α6β6.
Nella maggior parte dei procarioti e nelle piante l’acido grasso sintasi, detta di tipo II, non è un enzima multifunzionale ma un complesso multienzimatico essendo composto da enzimi distinti più ACP.
PRA isomerasi:IGP sintasi
La sintesi dell’aminoacido triptofano a partire dal corismato coinvolge diversi passaggi, di seguito brevemente descritti.
Nel primo, la glutammina dona un atomo di azoto all’anello indolico del corismato, che è convertito in antranilato, e la glutammina in glutammato; la reazione è catalizzata dalla antranilato sintasi (EC 4.1.3.27).
L’antranilato è fosforibosilato a spese del 5-fosforibosil-1-pirofosfato o PRPP a dare N-(5’-fosforibosil)-antranilato o PRA, nella reazione catalizzata dalla antranilato fosforibosiltransferasi (EC 2.4.2.18).
Nel passaggio successivo, catalizzato dalla PRA isomerasi (EC 5.3.1.24), PRA è isomerizzato a dare enol-1-o-carbossifenilamino-1-desossiribulosio fosfato o CdRP. PRA e CdRP sono un esempio di isomeria di struttura.
CdRP è convertito in indolo-3-glicerolo fosfato o IGP nella reazione catalizzata dalla indolo-3-glicerofosfato sintasi o IGP sintasi (EC 4.1.1.48).
Infine, la triptofano sintasi (EC 4.2.1.20) catalizza gli ultimi due passaggi della via: la conversione di IGP in indolo, una idrolisi, e la reazione dell’indolo con una serina a dare il triptofano.
In E. coli, PRA isomerasi e IGP sintasi sono presenti su un’unica catena polipeptidica, che è quindi un enzima bifunzionale. In altri microorganismi, come Bacillus subtilis, Salmonella typhimurium e Pseudomonas putida le due attività enzimatiche sono presenti su catene polipeptidiche distinte.
La triptofano sintasi è invece un esempio di complesso multienzimatico, e uno degli esempi meglio caratterizzati di canalizzazione del flusso dei metaboliti.
Glutammina-PRPP amido transferasi
La glutammina-PRPP ammidotransferasi (EC 2.4.2.14) catalizza la prima della 10 tappe che portano alla sintesi de novo delle purine, ossia la formazione della 5-fosforibosilammina a seguito del trasferimento dell’azoto ammidico della glutammina, che funge quindi da fonte di azoto, al PRPP.
La reazione procede in due tappe, catalizzate nei due siti attivi dell’enzima, un sito N-terminale e uno C-terminale.
Nella prima tappa, il sito attivo N-terminale catalizza l’idrolisi dell’azoto ammidico della glutammina a dare ammoniaca e glutammato.
Nella seconda tappa, catalizzata dal sito attivo C-terminale, che ha attività fosforibosiltransferasica, l’ammoniaca precedentemente rilasciata viene legata al C-1 del PRPP a dare la 5-fosforibosilammina. In questa reazione si verifica l’inversione della configurazione del C-1 del ribosio, da α a β, stabilendo la forma anomerica del nucleotide in via di sintesi.
Ci sono tre punti di controllo che cooperano nella regolazione della sintesi de novo dei nucleotidi purinici, e la reazione catalizzata dalla glutammina-PRPP ammidotransferasi, che è anche la prima reazione esclusiva della via, è il primo.
Al pari del complesso della carbamil fosfato sintetasi batterica, anche i siti attivi di questo enzima multifunzionale sono connessi attraverso un canale intramolecolare. Tuttavia questo canale è più corto, essendo lungo circa 20 Å, ed è delimitato da residui amminoacidici non polari, quindi è altamente idrofobico. Mancando gruppi in grado di stabilire legami idrogeno, il tunnel permette la diffusione dell’ammoniaca verso il secondo sito attivo.
CAD
La sintesi de novo dei nucleotidi pirimidinici avviene attraverso una serie di reazioni enzimatiche che, a differenza di quanto accade nella sintesi de novo dei nucleotidi purinici, comporta dapprima la formazione dell’anello pirimidinico e quindi il suo legame al ribosio-5-fosfato. Le prime tre tappe della via sono catalizzate in sequenza dalla carbamil fosfato sintetasi (EC 6.3.4.16), aspartato transcarbamilasi (EC 2.1.3.2) e diidroorotasi (EC 3.5.2.3), e sono comuni a tutte le specie.
Nella prima tappa, la carbamil fosfato sintetasi, che presenta due attività enzimatiche, ossia una attività amidotransferasica glutammina dipendente e un’attività sintasica, catalizza la sintesi del carbamil fosfato a partire da glutammina, ione bicarbonato e ATP.
Nella seconda tappa, che è quella di comando della via metabolica ed è catalizzata dalla aspartato transcarbamilasi, il carbamil fosfato reagisce con l’aspartato a dare l’N-carbamil aspartato.
Infine la diidroorotasi, catalizzando la rimozione di una molecola d’acqua dall’N-carbamil aspartato, porta alla chiusura dell’anello pirimidinico a formare L-diidroorotato.
Negli eucarioti, in particolare nei mammiferi, in Drosofila e Dictyostelium, un genere di amebe, le tre attività sono presenti su una singola catena polipeptidica, codificata da un singolo gene derivante da una fusione avvenuta almeno 100 milioni di anni fa. L’enzima multifunzionale, abbreviato come CAD, è un omomultimero composto da tre subunità o più.
Nei procarioti invece i tre enzimi sono indipendenti, e la carbamil fosfato sintetasi è un esempio di complesso multienzimatico.
Nei lieviti l’attività diidroorotasica è presente su una proteina separata.
Studi sull’attività enzimatica hanno rivelato l’esistenza di un incanalamento dei substrati, più efficace nella proteina del lievito, riguardo ai primi due passaggi, rispetto a quella dei mammiferi.
Bibliografia
Alberts B., Johnson A., Lewis J., Morgan D., Raff M., Roberts K., Walter P. Molecular Biology of the Cell. 6th Edition. Garland Science, Taylor & Francis Group, 2015
Eriksen T.A., Kadziola A., Bentsen A-K., Harlow K.W. & Larsen S. Structural basis for the function of Bacillus subtilis phosphoribosyl-pyrophosphate synthetase. Nat Struct Biol 2000:7;303-308. doi:10.1038/74069
Hyde C.C., Ahmed S.A., Padlan E.A., Miles E.W., and Davies D.R. Three-dimensional structure of the tryptophan synthase multienzyme complex from Salmonella typhimurium. J Biol Chem 1988:263(33);17857-17871. doi:10.1016/S0021-9258(19)77913-7
Hyde C.C., Miles E.W. The tryptophan synthase multienzyme complex: exploring structure-function relationships with X-ray crystallography and mutagenesis. Nat Biotechnol 1990:8;27-32. doi:10.1038/nbt0190-27
Michal G., Schomburg D. Biochemical pathways. An atlas of biochemistry and molecular biology. 2nd Edition. John Wiley J. & Sons, Inc. 2012
Muchmore C.R.A., Krahn J.M, Smith J.L., Kim J.H., Zalkin H. Crystal structure of glutamine phosphoribosylpyrophosphate amidotransferase from Escherichia coli. Protein Sci 1998:7;39-51. doi:10.1002/pro.5560070104
Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
Yon-Kahn J., Hervé G. Molecular and cellular enzymology. Springer, 2009
I complessi multienzimatici sono strutture discrete e stabili formate da enzimi associati in modo non covalente che catalizzano due o più reazioni consecutive di una via metabolica.[7]
Possono essere considerati come un passo in avanti nell’evoluzione dell’efficienza catalitica in quanto assicurano vantaggi che i singoli enzimi, anche quelli che hanno raggiunto la perfezione catalitica, da soli non avrebbero.[1][12]
Quali vantaggi offrono i complessi multienzimatici?
Nel corso dell’evoluzione alcuni enzimi si sono evoluti sino al raggiungimento della perfezione catalitica. Si tratta cioè di enzimi per i quali quasi ogni collisione con il proprio substrato ne determina la conversione in prodotto. Esempi sono:
la fumarasi (EC 4.2.1.2), che catalizza la settima tappa del ciclo dell’acido citrico, ossia l’idratazione/deidratazione reversibile del doppio legame del fumarato a dare malato;
l’acetilcolinesterasi (EC 3.1.1.7), che catalizza l’idrolisi dell’acetilcolina, un neurotrasmettitore, in colina e acido acetico, che a sua volta si dissocia a dare acetato e ioni idrogeno;
la superossido dismutasi (EC 1.15.1.1), che catalizza la conversione, e quindi l’inattivazione, del radicale superossido (O2.-), una specie molto reattiva, in perossido d’idrogeno od acqua ossigenata (H2O2) e acqua;
la catalasi (EC 1.11.1.6), che catalizza la degradazione di H2O2 in acqua e ossigeno.
La velocità con cui avviene una reazione enzimatica è dunque in parte è determinata dalla frequenza con cui i substrati e gli enzimi stessi collidono. Ne deriva che un modo semplice per aumentarla sia quello di aumentare le concentrazioni di enzima e substrato. Tuttavia, visto l’enorme numero di reazioni che si verificano all’interno della cellula le loro concentrazioni non potranno essere elevate. E infatti nella cellule la concentrazione della maggior parte dei metaboliti è dell’ordine delle micromoli (10-6 M), e per la maggior parte degli enzimi anche più bassa.[1]
L’evoluzione ha quindi imboccato strade differenti per aumentare la velocità di reazione. Una delle quali è stata quella di ottimizzare l’organizzazione spaziale degli enzimi con la formazione dei complessi multienzimatici e degli enzimi multifunzionali, ossia strutture che permettono di minimizzare la distanza che il prodotto di una reazione deve percorrere per passare al sito attivo che catalizza la reazione successiva, essendo i siti attivi vicini gli uni agli altri.[2][7]Si verifica cioè l’incanalamento dei substrati, in inglese substrate o metabolic channeling,[8][9][14] incanalamento che può avvenire anche attraverso veri e propri canali intramolecolari che connettono i siti attivi, come nel caso, tra i complessi enzimatici, del complesso della triptofano sintasi (EC 4.2.1.20),[3][4]il cui tunnel fu il primo a essere scoperto, e quello della carbamil fosfato sintetasi batterica (EC 6.3.4.16).[5][13]
L’incanalamento dei substrati è in grado di aumentare la velocità di reazione, ma più in generale l’efficienza catalitica, in più modi, di seguito brevemente descritti.[8][12]</sup>
Viene limitata al minimo la diffusione di substrati e prodotti nel mezzo circostante, quindi la loro diluizione e riduzione della concentrazione, producendo anzi elevate concentrazioni locali anche quando la loro concentrazione cellulare è bassa, il che porta a un aumento della frequenza delle collisioni enzima-substrato.
Viene ridotto il tempo di transito dei substrati da un sito attivo al sito attivo successivo.
Viene ridotta la probabilità che si verifichino reazioni collaterali.
Gli intermedi chimicamente labili sono protetti dalla degradazione da parte del solvente.
Un altro vantaggio metabolico apportato dai complessi multienzimatici, analogamente a quanto accade con gli enzimi multifunzionali, è che consentono di controllare in modo coordinato l’attività catalitica degli enzimi che lo compongono. E se si aggiunge il fatto che spesso l’enzima che catalizza la prima reazione della sequenza è l’enzima regolatorio, è possibile evitare:
la sintesi di intermedi non necessari, altrimenti prodotti se la sequenza di reazioni fosse regolata a valle della prima reazione;
la sottrazione di metaboliti ad altre vie come pure uno spreco di energia.
Esempi
Da quanto detto in precedenza non sorprende che, specialmente nelle cellule eucariote, i complessi multienzimatici, al pari degli enzimi multifunzionali, siamo comuni e coinvolti in differenti vie metaboliche, sia anaboliche che cataboliche, mentre sono pochi gli enzimi liberamente diffusibili. Di seguito alcuni esempi.
Alfa-chetoacido deidrogenasi
Esempi classici di complessi multienzimatici sono i tre complessi appartenenti alla famiglia delle alfa-chetoacido deidrogenasi o 2-ossiacido deidrogenasi, ossia:
il complesso dell’α-chetoacido deidrogenasi a catena ramificata o BCKDH , acronimo dell’inglese branched-chain α-keto acid dehydrogenase complex;
il complesso della α-chetoglutarato deidrogenasi, o 2-oxoglutarato deidrogenasi o OGDH, acronimo dell’inglese 2-oxoglutarate dehydrogenase.[2][6][7][10]
Alfa-Chetoglutarato Deidrogenasi
I tre complessi sono correlati sia dal punto di vista strutturale che funzionale.
Considerando ad esempio il complesso della piruvato deidrogenasi questi è formato da copie multiple di tre enzimi differenti:
piruvato deidrogenasi o E1 (EC 1.2.4.1);
diidrolipoil transacetilasi o E2 (EC 2.3.1.12);
diidrolipoil deidrogenasi o E3 (EC 1.8.1.4).
PDC, sia nei procarioti che negli eucarioti presenta quindi una struttura di base E1-E2-E3, struttura che si ritrova anche negli altri due complessi.[15] In aggiunta, all’interno di una data specie:
la diidrolipoil deidrogenasi è identica;
la piruvato deidrogenasi e la diidrolipoil transacetilasi sono omologhe
E, sebbene questi enzimi siamo specifici per i rispettivi substrati, utilizzano gli stessi cofattori, ossia il coenzima A, il NAD, al tiamina pirofosfato, il FAD e la lipoamide.
Al fine di differenziarle vengono indicate, per il complesso della piruvato deidrogenasi, della α-chetoglutarato deidrogenasi e della α-chetoacido deidrogenasi a catena ramificata, rispettivamente come:
E1p, E1o ed E1b (EC 1.2.4.4);
E3p, E3o, and E3b (EC 1.8.1.4).</li>
Nota: il complesso della piruvato deidrogenasi degli eucarioti è il più grande complesso multienzimatico conosciuto, più grande di un ribosoma e visibile anche al microscopio elettronico.[2]
Il complesso della piruvato deidrogenasi, che rappresenta il ponte di collegamento tra glicolisi e ciclo dell’acido citrico, catalizza la decarbossilazione ossidativa del piruvato, che è la base coniugata dell’acido piruvico e fa parte del gruppo dei chetoacidi, in particolare è un alpha-chetoacido. Nel corso delle reazioni il gruppo carbossilico del piruvato viene rilasciato in forma di anidride carbonica (CO2) e si verifica il trasferimento del gruppo acetilico risultante al coenzima A a dare acetil-coenzima A. Inoltre sono rilasciati due elettroni che sono trasferiti al NAD+.
Anche nel corso delle reazioni catalizzate dal complesso della α-chetoglutarato deidrogenasi e dal complesso della α-chetoacido deidrogenasi a catena ramificata, rispettivamente quarta reazione del ciclo dell’acido citrico, ossia l’ossidazione dell’α-chetoglutarato a succinil-CoA, e l’ossidazione degli α-chetoacidi derivanti dal catabolismo degli amminoacidi a catena ramificata valina, leucina e isoleucina, si verifica:
la liberazione del carbonio carbossilico dell’alfa-chetoacido in forma di CO2;
il trasferimento del gruppo acilico risultante al coenzima A a dare l’acil-CoA corrispondente;
la riduzione del NAD+ a NADH.
Dunque, la notevole somiglianza esistente tra le strutture proteiche, i cofattori richiesti e i meccanismi di reazione riflettono senza dubbio una origine evolutiva comune.
Triptofano sintasi
Il complesso della triptofano sintasi è uno degli esempi di incanalamento dei substrati meglio studiati.[3][12][14] Presente nei batteri e nelle piante ma non negli animali, nei batteri è formato da due subunità α e due β assemblate in unità dimeriche αβ, che sembrano esser l’unità funzionale del complesso, a dare una struttura tetramerica αββα.[4][5][8]
Il complesso catalizza gli ultimi due passaggi della sintesi del triptofano: dall’indolo-3-glicero fosfato, per azione di una liasi (EC 4.1.2.8) presente sulle subunità α, viene liberata una molecola di gliceraldeide-3-fosfato e l’indolo. L’indolo diffonde quindi verso il sito attivo presente sulla subunità β sfruttando un tunnel idrofobico di circa 30 Å che, in ciascuna coppia αβ, connette i due siti attivi. Nel sito attivo della subunità β avviene, in presenza del piridossal-5-fosfato, la condensazione tra l’indolo e una serina a dare il triptofano.
Acetil-CoA carbossilasi
L’acetil-CoA carbossilasi o ACC (EC 6.4.1.2), membro della famiglia delle carbossilasi biotina-dipendenti, catalizza la prima tappa di comando della sintesi degli acidi grassi, ossia la carbossilazione dell’acetil-CoA a malonil-CoA, che è utilizzato come donatore di unità bicarboniose dalla acido grasso sintasi (EC 2.3.1.85) durante il processo di allungamento che porterà alla sintesi dell’acido palmitico.[9][11][12]
Nei batteri ACC è un complesso multienzimatico composto da due enzimi, la biotina carbossilasi (EC 6.3.4.14) e una carbossitransferasi, cui si aggiunge una proteina trasportatrice della biotina o BCCP, acronimo dell’inglese biotin carboxyl-carrier protein.
Nei mammiferi e negli uccelli è invece un enzima multifunzionale, in quanto le due attività enzimatiche sono presenti su una stessa catena polipeptidica, nella quale si ritrova anche BCCP.[2]
Nelle piante superiori sono presenti entrambe le forme.
Carbamil fosfato sintetasi
Altro esempio ben caratterizzato di incanalamento dei substrati è il complesso della carbamil fosfato sintetasi dei batteri,[9][12][14] che catalizza la sintesi del carbamil fosfato, necessario sia per la sintesi delle pirimidine che per quella dell’arginina. Il complesso presenta un tunnel lungo circa 100 Å all’interno del quale si muovono i prodotti delle sue tre attività enzimatiche.[13]
La prima catalizza la cessione dell’azoto ammidico della glutammina in forma di ione ammonio, il quale entra nel tunnel dove nel secondo sito attivo si combina con il bicarbonato, a spese di una molecola di ATP, a dare carbamato, che infine nell’ultimo sito attivo viene fosforilato a carbamil fosfato.
Bibliografia
^ ab Alberts B., Johnson A., Lewis J., Morgan D., Raff M., Roberts K., Walter P. Molecular biology of the cell. 6th Edition. Garland Science, Taylor & Francis Group, 2015
^ ab Hilario E., Caulkins B.G., Huang Y-M. M., You W., Chang C-E. A., Mueller L.J., Dunn M.F., and Fan L. Visualizing the tunnel in tryptophan synthase with crystallography: insights into a selective filter for accommodating indole and rejecting water. Biochim Biophys Acta 2016;1864(3):268-279. doi:10.1016/j.bbapap.2015.12.006
^ ab Hyde C.C., Ahmed S.A., Padlan E.A., Miles E.W., and Davies D.R. Three-dimensional structure of the tryptophan synthase multienzyme complex from Salmonella typhimurium. J Biol Chem 1988;263(33):17857-17871. doi:10.1016/S0021-9258(19)77913-7
^ ab Hyde C.C., Miles E.W. The tryptophan synthase multienzyme complex: exploring structure-function relationships with X-ray crystallography and mutagenesis. Nat Biotechnol 1990:8;27-32. doi:10.1038/nbt0190-27
^ Koolman J., Roehm K-H. Color atlas of Biochemistry. 2nd Edition. Thieme, 2005
^ abc Michal G., Schomburg D. Biochemical pathways. An atlas of biochemistry and molecular biology. 2nd Edition. John Wiley J. & Sons, Inc. 2012
^ abc Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ ^ Perham R.N., Jones D.D., Chauhan H.J., Howard MJ. Substrate channeling in 2-oxo acid dehydrogenase multienzyme complexes. Biochem Soc Trans 2002;30(2):47-51. doi:10.1042/bst0300047
Il complesso della piruvato deidrogenasi o PDC è uno dei complessi multienzimatici mitocondriali, ed è composto da tre differenti enzimi:
piruvato deidrogenasi o E1 (EC 1.2.4.1);
diidrolipoil transacetilasi o E2 (EC 2.3.1.12);
diidrolipoil deidrogenasi o E3 (EC 1.8.1.4).
Ognuno di questi enzimi è presente in più copie il cui numero, e quindi le dimensioni del complesso stesso, varia da specie a specie, con una massa molecolare che oscilla da 4 a 10 milioni di Dalton.
Fanno parte del complesso multienzimatico anche:
cinque differenti coenzimi;
nelle piante, nei funghi e, tra gli animali, nei volatili e nei mammiferi, due enzimi con attività regolatoria: la piruvato deidrogenasi chinasi (EC 2.7.1.99), enzima Mg2+-dipendente, e la piruvato deidrogenasi fosfatasi) (EC 3.1.3.43), enzima attivato dagli ioni calcio (Ca2+);
negli eucarioti, una proteina con funzione di legame, E3BP.
Il complesso della piruvato deidrogenasi catalizza, attraverso una sequenza di cinque reazioni, la decarbossilazione ossidativa del piruvato, la base coniugata dell’acido piruvico, a dare il gruppo il gruppo acetilico dell’acetil-coenzima A o acetil-CoA, una molecola di CO2, e due elettroni trasportati dal NAD. La stechiometria complessiva della sequenza delle cinque reazioni è:
PDC: Reazione Complessiva
La reazione complessiva è essenzialmente irreversibile, con un ΔG°’ pari a -8,0 kcal/mol (-33,4 kJ/mol), e richiede l’intervento sequenziale di tutti e tre gli enzimi, le cui attività risultano coordinate. Nel corso delle reazioni i prodotti intermedi rimangono legati agli enzimi e al termine della sequenza catalitica il complesso multienzimatico è pronto per il ciclo successivo.
Le Cinque Reazioni Catalizzate da PDC
Nota: il complesso della piruvato deidrogenasi catalizza le stesse reazioni attraverso meccanismi simili in tutti gli organismi in cui è presente.
Coenzimi del complesso della piruvato deidrogenasi
Cinque coenzimi partecipano alle reazioni catalizzate dal complesso della piruvato deidrogenasi: la tiamina pirofosfato (TPP), la flavina adenina dinucleotide (FAD), il coenzima A (CoA), la nicotinammide adenina dinucleotide (NAD), e l’acido lipoico.
La tiamina pirofosfato deriva dalla tiamina o vitamina B1 di cui rappresenta la forma biologicamente attiva. TPP è il coenzima della piruvato deidrogenasi cui è strettamente, ma non covalentemente, legata. Il suo ruolo è quello di trasportatore di gruppi idrossietilici o “aldeidici attivati”.
La flavina adenina dinucleotide deriva dalla riboflavina o vitamina B2, di cui rappresenta una delle forme attive; l’altra è la flavina mononucleotide o FMN.
Forma Ridotta e Ossidata della Flavina Adenina Dinucleotide
Il FAD è il coenzima della diidrolipoil deidrogenasi cui è saldamente legato. Il suo ruolo, analogamente al NAD, è quello di trasportatore di elettroni o ioni idruro (:H– o H+ + 2e–).
Il coenzima A è formato da una β-mercaptoetilamina legata attraverso legame ammidico all’acido pantotenico o vitamina B5. A sua volta la vitamina è unita, attraverso un ponte pirofosfato, a una 3’-fosfoadenosina.
Il CoA partecipa alla reazione catalizzata dalla diidrolipoil transacetilasi. Il suo ruolo è quello di trasportatore di gruppi acilici.
CoA e Acetil-CoA
La porzione del coenzima A corrispondente alla β-mercaptoetilamina termina con un gruppo sulfidrilico (–SH). Si tratta di un tiolo reattivo, cruciale per il ruolo svolto dal coenzima in quanto i gruppi acilici trasportati, legati attraverso un legame tioestere, hanno una elevata energia libera standard di idrolisi. Ciò fornisce loro un elevato potenziale di trasferimento, pari a -31,5 kcal/mol (-7,5 kJ/mol). Degno di nota che questo potenziale di trasferimento è più esoergonico, anche se di solo 0,2 kcal/mol (1 kJ/mol), rispetto a quello per l’idrolisi dell’ATP ad ADP e Pi. Pertanto i tioesteri hanno un elevato potenziale di trasferimento del gruppo acilico e sono in grado di donarlo a numerose molecole accettrici. In altre parole il gruppo acilico legato al coenzima A può essere considerato come una forma attivata pronta per il trasferimento.
E’ anche possibile affermare che la formazione del legame tioestere permette di conservare parte dell’energia metabolica derivante dall’ossidazione del carburante metabolico. E va sottolineato che il coenzima A è anche abbreviato come CoA-SH, proprio per enfatizzare il ruolo svolto dal gruppo tiolico.
Nota: nel legame tioestere è presente un atomo di zolfo nella posizione in cui nel legame estere è presente un atomo di ossigeno.
La nicotinammide adenina dinucleotide può essere prodotta a partire dal triptofano, un aminoacido essenziale, o dalla niacina o vitamina B3 o vitamina PP, da Pellagra-Preventing, la fonte della parte nicotinammidica.
Forma Ridotta e Ossidata della Nicotinammide Adenina Dinucleotide
Il NAD è coinvolto nella reazione catalizzata dalla diidrolipoil deidrogenasi. Il suo ruolo, analogamente al FAD, è quello di trasportatore di elettroni, o ioni idruro.
A differenza degli altri coenzimi del complesso della piruvato deidrogenasi, l’acido lipoico non deriva, direttamente o indirettamente, da vitamine e/o aminoacidi essenziali, ossia da precursori che non possono essere sintetizzati de novo dall’organismo e devono essere assunti con la dieta. L’acido lipoico è il coenzima della diidrolipoil transacetilasi. Il coenzima è legato all’enzima a mezzo di un legame ammidico con l’ε-ammino gruppo di un residuo di lisina a formare un residuo di lipoil-lisina o lipoammide. Il coenzima accoppia il trasferimento di elettroni con quello di gruppi acilici.
Lipoammide o Lipoil-lisina
L’acido lipoico possiede due gruppi tiolici che possono andare incontro a ossidazione intramolecolare reversibile a dare un ponte disolfuro (-S-S-), similmente a quanto può accadere tra due residui di cisteina (Cys) di una proteina. Poiché il ponte disolfuro (nota: un disolfuro ciclico) può andare incontro a reazioni di ossidoriduzione, nel corso delle reazioni catalizzate dal complesso è dapprima ridotto a diidrolipoammide, un ditiolo o la forma ridotta del gruppo prostetico, e quindi riossidato nel disolfuro ciclico.
Molti enzimi necessitano per la loro attività catalitica di piccoli componenti non proteici chiamati cofattori. I cofattori possono essere ioni metallici o piccole molecole organiche o metallo-organiche e sono classificati come coenzimi e gruppi prostetici.
Un gruppo prostetico è un cofattore che si lega in odo permanente alla proteina, a mezzo di un legame non covalente o covalente.
Un coenzima è invece un cofattore che non è legato in modo permanente all’enzima.
Localizzazione del complesso della piruvato deidrogenasi
Negli eucarioti il complesso della piruvato deidrogenasi, al pari degli enzimi del ciclo dell’acido citrico e di quelli per l’ossidazione degli acidi grassi, si trova nel mitocondrio, associato alla superficie della membrana mitocondriale interna che guarda la matrice.
Nei procarioti si trova nel citosol.
Funzioni del complesso della piruvato deidrogenasi
Il complesso della piruvato deidrogenasi ha essenzialmente due funzioni: produrre acetil-CoA e NADH.
Il gruppo acetilico legato al coenzima A, un acetato attivato, a seconda delle condizioni metaboliche della cellula e/o del tipo di cellula, potrà andare incontro a due differenti destini.
Può essere ossidato a due molecole di anidride carbonica attraverso le reazioni del ciclo dell’acido citrico, il che permette la “raccolta” di una parte dell’energia potenziale immagazzinata in forma di ATP o GTP.
Può essere utilizzato per la sintesi di acidi grassi, colesterolo, steroidi, isoprenoidi, corpi chetonici e acetilcolina.
E’ quindi possibile affermare che, a seconda delle condizioni metaboliche e/o del tipo di cellula, il complesso della piruvato deidrogenasi indirizza intermedi carboniosi dal catabolismo degli aminoacidi e del glucosio verso:
il ciclo dell’acido citrico, e quindi la produzione di energia, come ad esempio nel muscolo scheletrico in condizioni aerobiche, e, sempre, nel muscolo cardiaco;
la sintesi dei lipidi e di acetilcolina.
Negli organismi aerobi il NADH potrà essere ossidato a NAD+ a seguito della cessione di uno ione idruro alla catena di trasporto degli elettroni mitocondriale. Quest’ultima a sua volta trasferisce i due elettroni all’ossigeno molecolare (O2), trasferimento che permette la produzione di 2,5 molecole di ATP per coppia di elettroni. Nota: negli organismi anaerobi è presente un accettore di elettroni alternativo all’ossigeno, come ad esempio un nitrato o un solfato.
Dal punto di vista concettuale il complesso della piruvato deidrogenasi rappresenta il ponte di collegamento tra glicolisi e ciclo dell’acido citrico. Tuttavia, vista l’irreversibilità della reazione complessiva catalizzata dal complesso, questo ponte è “a senso unico”. Il piruvato può essere decarbossilato, ossidato e l’unità acetilica rimanente legata al CoA. Non è invece possibile compiere il percorso inverso, ossia convertire acetil-CoA in piruvato.
L’irreversibilità di questa reazione e l’assenza di vie metaboliche alternative spiega perché non è possibile utilizzare l’acetil-CoA, e quindi gli acidi grassi, per la gluconeogenesi.
Altre fonti di acetil-CoA
Il gruppo acetilico dell’acetil-CoA può derivare, oltre che dal piruvato, dall’ossidazione degli acidi grassi e dal catabolismo di molti aminoacidi. Tuttavia, a prescindere dalla sua origine, l’acetil-CoA rappresenta un veicolo di ingresso di nuove unità carboniose nel ciclo dell’acido citrico. E’ anche possibile affermare che il gruppo acetilico dell’acetil-CoA rappresenta la forma in cui la maggior parte del carbonio entra nel ciclo.
Trasporto del piruvato nella matrice mitocondriale
Negli eucarioti, la glicolisi avviene nel citosol mentre tutti i passaggi successivi del metabolismo aerobico, quindi le reazioni catalizzate dal complesso della piruvato deidrogenasi, il ciclo dell’acido citrico, la catena di trasporto degli elettroni e la fosforilazione ossidativa, si svolgono nel mitocondrio.
Similmente a quanto accade per la maggior parte degli altri anioni e metaboliti, il passaggio del piruvato attraverso la membrana mitocondriale esterna è probabilmente mediato da un canale anionico voltaggio-dipendente relativamente non specifico.
Il passaggio attraverso la membrana mitocondriale interna è invece assicurato da uno specifico trasportatore formato da due proteine denominate MPC1 e MPC2 che vanno a formare un complesso etero-oligomerico nella membrana.
Struttura del complesso della piruvato deidrogenasi
Sebbene il complesso della piruvato deidrogenasi sia formato da copie multiple di tre enzimi differenti e catalizzi le medesime reazioni tramite meccanismi simili in tutti gli organismi in cui è presente, ha una struttura quaternaria molto varia.
Il primo complesso di cui fu analizzata la struttura fu quello di E. coli, grazie al lavoro di Lester Reed. Nel complesso multienzimatico, che ha un peso di circa 4600 kD e un diametro di circa 300 Å, 24 unità di diidrolipoil transacetilasi vanno a formare una struttura con simmetria cubica, ossia, gli enzimi, associati in trimeri, sono disposti agli angoli del cubo. Dimeri di piruvato deidrogenasi si associano al nucleo di diidrolipoil transacetilasi, al centro di ognuno dei 12 bordi del cubo, per un totale 24 unità. Infine, dimeri di diidrolipoil deidrogenasi si vanno a disporre al centro di ognuna delle sei facce del cubo, per un totale di 12 unità. Si noti che l’intero complesso è costituito da 60 unità. Anche nella maggior parte degli altri batteri Gram-negativi si osserva una analoga struttura con simmetria cubica.
In alcuni batteri Gram-positivi e negli eucarioti il complesso della piruvato deidrogenasi presenta una struttura dodecaedrica, ossia quella di un poliedro regolare con 20 vertici, 12 facce pentagonali e 30 bordi, con simmetrica icosaedrica, anche detta simmetria I. Se si considera ad esempio il complesso presente nei mitocondri, questi è il più grande complesso multienzimatico conosciuto. Ha un peso di circa 1000 kD e un diametro di circa 500 Å, dunque ben 5 volte le dimensioni di un intero ribosoma. Inoltre è abbastanza grande da poter essere visualizzato con il microscopio elettronico.
Il complesso è costituito da un nucleo dodecaedrico avente un diametro di circa 25 nm e formato, come nei batteri Gram-negativi, dalla diidrolipoil transacetilasi, ma composto da 20 trimeri dell’enzima, per un totale di 60 unità, disposti ai vertici della struttura. Il nucleo è circondato da 30 unità di piruvato deidrogenasi, una centrata su ogni bordo, e 12 unità di diidrolipoil deidrogenasi, una centrata su ogni faccia. L’intero complesso è quindi costituito da 102 unità.
La struttura quaternaria del complesso è ulteriormente complicata dalla presenza di tre subunità addizionali: la piruvato deidrogenasi chinasi, la piruvato deidrogenasi fosfatasi, e la E3BP.
La chinasi e la fosfatasi sono legate al nucleo di diidrolipoil transacetilasi.
E3BP è legata a ognuna della 12 facce pentagonali, e dunque presente in circa dodici copie. La proteina è necessaria per il legame della diidrolipoil deidrogenasi al nucleo formato dalla diidrolipoil transacetilasi, come dimostrato dal fatto che la sua proteolisi parziale riduce la capacità di legame della deidrogenasi. Nella E3-bindign protein è possibile individuare un dominio C-terminale, privo di attività catalitica, e un dominio che contiene una lipoil-lisina, simile a quella della diidrolipoil transacetilasi, in grado anche di accettare un gruppo acetilico. Tuttavia, la rimozione di questo dominio non comporta alcuna riduzione dell’attività catalitica del complesso multienzimatico.
Struttura della piruvato deidrogenasi o E1
La piruvato deidrogenasi degli eucarioti e di alcuni batteri Gram-positivi è composta da due differenti catene polipeptide, indicate come α e β, disposte a formare un eterotetramero α2β2 simmetrico. Di contro in E. coli e in altri batteri Gram-negativi le subunità sono fuse a formare un’unica catena polipeptidica e l’enzima risulta un omodimero.
L’enzima presenta due siti attivi.
Considerando la struttura eterotetramerica della piruvato deidrogenasi di Bacillus stearothermophilus, un batterio Gram-positivo, ogni tiamina pirofosfato si lega tra i domini N-terminali di una subunità α e di una β, al termine di un canale a forma di imbuto profondo circa 21 Å che porta al sito attivo, con l’anello tiazolico, il suo gruppo reattivo, posizionato vicino all’ingresso del canale stesso.
All’ingresso del canale sono inoltre presenti due anse peptidiche conservate, essenziali sia per l’attività catalitica dell’enzima che per la sua regolazione.
L’analisi ai raggi X dell’enzima di B. stearothermophilus, quando lega sia la tiamina pirofosfato che PSBD della diidrolipoil transacetilasi, che si lega al dominio C-terminale delle subunità β, ha evidenziato, oltre a un eterotetramero con un struttura estremamente compatta, che i due siti attivi hanno una struttura differente, in particolare riguardo alla disposizione delle due anse peptidiche conservate. Infatti, in una subunità dell’enzima, in presenza della forma attivata della tiamina pirofosfato:
l’ansa più interna è disposta in modo da bloccare l’ingresso al sito attivo;
l’ansa all’ingresso dell’altro sito attivo ha una conformazione tale da non bloccarne l’ingresso.
Questo spiega dal punto di vista strutturale le differenze osservate nella velocità di legame del substrato esibite dai due siti attivi. Una disposizione e una asimmetria simili sono state osservate anche in tutti gli altri enzimi tiamina pirofosfato dipendenti di cui sia nota la struttura.
Oltre alla tiamina pirofosfato e a uno ione magnesio (Mg2+), localizzati in ciascuno dei due siti attivi dell’enzima, un terzo Mg2+ è posizionato al centro del tetramero, all’interno di un tunnel lungo circa 20 Å, riempito di solvente, che collega i due siti attivi. Il tunnel è in gran parte delimitato da 10 residui amminoacidici conservati, rispettivamente sei residui di glutammato (Glu) e quattro di aspartato (Asp), che provengono da tutte e quattro le subunità, più altri residui acidi attorno all’anello amminopirimidinico della TPP. Va invece sottolineata l’assenza di residui basici che neutralizzino i precedenti. Tunnel simili sono stati osservati in tutti gli enzimi tiamina pirofosfato dipendenti con struttura dimerica o tetramerica di cui sia nota la struttura cristallina. Un esempio è la transchetolasi, enzima della via del pentoso fosfato.
Nota: Bacillus stearothermophilus è un membro del phylum Firmicutes ed è stato da poco rinominato Geobacillus stearothermophilus.
Qual’è la funzione del tunnel acido?
Attraverso esperimenti di mutagenesi condotti sulla piruvato deidrogenasi di B. stearothermophilus è stato dimostrato che il tunnel ha un ruolo nel meccanismo catalitico.
La sostituzione di alcuni dei suddetti residui acidi con amminoacidi neutri non alterava, rispetto alla forma wild-type:
l’efficienza dell’incorporazione dell’enzima modificato nel complesso multienzimatico;
la struttura dei siti attivi;
la struttura quaternaria dell’enzima.
Tuttavia, la velocità di decarbossilazione risultava ridotta di oltre il 70% rispetto all’enzima wild-type, come pure, una volta che l’enzima mutante era inserito nel complesso della piruvato deidrogenasi, l’attività del complesso stesso di oltre l’85%, sempre in confronto alla forma wild-type. Ma a che cosa sono dovute queste riduzioni dell’efficienza catalitica?
Poiché la distanza tra gli amminoacidi sostituiti e i siti attivi dell’enzima è di 7 o più Å, questi amminoacidi sono troppo lontani dai siti attivi per poterne influenzare direttamente l’attività catalitica. E’ stato quindi proposto il meccanismo di seguito descritto.
Considerando l’apoenzima, la TPP si lega velocemente e fortemente al primo sito attivo, viene attivata, e il sito attivo viene chiuso. In questo modo lo zwitterione tiazolico risulta protetto dall’ambiente esterno.
Nel secondo sito attivo invece la tiamina pirofosfato si lega, ma non viene attivata, e quindi il sito attivo rimane in conformazione aperta.
Nel primo sito attivo il piruvato reagisce con il C-2 tiazolico e la tiamina pirofosfato del secondo sito attivo, che è un acido generale, dona un protone al primo sito. Il risultato è una reazione di decarbossilazione nel primo sito attivo e l’attivazione del coenzima nel secondo sito, che quindi viene chiuso.
Si noti che mentre l’attivazione della prima tiamina pirofosfato è conseguente al legame al sito attivo, l’attivazione della seconda, e quindi del secondo sito attivo, è accoppiata alla decarbossilazione del piruvato nel primo sito attivo. O, da un altro punto di vista, mentre un sito attivo richiede un acido generale, l’altro richiede una base generale.
I protoni sono necessari per l’attività catalitica, e il loro trasferimento tra i siti attivi avviene attraverso il tunnel acido. Si verifica cioè il loro trasporto reversibile lungo una catena di gruppi accettori-donatori forniti dai residui di glutammato e aspartato e dalle molecole d’acqua presenti nel tunnel, che nell’insieme agiscono come un vero e proprio filo protonico, in inglese proton wire.
Sembra quindi che, a differenza di quanto accade in molti altri enzimi, in cui la comunicazione tra i siti attivi avviene attraverso modificazioni conformazionali e riarrangiamenti delle subunità che costituiscono l’enzima stesso, nella piruvato deidrogenasi e negli altri enzimi tiamina pirofosfato dipendenti il “proton wire” sia la base molecolare di tale comunicazione.
A questo punto l’oloenzima è formato e i siti attivi si trovano in un equilibrio dinamico, passando vicendevolmente dallo stato dormiente a quello attivato. Questo sembra essere lo stato in cui si trova l’enzima in vivo all’inizio di ogni ciclo catalitico.
Una conseguenza di un tale meccanismo è che, a mano a mano che i cicli catalitici si susseguono, i due siti attivi sono fuori fase tra di loro, ossia mentre uno richiede una base generale, l’altro necessita di un acido generale, e vice versa.
Da notare infine che un tale meccanismo permette anche il cambio di conformazione delle anse che chiudono i siti attivi, in modo da:
coordinare l’uptake dei substrati e il rilascio dei prodotti:
spiegare l’asimmetria esistente tra i due siti attivi.
Nota: un apoenzima è un enzima privo dei suoi cofattori, mentre un oloenzima è un enzima che lega tutti i suoi cofattori. L’apoenzima è la forma cataliticamente inattiva dell’enzima, mentre l’oloenzima ne è la forma cataliticamente attiva.
Struttura della diidrolipoil transacetilasi o E2
Nella struttura della diidrolipoil transacetilasi è possibile individuare tre domini funzionalmente distinti: un dominio lipoilico N-terminale, un dominio centrale di legame o PSBD, e un dominio catalitico C-terminale o acetiltransferasico.
Questi domini sono connessi da sequenze di circa 20-40 residui amminoacidici ricche in alanina e prolina, aminoacidi idrofobici che sono inframezzati da residui dotati di carica. Queste sequenze di collegamento sono altamente flessibili ed estese, il che permette di tenere lontani gli uni dagli altri i tre domini.
Domini di E2
Nota: analoghe sequenze di collegamento flessibili sono presenti anche in E3BP.
Il dominio lipoilico N-terminale, composto da circa 80 residui amminoacidici, è così chiamato in quanto lega l’acido lipoico. Il numero di questi domini dipende dalla specie considerata, variando da uno a tre. Ad esempio è presente con una copia in B. stearothermophilus e nei lieviti, con due nello Streptococcus faecalis e nei mammiferi, e con tre in Azotobacter vinelandii ed E. coli.
Il legame tra l’ɛ-amino gruppo della catena laterale di una lisina e l’acido lipoico porta alla formazione di un braccio flessibile, la lipoil-lisina o lipoammide, che alla sua massima estensione ha una lunghezza di circa 14 Å. Se a questo si aggiunge la sequenza che collega il dominio N-terminale al dominio adiacente, la cui lunghezza è superiore ai 140 Å, si ottiene un braccio flessibile in grado di far oscillare il gruppo lipoilico tra i siti attivi della piruvato deidrogenasi e della diidrolipoil deidrogenasi, nonché di interagire con le unità di diidrolipoil transacetilasi vicine. Si noti che il numero di queste braccia flessibili in E. coli è pari a 3 x 24 = 72, mentre nei mammiferi a 2 x 60 = 120, sulla base del numero dei domini N-terminali e delle unità di diidrolipoil transacetilasi.
Da tutto ciò consegue che una piruvato deidrogenasi può acetilare numerose diidrolipoil transacetilasi, e una diidrolipoil deidrogenasi può riossidare diversi gruppi diidrolipoamidici.
Inoltre, si verifica anche:
un interscambio di gruppi acetilici tra i gruppi lipoilici del nucleo di diidrolipoil transacetilasi;
lo scambio sia di gruppi acetilici che di disolfuri tra le braccia flessibili sopracitate.
PSBD è composto da circa 35 residui amminoacidici disposti a formare una struttura globulare che si lega sia alla piruvato deidrogenasi che alla diidrolipoil deidrogenasi, ossia tiene insieme il complesso multienzimatico.
Il dominio catalitico C-terminale, che, ovviamente, contiene il sito attivo, è composto da circa 250 residui amminoacidici. La disposizione dei residui aminoacidici è tale da formare una struttura simile a una gabbia vuota che contiene canali sufficientemente larghi da permettere ai substrati e prodotti di diffondere fuori e dentro. Ad esempio, la lipoamide e il coenzima A, i due substrati della diidrolipoil transacetilasi, si legano in conformazione estesa alle estremità opposte di un canale che si trova localizzato all’interfaccia tra ogni paio di subunità di ciascun trimero.
Struttura della diidrolipoil deidrogenasi o E3
La struttura della diidrolipoil deidrogenasi è stata desunta dalla studio dell’enzima in numerosi microrganismi.
Presenta una struttura omodimerica, con ciascuna catena polipeptidica, formata da circa 470 residui amminoacidici, ripiegata a dare quattro domini, dall’estremità N-terminale a quella C-terminale:
un dominio legante il FAD;
uno legante il NAD;
un dominio centrale;
un dominio di interfaccia.
Tutti prendono parte alla formazione del sito attivo.
Il FAD si trova quasi completamente all’interno della proteina. Ciò è dovuto al fatto che, a differenza del NADH o dei tioli, è facilmente ossidabile e quindi deve essere protetto dalla soluzione circostante, ossia dall’ossidazione ad opera di O2. E infatti, in assenza del NAD+, la catena laterale fenolica di uno specifico residuo di tirosina (Tyr), ad esempio nel batterio Gram-negativo Pseudomonas putida la Tyr181, va a coprire la tasca dove si va a legare la nicotinamide proteggendo il FADH2 dal contatto con la soluzione.
Quando invece il NAD+ è in posizione nel sito attivo, la catena laterale fenolica della suddetta tirosina si va a disporre tra l’anello nicotinamidico e quello flavinico.
Nel sito attivo della forma ossidata dell’enzima è presente anche un ponte disolfuro redox-attivo che si forma tra due residui di cisteina presenti in un segmento della catena polipeptidica altamente conservato, ad esempio in P. putida tra la Cys43 e la Cys48. Il disolfuro è localizzato dal lato dell’anello flavinico opposto rispetto a quello che guarda l’anello nicotinammidico, e lega due giri consecutivi di un segmento di un’α-elica distorta.
Da notare che in assenza della distorsione dell’ α-elica i Cα delle due cisteine sarebbero troppo distanti per poter permettere la formazione del ponte disolfuro.
La diidrolipoil deidrogenasi possiede quindi due accettori di elettroni: il FAD e il ponte disolfuro redox-attivo.
Nota: i due anelli eterociclici del NAD e del FAD sono paralleli e in contatto attraverso interazioni di van der Waals; anche S48 è in contatto di van der Waals con l’anello flavinico, dal lato opposto rispetto all’anello nicotinamidico.
Reazione della piruvato deidrogenasi o E1
La piruvato deidrogenasi, nella sequenza delle cinque reazioni catalizzate dai componenti del complesso della piruvato deidrogenasi, ne catalizza le prime due, ovvero:
la decarbossilazione del piruvato a dare CO2 e l’intermedio idrossietil-TPP;
l’acetilazione riduttiva del gruppo lipoilico della diidrolipoil transacetilasi.
La prima reazione è essenzialmente identica alla reazione catalizzata dalla piruvato decarbossilasi (EC 4.1.1.1), che opera una decarbossilazione non ossidativa nel corso della fermentazione del glucosio a etanolo. Quello che differisce è il destino del gruppo idrossietilico legato alla tiamina pirofosfato:
nella reazione catalizzata dalla piruvato deidrogenasi viene trasferito all’enzima successivo della sequenza, la diidrolipoil transacetilasi;
nella reazione catalizzata dalla piruvato decarbossilasi è convertito in acetaldeide.
Meccanismo di reazione della piruvato deidrogenasi o E1
Nelle reazioni catalizzate da enzimi TPP-dipendenti, l’anello tiazolico rappresenta la parte attiva, ma solo in forma di carbanione dipolare o ilide, ossia uno ione dipolare o zwitterione, con carica positiva sull’N-3 e carica negativa sul C-2. Di contro, l’anello tiazolico con carica positiva, ossia una carica positiva sull’azoto e nessuna carica sul C-2, può essere definito come la forma “dormiente” o inattiva.
La reazione ha inizio con l’attacco nucleofilo da parte del carbanione sul C-2 al carbonio carbonilico del piruvato, che ha lo stato di ossidazione di un’aldeide. L’attacco porta alla formazione di un legame covalente tra il coenzima e il piruvato.
Segue la scissione del legame tra il C-1 e il C-2 del piruvato. Questo porta al distacco del gruppo carbossilico, ossia del C-1, in forma di CO2, mentre i due restanti atomi di carbonio, il C-2 e il C-3, rimangono legati alla tiamina pirofosfato in forma di gruppo idrossietilico.
La scissione del legame C-1–C-2, e quindi la decarbossilazione del piruvato, è favorita dal fatto che la carica negativa sul C-2, che è instabile, viene stabilizzata grazie alla presenza nell’anello tiazolico dell’azoto imminico carico positivamente, N-3, (C=N+), ossia grazie alla presenza di una struttura elettrofila o elettron deficiente che funge da “pozzo” o “trappola” per gli elettroni, nella quale gli elettroni dei carbanioni possono delocalizzarsi per risonanza.
A questo punto l’intermedio stabilizzato per risonanza può essere protonato a dare idrossietil-TPP.
Nota: questo primo passaggio della reazione catalizzata dalla piruvato deidrogenasi è quello dove il complesso della privato deidrogenasi esercita la sua specificità di substrato. Inoltre è anche il più lento dell’intera sequenza, ossia è quello che limita la velocità della reazione complessiva.
Meccanismo Catalitico della Piruvato Deidrogenasi
Di seguito, l’enzima catalizza l’ossidazione del gruppo idrossietilico a gruppo acetilico e il suo trasferimento sul braccio lipoamidico della diidrolipoil transacetilasi.
La reazione ha inizio con la formazione di un carbanione sul carbonio idrossilico della idrossietil-TPP, a seguito della rimozione del protone legato al carbonio stesso da parte di una base dell’enzima.
Segue l’attacco nucleofilo del carbanione al disolfuro della lipoamide, con formazione di un legame acetil-tioestere ad alta energia con uno dei due tioli. In questa reazione l’ossidazione del gruppo idrossietilico a gruppo acetilico è accompagnata dalla concomitante riduzione del ponte disolfuro della lipoamide. I due elettroni rimossi dal gruppo idrossietilico sono utilizzati per ridurre il ponte disolfuro.
Questa reazione è quindi una acetilazione riduttiva, cui si accoppia la rigenerazione della forma attiva della piruvato deidrogenasi, ossia l’enzima con il C-2 dell’anello tiazolico nella forma deprotonata, la forma ilide o carbanione dipolare.
Si noti che l’energia derivante dall’ossidazione del gruppo idrossietilico a gruppo acetilico permette la formazione del legame tioestere tra il gruppo acetilico e il coenzima A.
Nota: il braccio di lipoil-lisina, presente in conformazione estesa nel canale dove si trova anche la TPP, permette il trasferimento dell’idrossietile dalla idrossietil-TPP al coenzima A. Dunque il braccio di lipoil-lisina si può spostare dal sito attivo della piruvato deidrogenasi a quelli della diidrolipoil transacetilasi e quindi della diidrolipoil deidrogenasi.
Proprietà della tiamina pirofosfato
Nella molecola della tiamina pirofosfato è possibile individuare tre parti distinte, da cui dipendono le sue proprietà chimiche ed enzimologiche: l’anello tiazolico, l’anello 4’-amminopirimidinico, e il gruppo difosfato.
Il difosfato lega il cofattore all’enzima attraverso la formazione di legami elettrostatici tra le cariche negative portate dai suoi gruppi fosforici e quelle positive degli ioni Ca2+ e Mg2+. I due ioni sono a loro volta legati a sequenze altamente conservate, rispettivamente GlyAspGly (GDG) per il Ca2+ e GlyAspGly-X26-AsnAsn (GDG-X26-NN) per l’Mg2+.
L’anello tiazolico ha un ruolo centrale nella catalisi grazie alla capacità di formare un carbanione, ossia un centro nucleofilo sul C-2.
Nota: una volta legatasi all’enzima, la TPP si dispone nel sito attivo in modo che l’anello tiazolico sia posizionato vicino all’ingresso del canale che porta al sito attivo.
L’anello amminopirimidinico ha una duplice funzione.
Ancora il coenzima tenendolo in posizione.
Ha uno specifico ruolo catalitico, partecipando a una catalisi acido/base. Questo è evidenziato da studi condotti utilizzando analoghi della tiamina pirofosfato in cui, a turno, erano stati sostituiti i tre atomi di azoto dell’anello. Gli studi hanno dimostrato che l’atomo N-1’ e il gruppo amminico legato a N-4’ sono necessari per l’attività del coenzima, mentre l’atomo N-3’ lo è meno.
Come si forma il carbanione dipolare della tiamina pirofosfato?
E’ possibile individuare tre forme tautomeriche in cui si presenta l’anello amminopirimidinico della tiamina pirofosfato legata allo specifico enzima ancora non impegnato nella reazione:
la forma canonica, ossia la 4’-amminopirimidina;
la forma protonata su N-1’, ossia lo ione 4’-amminopirimidinio;
l’1’,4’-imminopirimidina.
E sembra che la 1’,4’-imminopirimidina sia il tautomero che, prima dell’arrivo del substrato nel sito attivo, subisce la deprotonazione.
Il C-2 dell’anello tiazolico è assai più acido rispetto alla maggior parte degli altri gruppi =C-H presenti in altre molecole. La maggior acidità, ossia il fatto che il protone legato al C-2 sia facilmente dissociabile, è dovuta alla presenza dell’atomo di azoto quaternario con carica positiva dell’anello tiazolico che è in grado di stabilizzare elettrostaticamente il carbanione risultante.
Nel processo di deprotonazione sembra avere un ruolo essenziale il gruppo amminico dell’anello amminopirimidinico. Il gruppo funge infatti da base ed è posizionato opportunamente per accettare il protone.
Tuttavia, nella forma canonica, la 4’-amminopirimidina, uno dei suoi protoni collide stericamente con il protone legato al C-2. In aggiunta, anche il suo pK è troppo basso per operare la deprotonazione in modo efficiente. E’ stato quindi proposto un meccanismo in cui la catena laterale di un residuo conservato di glutammato, ad esempio βGlu59 in B. stearothermophilus, o Glu51 nella piruvato decarbossilasi di Saccharomyces uvarum, donando un protone all’amminopirimidina la converte nella sua forma immino tautomerica, la 1’,4’-imminopirimidina, che, accettando il protone dal C-2, torna alla forma canonica 4’-amminopirimidinica e permette la formazione del carbanione.
Nota: la formazione del carbanione sul C-2 è quindi conseguenza di un trasferimento protonico intramolecolare.
Deprotonazione della tiamina pirofosfato e chiusura del sito attivo
La perdita del protone dal C-2 dell’anello tiazolico porta, da una situazione con carica positiva sull’anello, alla formazione di uno ione dipolare o zwitterione.
Questo cambio nello stato di carica innesca una modificazione conformazionale in una delle due anse peptidiche conservate presenti all’ingresso del canale che immette al sito attivo, nello specifico la più interna, modificazione che, a sua volta, porta alla chiusura dello canale rispetto all’ambiente acquoso circostante. In questa conformazione chiusa il carbanione tiazolico è protetto dall’attacco di reagenti vari, come elettrofili.
Riassumendo, la deprotonazione della tiamina pirofosfato determina la chiusura del sito attivo e la protezione del carbanione dipolare neoformato; in altri termini, gli enzimi TPP-dipendenti sarebbero attivi solo nella conformazione chiusa.
Se invece si considera l’altro sito attivo, la sua tiamina pirofosfato non è in forma di ilide, il è canale aperto, e il sito stesso risulta inattivo.
Reazione della diidrolipoil transacetilasi o E2
La diidrolipoil transacetilasi catalizza la terza reazione della sequenza delle cinque catalizzate dai componenti del complesso della piruvato deidrogenasi. La reazione comporta il trasferimento del gruppo acetilico dall’acetil-diidrolipoammide al coenzima A, a dare acetil-CoA e la forma completamente ridotta della lipoammide, la diidrolipoammide, il ditiolo.
Va notato che il gruppo acetilico, inizialmente legato tramite legame estere a uno dei gruppi –SH della lipoammide è di seguito legato al gruppo –SH del coenzima A, di nuovo attraverso legame estere, da cui il termine di transesterificazione.
Meccanismo di reazione della diidrolipoil transacetilasi o E2
Nel corso della reazione, il gruppo sulfidrilico del coenzima A porta un attacco nucleofilo al carbonio carbonilico del gruppo acetilico dell’acetil diidrolipoammide-diidrolipoil transacetilasi a formare un intermedio tetraedrico transiente che si “decompone” a dare acetil-CoA e diidrolipoammide-diidrolipoil transacetilasi.
Meccanismo Catalitico della Diidrolipoil Transacetilasi
Come già detto, nel meccanismo di reazione gioca un ruolo centrale la mobilità della lipoammide.
Reazione della diidrolipoil deidrogenasi o E3
La diidrolipoil deidrogenasi catalizza la quarta e la quinta reazione della sequenza delle cinque catalizzate dai componenti del complesso della piruvato deidrogenasi.
L’enzima catalizza trasferimenti di elettroni necessari per rigenerare il ponte disolfuro della lipoammide della diidrolipoil transacetilasi, ossia per rigenerare la forma ossidata del gruppo prostetico e completare così il ciclo catalitico della transacetilasi.
La reazione segue un meccanismo a ping-pong, procedendo attraverso due semireazioni consecutive nelle quali i due substrati, la diidrolipoammide e il NAD+, reagiscono uno in assenza dell’altro. Inoltre durante la prima semireazione, il rilascio del primo prodotto e la formazione di un complesso enzima-intermedio si verificano prima che il secondo substrato si leghi, mentre l’enzima va incontro a un cambio strutturale. Nella seconda semireazione si verificano il rilascio del secondo prodotto e il ritorno dell’enzima allo stato iniziale, di nuovo attraverso una modificazione strutturale.
Considerando la cinetica del meccanismo a ping-pong della diidrolipoil deidrogenasi:
nella prima semireazione si verifica l’ossidazione della diidrolipoamide a lipoammide;
nella seconda semireazione si verifica la riduzione del NAD+ a NADH.
Meccanismo di reazione della diidrolipoil deidrogenasi o E3
Di seguito viene descritto il meccanismo di reazione della diidrolipoil deidrogenasi di P. putida.
Nella prima semireazione l’enzima in forma ossidata (E), ossia con il ponte disolfuro tra la Cys43 e la Cys48, lega la diidrolipoammide (LH2) a formare il complesso enzima-diidrolipoammide (E●LH2). A questo punto un atomo di zolfo della diidrolipoammide porta un attacco nucleofilo allo zolfo della Cys43, con formazione di un ponte disolfuro lipoammide-Cys43, (E–S–S–L), mentre lo zolfo della Cys48 viene rilasciato in forma di ione tiolato (S–48).
Il protone che è rimasto sul secondo gruppo tiolico della lipoammide è quindi estratto ad opera dell’istidina (Hys) 451. L’aminoacido agisce come catalizzatore acido-base generale, il che porta alla formazione di un secondo ione tiolato, stavolta sulla lipoamide (E–S–S–L●S–), il quale, attraverso un attacco nucleofilo, va a sostituire lo zolfo della Cys43, S43, aiutato in questo attacco dalla catalisi acida generale ad opera della Hys451 che dona un protone a S43. L’azione catalitica della Hys451 è essenziale, come dimostrato da studi di mutagenesi in cui la sua sostituzione con un residuo di glutammina fa si che l’enzima mantenga solamente circa lo 0,4% dell’attività catalitica rispetto alla forma wild-type.
L’anione tiolato S–48 entra quindi in contatto, mediante interazioni non covalenti, con l’anello flavinico in vicinanza della sua posizione 4a, ossia una coppia di elettroni di S–48, che funge da donatore di elettroni, è parzialmente trasferita all’anello flavinico ossidato, che a sua volta funge da accettore di elettroni. La struttura derivante è chiamata complesso a trasferimento di carica.
Nel frattempo la catena laterale fenolica della Tyr181 continua a bloccare l’accesso all’anello flavinico, proteggendolo così dall’ossidazione da parte di O2.
Ossidazione della Diidrolipoammide da Parte di E3
Quello che si verifica è quindi una reazione di interscambio di ponti disolfuro. I prodotti sono la forma ossidata della lipoammide, il primo prodotto, che è rilasciato, e la forma ridotta della diidrolipoil deidrogenasi.
La seconda semireazione comporta la riduzione del NAD+ a NADH + H+ mediante il trasferimento di elettroni dal disolfuro reattivo dell’enzima via FAD.
Ossidazione di E3 Ridotta
Il tutto ha inizio con l’ingresso nel sito attivo del NAD+ e il suo legame a formare il complesso EH2●NAD+. Si noti che l’ingresso del coenzima determina lo spostamento a lato della catena laterale fenolica della Tyr181 ad opera dell’anello nicotinammidico.
A seguito del collasso del complesso a trasferimento di carica si forma un legame covalente tra il C-4a dell’anello flavinico e S48. A ciò si accompagna l’estrazione di un protone da parte dell’N-5 flavinico, con formazione del corrispondente anione tiolato S–43.
S–43 porta un attacco nucleofilo a S48. L’attacco determina la formazione del ponte disolfuro redox-attivo tra la Cys43 e la Cys48, cui segue la rottura del legame covalente tra S48 e il C-4a dell’anello flavinico a dare l’anione FADH–, con carica negativa su N-1. Si noti che la diidrolipoil deidrogenasi è nuovamente nella forma ossidata (E).
Il FADH– ha un’esistenza transitoria in quanto si verifica il trasferimento praticamente immediato del protone legato a N-5, in forma di ione idruro, al C-4 dell’anello nicotinamidico, che è giustapposto all’N-5 flavinico. Questo porta alla formazione di FAD e del secondo prodotto della reazione, il NADH, che lascia l’enzima.
In definitiva gli elettroni che sono stati rimossi dal gruppo idrossietilico derivato dal piruvato passano, via FAD, al NAD+. In questo modo si è completato il ciclo catalitico della diidrolipoil deidrogenasi, essendo l’enzima e il suo coenzima nella loro forma ossidata.
A questo punto anche il ciclo catalitico dell’intero complesso della piruvato deidrogenasi si è completato, e il complesso stesso è pronto per un altro ciclo di reazioni.
Nota: a differenza dell’anello tiazolico della tiamina pirofosfato, il FAD non ha la funzione di “trappola” o “pozzo” per gli elettroni quanto piuttosto di condotta per gli elettroni tra il disolfuro redox-attivo, nella sua forma ridotta, e il NAD+.
Il meccanismo catalitico della diidrolipoil deidrogenasi è stato in gran parte determinato in analogia con quello della glutatione reduttasi (EC 1.8.1.7), al 33% identica ma strutturalmente molto meglio caratterizzata. Va tuttavia notato che, sebbene i due enzimi catalizzino reazioni simili, queste normalmente corrono in direzione opposta:
la diidrolipoil deidrogenasi utilizza il NAD+ per ossidare due gruppi –SH a dare un disolfuro (–S–S–);
la glutatione reduttasi utilizza il NADPH per ridurre un –S–S– a due gruppi tiolici.
Nonostante ciò, i siti attivi dei due enzimi sono strettamente sovrapponibili.
Regolazione dell’attività del complesso della piruvato deidrogenasi
Nei mammiferi, la regolazione dell’attività del complesso della piruvato deidrogenasi è essenziale, sia nello stato di digiuno che in quello alimentato. Infatti, il complesso multienzimatico svolge un ruolo centrale nel metabolismo in quanto, catalizzando la decarbossilazione ossidativa irreversibile del piruvato, rappresenta la porta di ingresso dello scheletro carbonioso dei carboidrati e di circa il 50% dello scheletro carbonioso degli amminoacidi glucogenici, che nell’insieme costituiscono circa il 60% dell’apporto calorico giornaliero, verso:
il ciclo dell’acido citrico, e quindi alla completa ossidazione a CO2;
la sintesi degli lipidi, nello stato alimentato, e dell’acetiolcolina.
L’importanza della regolazione della conversione del piruvato in acetil-CoA è sottolineata anche dal fatto che i mammiferi, sebbene siano in grado di produrre glucosio dal piruvato, non sono in grado di farlo dall’acetil-CoA. Ciò è conseguenza sia della irreversibilità della reazione catalizzata dalla piruvato deidrogenasi che dell’assenza di vie metaboliche alternative in grado di farlo.
L’inibizione dell’attività del complesso permetterà quindi di risparmiare glucosio e gli aminoacidi che possono essere convertiti in piruvato, come l’alanina, quando sono disponibili altri carburanti, ad esempio l’acetil-CoA derivante dall’ossidazione degli acidi grassi.
Per questi motivi l’attività del complesso è finemente regolata attraverso:
inibizione a feed-back, anche nota come inibizione da prodotto finale;
nucleotidi;
modificazioni covalenti, ossia fosforilazioni e defosforilazioni di specifiche proteine bersaglio.
Regolazione attraverso inibizione da prodotto finale e stato energetico della cellula
L’attività della forma defosforilata del complesso della piruvato deidrogenasi è regolata attraverso inibizione a feed-back o inibizione da prodotto finale.
Acetil-CoA e NADH inibiscono allostericamente gli enzimi che ne catalizzano la sintesi, rispettivamente diidrolipoil transacetilasi e la diidrolipoil deidrogenasi.
Inoltre, il CoA e l’acetil-CoA, così come il NAD+ e il NADH, competono per i siti di legame sui rispettivi enzimi, i quali catalizzano reazioni reversibili.
Questo significa che, in presenza di elevati valori dei rapporti [NADH]/[NAD+] e [Acetil-CoA]/[CoA], le reazioni di transacetilazione e deidrogenazione vanno nella direzione opposta rispetto a quella della formazione dell’acetil-CoA. Di conseguenza la diidrolipoil transacetilasi non può accettare il gruppo idrossietilico dalla TPP in quanto è mantenuta nella forma acetilata. Questo fa si che la tiamina pirofosfato rimanga legata alla piruvato deidrogenasi nella sua forma idrossietilica. Questo a sua volta riduce la velocità di decarbossilazione del piruvato. Quindi, elevati valori dei rapporti [NADH]/[NAD+] e [Acetil-CoA]/[CoA] influenzano indirettamente l’attività della piruvato deidrogenasi.
PDC: Inibizione a Feed-back
Acetil-CoA e NADH sono prodotti anche durante l’ossidazione degli acidi grassi. Anche questa via metabolica che, al pari delle reazioni catalizzate dal complesso della piruvato deidrogenasi, si verifica nel mitocondrio.
Ciò significa che la cellula, attraverso la regolazione dell’attività del complesso multienzimatico, è in grado di preservare le riserve di carboidrati quando sono disponibili acidi grassi per la produzione di energia. Questo per esempio è quanto accadde nel digiuno, quando il fegato, il muscolo scheletrico e molti altri organi e tessuti si affidano principalmente all’ossidazione degli acidi grassi per la produzione di energia.
Di contro, l’attività del complesso è aumentata nello stato alimentato. In questa condizione molti differenti tipi di cellule e tessuti utilizzano in prevalenza il glucosio come fonte di energia.
Più in generale, quando la produzione di NADH e/o acetil-CoA supera la capacità della cellula di utilizzarli per la produzione di ATP, l’attività del complesso della piruvato deidrogenasi è inibita.
Analogo discorso vale nella condizione in cui non ci sia necessità di produrre ulteriore ATP. Infatti l’attività catalitica del complesso multienzimatico è sensibile anche allo stato energetico della cellula. Attraverso meccanismi allosterici:
livelli di ATP elevati inibiscono l’attività della componente piruvato deidrogenasi del complesso multienzimatico;
elevati livelli di ADP, che segnalano che la cellula potrebbe entrare in una fase di carenza di energia, lo attivano, indirizzando quindi lo scheletro carbonioso dei carboidrati e di alcuni aminoacidi verso la produzione di energia.
Nota: nel muscolo scheletrico l’attività del complesso della piruvato deidrogenasi aumenta con l’aumento dell’attività aerobica. Questo si traduce in una maggiore dipendenza del muscolo dal glucosio come fonte di energia.
Regolazione attraverso fosforilazione/defosforilazione
A differenza di quanto accade nei procarioti, nei mammiferi l’attività del complesso della piruvato deidrogenasi è regolata anche attraverso modificazioni covalenti. Nello specifico si tratta di fosforilazioni e defosforilazioni di tre specifici residui di serina presenti sulla subunità α della piruvato deidrogenasi, l’enzima che catalizza il primo passaggio, irreversibile, dell’intera sequenza di reazioni.
Nota: poiché la piruvato deidrogenasi dei mammiferi è un eterotetramero, ci sono sei potenziali siti di fosforilazione.
PDC: Regolazione Mediante Modificazione Covalente
La fosforilazione, che inattiva la piruvato deidrogenasi, e quindi blocca l’intera sequenza di reazioni, è catalizzata dalla piruvato deidrogenasi chinasi. Due delle suddette serine si trovano su una delle due anse presenti all’ingresso del canale per il substrato che porta al rispettivo sito attivo, quella più vicina all’estremità C-terminale. La fosforilazione di uno solo di questi residui inattiva la piruvato deidrogenasi, dimostrando così la l’accoppiamento fuori fase tra i due siti attivi.
Nello stato defosforilato invece il complesso risulta attivo. La defosforilazione è catalizzata da una specifica protein fosfatasi, la piruvato deidrogenasi fosfatasi.
Le attività della piruvato deidrogenasi chinasi e della piruvato deidrogenasi fosfatasi sono a loro volta soggette a regolazione allosterica da parte di numero effettori.
Regolazione della piruvato deidrogenasi chinasi
L’attività della piruvato deidrogenasi chinasi dipende dal valore dei rapporti [NADH]/[NAD+], [acetil-CoA]/[CoA], e [ATP]/[ADP], come anche dalla concentrazione del piruvato, nella matrice mitocondriale.
Elevati valori dei rapporti [NADH]/[NAD+] e [acetil-CoA]/[CoA], come nel corso dell’ ossidazione degli acidi grassi e dei corpi chetonici, attivano la chinasi. La piruvato deidrogenasi viene quindi fosforilata, e il complesso multienzimatico risulta inibito. Questo permette ai tessuti, come ad esempio il muscolo cardiaco, di risparmiare glucosio quando si stanno utilizzando acidi grassi e/o corpi chetonici per la produzione di energia, in quanto la sintesi di acetil-CoA dal piruvato, e quindi dai carboidrati (e da alcuni aminoacidi) e bloccata. Quando invece le concentrazioni di NAD+ e coenzima A sono elevate, l’attività catalitica della chinasi è inibita e il complesso risulta attivo. Quindi, acetil-CoA e NADH, due dei tre prodotti finali delle reazione catalizzate dal complesso della piruvato deidrogenasi, controllano allostericamente la propria sintesi regolando direttamente e indirettamente, tramite la regolazione dell’attività della piruvato deidrogenasi chinasi, l’attività del complesso multienzimatico.
Elevati valori del rapporto [ATP]/[ADP] attivano la chinasi, e quindi inibiscono il complesso multienzimatico. Nota: a differenza di molte altre chinasi, come quelle che intervengono nel controllo del metabolismo del glicogeno, la piruvato deidrogenasi chinasi non è regolata dai livelli di cAMP, bensì da molecole che segnalano variazioni nel livello della carica energetica della cellula e nella disponibilità di intermedi biosintetici, ossia rispettivamente ATP e NADH e acetil-CoA.
Il piruvato è un effettore allosterico negativo della piruvato deidrogenasi chinasi. Quando i suoi livelli sono elevati, il suo legame alla chinasi la inattiva, la piruvato deidrogenasi non viene fosforilata, e il complesso della piruvato deidrogenasi rimane attivo.
La piruvato deidrogenasi chinasi è attivata anche a seguito dell’interazione con la diidrolipoil transacetilasi nella sua forma acetilata, ossia quando è presente la acetil-diidrolipoamide.
Altri attivatori della piruvato deidrogenasi chinasi sono gli ioni potassio e magnesio.
Regolazione della piruvato deidrogenasi fosfatasi
L’attività della piruvato deidrogenasi fosfatasi dipende dal valore dei rapporti [NADH]/[NAD+] e [acetil-CoA]/[CoA], come anche dalla [Ca2+], nella matrice mitocondriale.
Bassi valori dei rapporti [NADH]/[NAD+] e [acetil-CoA]/[CoA] attivano la fosfatasi, la piruvato deidrogenasi viene defosforilata, e il complesso multienzimatico risulta attivo.
Al contrario, in presenza di elevati valori dei suddetti rapporti l’attività della fosfatasi si riduce, quella della chinasi aumenta, e il complesso multienzimatico viene inibito.
Lo ione calcio attiva la piruvato deidrogenasi fosfatasi.
Ca2+ è un importante secondo messaggero che segnala la necessità da parte della cellula di ulteriore energia. Quindi, quando è presente in elevate concentrazioni, come nelle cellule muscolari cardiache a seguito della stimolazione da parte dell’adrenalina, o nella cellule muscolari scheletriche nel corso della contrazione muscolare, la fosfatasi è attiva, il complesso è defosforilato, e quindi attivato.
Anche l’insulina interviene nel controllo dell’attività catalitica del complesso della piruvato deidrogenasi attraverso l’attivazione della piruvato deidrogenasi fosfatasi. L’ormone, in risposta all’aumento della glicemia, stimola sia la sintesi del glicogeno che dell’acetil-CoA, precursore nella sintesi degli lipidi.
Anche il digiuno e la successiva rialimentazione influiscono sull’attività del complesso multienzimatico.
In tessuti come il muscolo scheletrico, il muscolo cardiaco o il rene, il digiuno riduce in modo significativo l’attività del complesso, mentre la rialimentazione inverte la situazione.
Nel cervello invece non si osservano queste variazioni poiché l’attività del complesso della piruvato deidrogenasi è essenziale per la produzione di ATP.
Bibliografia
Frank R.A.W., Titman C.M., Pratap J.V., Luisi B.F., and Perham R.N. A molecular switch and proton wire synchronize the active sites in thiamine enzymes. Science 2004;306(5697):872-876. doi:10.1126/science.1101030
Gray L.R., Tompkins S.C., Taylor E.R. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-2604. doi:10.1007/s00018-013-1539-2
McCommis K.S. and Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J 2015;466(3):443-454. doi:10.1042/BJ20141171
Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
Nemeria N.S., Chakraborty S., Balakrishnan A., and Frank Jordan. Reaction mechanisms of thiamin diphosphate enzymes: defining states of ionization and tautomerization of the cofactor at individual steps. FEBS J 2009;276:2432-2446. doi:10.1111/j.1742-4658.2009.06964.x
Patel M.S. and Korotchkina L.G. Regulation of the pyruvate dehydrogenase complex. Biochem Soc T 2006;34(2):217-222. doi:10.1042/bst0340217
Patel M.S., Nemeria N.S., Furey W., and Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem 2014;289(24):16615-16623. doi:10.1074/jbc.R114.563148
Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009
Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011
Wang J., Nemeria N.S., Chandrasekhar K., Kumaran S., Arjunan P., Reynolds S., Calero G., Brukh R., Kakalis L., Furey W., and Jordan F. Structure and function of the catalytic domain of the dihydrolipoyl acetyltransferase component in Escherichia coli pyruvate dehydrogenase complex. J Biol Chem 2014;289(22):15215-15230. doi:10.1074/jbc.M113.544080
Zhou Z.H., McCarthy D.B., O’Connor C.M., Reed L.J., and J.K. Stoops. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA 2001;98(26):14802-14807. doi:10.1073/pnas.011597698
La via del pentoso fosfato, anche detta via del fosfogluconato, è una via metabolica, comune a tutti gli organismi viventi, per il metabolismo ossidativo del glucosio, alternativa alla glicolisi da cui si ramifica a livello del glucosio-6-fosfato, e le cui funzioni principali sono la produzione, in rapporti variabili, di NADPH, un coenzima ridotto, e ribosio-5-fosfato, uno zucchero fosforilato a cinque atomi di carbonio, ossia un pentoso fosfato, da cui il nome della via.[14]
La via del fosfogluconato, ramificandosi dalla glicolisi, è anche detta shunt dell’esoso monofosfato, dove shunt può essere tradotto come derivazione o smistamento.
Dal punto di vista concettuale, nella via del pentoso fosfato è possibile individuare due fasi: la fase ossidativa, nel corso della quale viene prodotto il NADPH, e la fase non ossidativa, nella quale si formano il ribosio-5-fosfato e altri zuccheri fosforilati.[16]
Via del Pentoso Fosfato
E’ stato stimato che più del 10% del glucosio metabolizzato è trasportato attraverso questa via metabolica che, degno di nota, pur ossidando il monosaccaride non comporta alcuna produzione diretta, ma neppure consumo, di ATP.[7][13]
Le prime evidenze dell’esistenza della via del fosfogluconato emersero negli anni trenta del secolo scorso dagli studi di Otto Warburg, premio Nobel per la Fisiologia o la Medicina nel 1931, che scoprì il NADP durante studi sull’ossidazione del glucosio-6-fosfato a 6-fosfogluconato.[22]
Ulteriori indicazioni emersero dall’osservazione che nei tessuti il glucosio continuava a essere metabolizzato anche in presenza di inibitori della glicolisi, quali gli ioni fluoruro e iodoacetato, inibitori rispettivamente della enolasi (EC 4.2.1.11) e della gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12).
Tuttavia la delucidazione completa della via avvenne solo negli anni cinquanta del secolo scorso grazie al lavoro di diversi ricercatori e principalmente dei gruppi di Efraim Racker, Fritz Lipmann, vincitore del premio Nobel per la Fisiologia o la Medicina nel 1953 anche grazie alla scoperta del coenzima A, Bernard Horecker e Frank Dickens.[8]
Funzione principale
La principale funzione della via del pentoso fosfato è la produzione di NADPH e ribosio-5-fosfato.[22]
Il NADPH è necessario per le biosintesi riduttive, quali la sintesi degli acidi grassi, del colesterolo, degli ormoni steroidei e degli aminoacidi non essenziali prolina e tirosina, rispettivamente dal glutammato e dalla fenilalanina, nonché per la riduzione del glutatione ossidato.[14] Nelle suddette reazioni il coenzima ridotto funge da donatore di elettroni, o meglio da donatore di uno ione idruro (:H–), equivalente a un protone e due elettroni.
Nicotinammide Adenina Dinucleotide Fosfato
Nota: nei vertebrati circa la metà del NADPH necessario per i passaggi riduttivi della sintesi degli acidi grassi deriva dalla via del pentoso fosfato, mentre la restante parte dalla reazione catalizzata dall’enzima malico (EC 1.1.1.40), di cui di seguito è indicata la reazione.[13]
Malato + NADP+ ⇄ Piruvato + NADPH + H++ HCO3–
Il ribosio-5-fosfato viene utilizzato nella sintesi dei nucleotidi e degli acidi nucleici, DNA e RNA, dell’ATP, di coenzimi come il coenzima A, il NAD, il NADP e il FAD, e degli aminoacidi essenziali triptofano e istidina.[5] Lo zucchero fosforilato non viene utilizzato direttamente come tale ma previa conversione, attivazione, a 5-fosforibosil-1-pirofosfato o PRPP, acronimo dell’inglese 5-phosphoribosyl 1-pyrophosphate, nella reazione catalizzata dalla ribosio fosfato pirofosfochinasi o PRPP sintetasi (EC 2.7.6.1).[13]
Ribosio-5-fosfato + ATP → 5-Fosforibosil-1-pirofosfato + AMP
Funzioni aggiuntive
In aggiunta alla produzione di NADPH e ribosio-5-fosfato, la via del pentoso fosfato ha altre funzioni, sia anaboliche che cataboliche.[18]
In molti batteri e nei lieviti è coinvolta nel catabolismo degli zuccheri a cinque atomi di carbonio ribosio, xilosio e arabinosio.
Anche nell’uomo questa è coinvolta nel catabolismo dei suddetti pentosi e dei meno comuni carboidrati a tre, quattro e sette atomi di carbonio assunti con la dieta, nonché per il catabolismo dei:
pentosi derivanti dal catabolismo dei carboidrati strutturali;
ribosio-5-fosfato derivante dal catabolismo dei nucleotidi.
Negli organismi fotosintetici contribuisce all’incorporazione dell’anidride carbonica (CO2) nella molecola del glucosio nel corso del ciclo di Calvin.[18]
Porta alla sintesi oltre che di ribosio-5-fosfato, anche di altri intermedi utilizzati in vari processi biosintetici come:
eritrosio-4-fosfato, utilizzato per la sintesi dei tre aminoacidi aromatici triptofano, tirosina e fenilalanina;
ribulosio-5-fosfato, utilizzato per la sintesi della riboflavina;
sedoeptulosio-7-fosfato che, nei batteri Gram-negativi, è utilizzato per la sintesi delle unità di eptosio dello strato lipopolisaccaridico della membrana esterna.[19]
Dove avviene la via del pentoso fosfato?
Nelle cellule animali la via del pentoso fosfato, al pari della glicolisi, della sintesi degli acidi grassi, e della maggior parte delle reazioni della gluconeogenesi, avviene nel citosol, analogamente a quanto accade nei batteri.[5] E, considerando glicolisi, gluconeogenesi e via del pentoso fosfato è possibile affermare che queste tre vie sono interconnesse tramite diversi enzimi e/o intermedi condivisi.
Nelle cellule vegetali la via del fosfogluconato avviene nei plastidi, e i suoi intermedi possono raggiungere il citosol attraverso i pori di membrana di questi organelli.[13]
Nell’uomo, i livelli di espressione degli enzimi della via variano da tessuto a tessuto. Livelli relativamente elevati si ritrovano nel fegato, nella corteccia surrenale, nei testicoli, nelle ovaie, nella tiroide, nelle ghiandole mammarie durante l’allattamento e nei globuli rossi.[14] In tutte queste sedi è richiesto un apporto continuo di NADPH per sostenere le biosintesi riduttive e/o contrastare gli effetti dei radicali liberi dell’ossigeno su strutture cellulari sensibili come il DNA, i lipidi di membrana e le proteine.[1]
Elevati livelli degli enzimi di questa via metabolica sono presenti anche in cellule in rapida divisione quali quelle dell’embrione nelle prime fasi di sviluppo, gli enterociti, le cellule della pelle e del midollo osseo e, in condizioni patologiche, le cellule tumorali, tutte cellule che richiedono elevate quantità di ribosio-5-fosfato per la sintesi dei nucleotidi.[16]
Di contro nel muscolo scheletrico sono presenti livelli estremamente bassi degli enzimi della via del pentoso fosfato, via che in questa sede è praticamente assente e dove il glucosio-6-fosfato è utilizzato principalmente per la produzione di energia attraverso la glicolisi e il ciclo dell’acido citrico.[5]
Fase ossidativa
La fase ossidativa della via del pentoso fosfatosi compone di tre tappe, due ossidazioni irreversibili, rispettivamente la prima e la terza reazione, e una idrolisi.
In questa fase una molecola di glucosio-6-fosfato viene convertita in una di ribulosio-5-fosfato, uno zucchero fosforilato a 5 atomi di carbonio e substrato di partenza per le reazioni della fase non ossidativa, con la concomitante produzione di due molecole di NADPH e la liberazione del C-1 del glucosio in forma di CO2.[7] L’equazione complessiva è:
Ossidazione del glucosio-6-fosfato a 6-fosfoglucono-delta-lattone
Nella prima tappa della fase ossidativa della via del pentoso fosfato, il glucosio-6-fosfato viene ossidato a 6-fosfoglucono-δ-lattone, un estere intramolecolare, reazione catalizzata dalla glucosio-6-fosfato deidrogenasi.
Nella reazione il NADP+ funge da agente riducente accettando uno ione idruro dal carbonio 1 del glucosio-6-fosfato, e riducendosi quindi a NADPH.
Si noti che questa è la prima molecola di NADPH prodotta nella via del pentoso fosfato.
La reazione catalizzata dalla glucosio-6-fosfato deidrogenasi o G6PD (EC 1.1.1.49) è una reazione “esclusiva” della via del pentoso fosfato.[1] Analogamente a quanto accade nella maggior parte delle altre vie metaboliche, anche in questo caso, la prima reazione esclusiva della via è essenzialmente irreversibile, con un ΔG nel fegato pari a -4,21 kcal/mol (-17,6 kJ/mol), e altamente regolata per via allosterica.[22] L’enzima infatti rappresenta il principale punto di controllo del flusso di metaboliti attraverso la via stessa.
Nell’uomo i livelli più elevati di G6PD sono presenti nei neutrofili e macrofagi, cellule fagocitarie in cui, durante l’infiammazione, il NADPH viene utilizzato per la produzione di radicali superossido (O2–.) dall’ossigeno molecolare nella reazione catalizzata dalla NADPH ossidasi (EC 1.6.3.1 ).[2][16]
2 O2 + NADPH → 2 O2–. + NADP+ + H+
Di seguito i radicali superossido potranno essere utilizzati per la sintesi, a scopi difensivi, ossia l’uccisione dei microrganismi fagocitati, di altri ROS come anche di radicali liberi dell’azoto o RNS, acronimo dell’inglese reactive nitrogen species, quali:
il perossido di idrogeno o acqua ossigenata (H2O2), nella reazione catalizzata dalla superossido dismutasi o SOD (EC 1.15.1.1)
2 O2–. + 2 H+ → H2O2 + O2
il perossinitrito (O=N–O–O–), a seguito della reazione con il monossido di azoto (•NO)
O2–. + •NO → O=N–O–O–
il radicale idroperossido (HOO•)
O2–. + H+ → HOO•
Meccanismo catalitico della glucosio-6-fosfato deidrogenasi
Il meccanismo catalitico dell’enzima è stato ampiamente studiato nel microorganismo Leuconostoc mesenteroides, la cui glucosio-6-fosfato deidrogenasi ha la peculiare caratteristica di poter utilizzare come coenzima il NAD+ e/o il NADP+.[12]
Meccanismo Catalitico della Glucosio-6-fosfato Deidrogenasi
L’enzima non necessita della presenza di ioni metallici per la propria attività; uno dei residui presenti nel sito attivo funge da base generale, ossia è in grado di estrarre uno ione idruro dal gruppo ossidrilico legato al C-1 del glucosio-6-fosfato.[4]
Nell’enzima batterico questo è effettuato dall’atomo Nɛ2 dell’anello imidazolico della catena laterale di un residuo di istidina, azoto che presenta un doppietto elettronico disponibile portare un l’attacco nucleofilo. Questa fa si che il glucosio-6-fosfato, un emiacetale ciclico con il C-1 nello stato di ossidazione aldeidico, sia ossidato a estere ciclico, ossia un lattone. Ciò consente il trasferimento dello ione idruro legato al C-1 del glucosio sul C-4 dell’anello nicotinamidico del NADP+ a dare NADPH.
Poiché l’istidina suddetta è conservata in molte delle glucosio-6-fosfato deidrogenasi sequenziate, è probabile che questo meccanismo catalitico sia generalizzabile a tutte le glucosio-6-fosfato deidrogenasi.
Regolazione della glucosio-6-fosfato deidrogenasi
La glucosio-6-fosfato deidrogenasi rappresenta l’enzima chiave nella regolazione del flusso metabolico attraverso la via del pentoso fosfato, e quindi il principale punto di controllo della velocità di produzione del NADPH.[2]
Nell’uomo l’enzima è presente in due forme: quella monomerica, inattiva, e in un equilibrio tra una forma dimerica e una tetramerica, le conformazioni attive.[1]
Uno dei principali modulatori della sua attività è il rapporto citosolico NADP+/NADPH.[15] Elevati livelli di NADPH inibiscono l’enzima poiché il coenzima ridotto è un potente inibitore competitivo della G6PD, mentre il NADP+ è necessario sia per la sua attività catalitica che per il mantenimento della conformazione attiva. Infatti il legame del coenzima ossidato a un sito specifico, distante dal sito attivo ma vicino all’interfaccia del dimero, è necessario per mantenerne la conformazione dimerica.
Regolazione dell’Attività della G6PD
Nella maggior parte delle condizioni metaboliche il rapporto NADP+/NADPH è basso, meno NADP+ è disponibile per legarsi all’enzima e di conseguenza si riduce l’attività dell’enzima stesso, a prescindere dai livelli della sua espressione genica. In queste condizioni la fase ossidativa è praticamente inattiva.
Se invece si considerano cellule in cui siano particolarmente attive vie metaboliche e/o reazioni che utilizzano NADPH, si verifica la riduzione della sua concentrazione citosolica, un aumento di quella del NADP+ e quindi una stimolazione dell’attività dell’enzima. Ne consegue un’attivazione della fase ossidativa della via dell’esoso monofosfato.
Quindi è anche possibile affermare che il destino del glucosio-6-fosfato, un intermedio comune alla glicolisi e alla via del fosfogluconato, dipende anche dal fabbisogno contingente di NADPH.[15]
Un secondo meccanismo di regolazione dell’attività della glucosio-6-fosfato deidrogenasi chiama in causa l’accumulo di acil-CoA, intermedi nella sintesi degli acidi grassi.[5] Queste molecole, legandosi alla forma dimerica dell’enzima ne causano la dissociazione nei due monomeri costitutivi, e quindi la perdita dell’attività catalitica.
L’insulina up-regola l’espressione del gene per la glucosio-6-fosfato deidrogenasi e per la 6-fosfogluconato deidrogenasi, per cui l’ormone, nello stato ben alimentato, concorre ad aumentare il flusso di carbonio attraverso la via del pentoso fosfato, promuovendo quindi la sintesi di NADPH.[7]
Nota: l’insulina promuove anche la sintesi degli acidi grassi.
Idrolisi del 6-fosfoglucone-delta-lattone a 6-fosfogluconato
Nella seconda tappa della fase ossidativa si verifica l’idrolisi del 6-fosfoglucone-δ-lattone a dare il 6-fosfogluconato, una molecola lineare. Il 6-fosfoglucone-δ-lattone è di per se idroliticamente instabile e va incontro a una idrolisi spontanea che apre l’anello, reazione che avviene a una velocità significativa.[22] Tuttavia nella cellula la reazione è accelerata dall’azione catalitica di una specifica lattonasi (EC 3.1.1.31).[3]
Decarbossilazione ossidativa del 6-fosfogluconato a ribulosio-5-fosfato
Nella terza tappa della fase ossidativa si verifica la decarbossilazione ossidativa del 6-fosfogluconato a dare ribulosio-5-fosfato, un cheto pentoso, una molecola di CO2 e una di NADPH. La reazione è catalizzata dalla 6-fosfogluconato deidrogenasi (EC 1.1.1.44), enzima che richiede la presenza di ioni magnesio, Mg2+.
6-Fosfogluconato + NADP+ → Ribulosio-5-fosfato + NADPH + CO2
Si noti che questa è la seconda molecola di NADPH prodotta nella via del pentoso fosfato.[15]
Meccanismo catalitico della 6-fosfogluconato deidrogenasi
Il meccanismo catalitico, simile a quello dell’enzima del ciclo dell’acido citrico isocitrico deidrogenasi (EC 1.1.1.41), consiste in una catalisi acido/base e procede attraverso tre passaggi in cui sono coinvolti in particolare due residui conservati presenti nel sito attivo, una lisina (Lys) e un glutammato (Glu), nell’uomo rispettivamente la Lys185 e il Glu192.[22] La lisina agisce sia come acido che come base mentre il glutammato come acido.[6][17]
Meccanismo Catalitico della 6-Fosfogluconato Deidrogenasi
Nel primo passaggio, il passaggio ossidativo, il 6-fosfogluconato viene ossidato in un β-cheto acido, il 3-cheto-6-fosfogluconato.
In questo passaggio l’ε-amino gruppo della catena laterale del suddetto residuo di lisina funge da base, da nucleofilo, estraendo un protone dal gruppo ossidrilico legato sul C-3, cui segue il trasferimento dello ione idruro legato al C-3 sul C-4 dell’anello nicotinamidico del NADP+. In questo modo si viene a formare il 3-cheto intermedio e una molecola di NADPH che lascia il sito attivo.
Nel secondo passaggio, il passaggio decarbossilativo, il 3-cheto-6-fosfogluconato, che è estremamente suscettibile a decarbossilazione, perde, in forma di CO2, il C-1 del glucosio-6-fosfato a dare il cis-1,2-enediolo del ribulosio-5-fosfato, un intermedio a elevata energia. In questo passaggio la lisina agisce come acido donando un protone all’ossigeno carbonilico legato al C-3.
Infine l’enzima catalizza una conversione stereospecifica enolo-chetone con formazione del ribulosio-5-fosfato. In questo passaggio il residuo di acido glutammico suddetto funge da acido donando un protone al C-1 del cis-1,2-enediolo, mentre l’ε-amino gruppo della lisina accetta un protone dal gruppo ossidrilico legato sul C-2. Il risultato è la formazione del ribulosio-5-fosfato.
Fase non ossidativa
Nella fase non ossidativa della via del pentoso fosfato viene prodotta una varietà di carboidrati fosforilati il cui destino dipende dalle necessità relative di NADPH, ribosio-5-fosfato e ATP da parte della cellula.[7]
Questa fase si compone di cinque tappe, tutte facilmente reversibili, nel corso delle quali si verifica una serie di interconversioni di zuccheri fosforilati.
La fase non ossidativa ha inizio con due possibili reazioni a carico del ribulosio-5-fosfato: una isomerizzazione e una epimerizzazione che portano alla formazione rispettivamente di ribosio-5-fosfato e xilulosio-5-fosfato.[14]
Le Epimerasi (EC 5.1), un sottogruppo delle Isomerasi (EC 5.), catalizzano l’inversione reversibile della configurazione di un atomo di carbonio asimmetrico, in genere attraverso la rimozione e l’aggiunta di un atomo di idrogeno.
Nelle reazioni di isomerizzazione lo scambio di gruppi chimici si verifica tra atomi di carbonio.
Nota: le reazioni enzimatiche di isomerizzazione ed epimerizzazione svolgono un ruolo importante nel metabolismo dei carboidrati.
Isomerizzazione del ribulosio-5-fosfato a ribosio-5-fosfato
Nella prima tappa della fase non ossidativa della via del pentoso fosfato, una reazione di isomerizzazione, il ribulosio-5-fosfato, un chetoso, viene convertito nell’aldoso corrispondente, il ribosio-5-fosfato. La reazione è catalizzata dalla fosfopentoso isomerasi o ribosio-5-fosfato isomerasi (EC 5.3.1.6).
Ribulosio-5-fosfato ⇄ Ribosio-5-fosfato
Le due molecole sono un esempio di isomeria di gruppo funzionale.
Meccanismo catalitico della ribosio-5-fosfato isomerasi
Il meccanismo catalitico della ribosio-5-fosfato isomerasi, simile a quello dell’enzima glicolitico fosfoglucosio isomerasi (EC 5.3.1.9), procede attraverso la formazione dell’intermedio ad alta energia cis-1,2-enediolo del ribulosio-5-fosfato, formazione conseguente a un meccanismo di trasferimento protonico comune alle isomerizzazione aldoso-chetoso.
Di seguito viene descritto il possibile meccanismo catalitico dell’enzima di E. coli nella direzione della formazione del ribulosio-5-fosfato dal ribosio-5-fosfato, che è anche la direzione seguita nel ciclo di Calvin della fotosintesi.[23]
Meccanismo Catalitico della Fosfopentoso Isomerasi
Il primo passaggio consiste nell’apertura dell’anello furanosico del ribosio-5-fosfato, apertura rara in soluzione acquosa (<0.5%), e indotta dall’interazione con un residuo di acido aspartico (Asp81) che accetta un protone dal gruppo idrossilico legato al C-1, mentre è probabile che sia l’acqua il donatore di un protone.
Una volta aperta la catena un residuo di acido glutammico (Glu103) funge da base generale, da nucleofilo, estraendo il protone legato sul C-2, mentre Asp81 dona un protone. Come risultato si forma il cis-1,2-enediolo.
Di seguito, la base iniziale, Glu103 adesso protonata, funge da acido e dona un protone al C-1 dell’intermedio enediolico, mentre Asp81 funge da base generale accettando un protone dal gruppo ossidrilico legato al C-2. Il risultato è la formazione del ribulosio-5-fosfato.
Il meccanismo di reazione per la sintesi del ribosio-5-fosfato dal ribulosio-5-fosfato è semplicemente l’inverso di quanto appena descritto.
Epimerizzazione del ribulosio-5-fosfato a xilulosio-5-fosfato
L’altro destino metabolico del ribulosio-5-fosfato nella via del pentoso fosfato è quello di essere epimerizzato a xilulosio-5-fosfato, un chetoso come il ribulosio-5-fosfato, nella reazione catalizzata dalla fosfopentoso epimerasi (EC 5.1.3.1).
Ribulosio-5-fosfato ⇄ Xilulosio-5-fosfato
Nota: lo xilulosio-5-fosfato, oltre a essere un intermedio della via del pentoso fosfato ha anche azione regolatoria: stimola la glicolisi e inibisce la gluconeogenesi, intervenendo nel controllo della concentrazione del fruttosio-2,6-bisfosfato nel fegato.[10]
Meccanismo catalitico della fosfopentoso epimerasi
Anche questa reazione, come quelle catalizzate dalla 6-fosfogluconato deidrogenasi e dalla ribosio-5-fosfato isomerasi, procede attraverso la formazione di un intermedio enediolico, ma con il doppio legame non tra i carboni 1 e 2 ma tra quelli 2 e 3.
Quello che accade nel corso della reazione è che un residuo aminoacidico presente nel sito attivo dell’enzima funge da base generale, da nucleofilo, ed estrae un protone dal C-3, il che porta alla formazione dell’intermedio cis-2,3-enediolico. Di seguito un residuo aminoacidico acido dona un protone al C-3, ma sul lato opposto, e dunque determinando l’inversione della configurazione sul C-3 a formare lo xilulosio-5-fosfato.[5][9]
Meccanismo Catalitico della Fosfopentoso Epimerasi
Arrivati a questo punto la via dell’esoso monofosfato ha portato alla formazione, per ogni molecola di glucosio-6-fosfato metabolizzata, di:
un pool di tre pentoso-5-fosfati, ossia ribulosio-5-fosfato, ribosio-5-fosfato e xilulosio-5-fosfato, che coesistono all’equilibrio;
2 molecole di NADPH.
Nelle tre tappe successive, dalla sesta all’ottava, la transchetolasi (EC 2.2.1.1) e la transaldolasi (EC 2.2.1.2), enzimi esclusivi della via del pentoso fosfato, catalizzano una serie di riarrangiamenti degli scheletri carboniosi che porteranno alla formazione di unità carboniose a tre, quattro, sei e sette atomi che potranno essere utilizzate per vari scopi metabolici, a seconda delle necessità della cellula.[2]
In termini di analisi dei flussi di metaboliti tra le differenti vie metaboliche, l’azione concertata della transchetolasi e della transaldolasi permette l’interazione della via del pentoso fosfato, in particolare la sua fase non ossidativa, con la glicolisi, e la gluconeogenesi, nonché con le vie che portano alla formazione di numerose vitamine, coenzimi e precursori degli acidi nucleici.
Transchetolasi
La transchetolasi è l’enzima che catalizza la tappa limitante della fase non ossidativa della via del pentoso fosfato, e il primo enzima che agisce a valle delle reazioni catalizzate dalla ribosio-5-fosfato isomerasi e della fosfopentoso epimerasi.[11]
Scoperta indipendentemente nel 1953 da Horecker e Racker, e così chiamata da quest’ultimo, catalizza, nella sesta e ottava tappa, il trasferimento di unità a due atomi di carbonio da un donatore chetoso, ossia lo xilulosio-5-fosfato, il sedoeptulosio-7-fosfato o il fruttosio-6-fosfato, a un aldoso accettore, ribosio-5-fosfato, gliceraldeide-3-fosfato o eritrosio-4-fosfato.[8]
Reazioni della Transchetolasi
Considerando come esempi le reazioni “in avanti”, nella sesta tappa il chetoso donatore è lo xilulosio-5-fosfato mentre l’aldoso accettore è il ribosio-5-fosfato, a formare gliceraldeide-3-fosfato, il frammento a tre atomi di carbonio rimanente dello xilulosio-5-fosfato, e sedoeptulosio-7-fosfato, uno zucchero fosforilato a sette atomi di carbonio che sarà utilizzato nel passaggio successivo, il settimo.
Nell’ottava tappa il chetoso donatore è lo xilulosio-5-fosfato mentre l’aldoso accettore è l’eritrosio-4-fosfato, a dare una seconda molecola di gliceraldeide-3-fosfato e una di fruttosio-6-fosfato.
Da notare che tre dei quattro prodotti delle reazioni catalizzate dalla transchetolasi, due molecole di gliceraldeide-3-fosfato e una di fruttosio-6-fosfato, sono anche intermedi della glicolisi.[11]
La transchetolasi può utilizzare come substrati, oltre allo xilulosio-5-fosfato, al sedoeptulosio-7-fosfato e al fruttosio-6-fosfato anche altri 2-chetozuccheri, nonché diversi altri aldoso fosfati.
Transchetolasi e tiamina pirofosfato
La transchetolasi appartiene al gruppo degli enzimi che richiedono la tiamina pirofosfato o TPP, acronimo dell’inglese thiamine pyrophosphate, come cofattore.
La tiamina pirofosfato è la forma biologicamente attiva della tiamina o vitamina B1, ed è saldamente legata all’enzima.
Tiamina Pirofosfato
Altri enzimi che richiedono la TPP come cofattore sono:
la piruvato decarbossilasi (EC 4.1.1.1), che interviene nella fermentazione alcolica;
la componente alfa-chetoacido deidrogenasi o subunità E1 (EC 1.2.4.4) del complesso della alfa-chetoacido deidrogenasi dei chetoacidi a catena ramificata;
la componente 2-chetoglutarato deidrogenasi o subunità E1 (EC 1.2.4.2) del complesso della alfa-chetoglutarato deidrogenasi, enzima del ciclo dell’acido citrico.
La funzione della tiamina pirofosfato è quella di contribuire al trasferimento di intermedi aldeidici attivati stabilizzando carbanioni a due atomi di carbonio che si forma durante la reazione.
Meccanismo catalitico della transchetolasi
L’atomo di carbonio compreso tra l’atomo di zolfo e quello di azoto dell’anello tiazolico della tiamina pirofosfato, ossia l’atomo C-2, è molto più acido rispetto alla maggior parte dei gruppi =C- presenti in altre molecole a causa dell’adiacente atomo di azoto caricato positivamente che stabilizza elettrostaticamente il carbanione derivante dalla dissociazione del protone. Questo fa si che l’atomo di idrogeno a esso legato sia facilmente dissociabile a formare un carbanione, ossia un atomo di carbonio con carica negativa. L’estrazione del protone è catalizzata dalla transchetolasi.[5][14][22]
I carbanioni reagiscono con il carbonio carbonilico del substrato chetonico, nella tappa 6 lo xilulosio-5-fosfato o il sedoeptulosio-7-fosfato nella reazione in direzione opposta, oppure, considerando la tappa 8, di nuovo lo xilulosio-5-fosfato o il fruttosio-6-fosfato nella reazione in direzione opposta.
Prendendo come esempio la reazione “in avanti” della tappa 6, il composto di addizione tra la tiamina pirofosfato e lo xilulosio-5-fosfato va incontro a frammentazione a seguito della scissione del legame C2-C3 dello xilulosio-5-fosfato, a dare gliceraldeide-3-fosfato, che è rilasciata, e un’unità a due atomi di carbonio, un gruppo idrossietilico con carica negativa, che rimane legato al C-2 dell’anello tiazolico.
Meccanismo Catalitico della Transchetolasi
La carica negativa presente sull’intermedio idrossietilico, ossia l’intermedio carbanionico, viene stabilizzata dall’anello tiazolico della tiamina pirofosfato grazie alla presenza dell’atomo di azoto carico positivamente che funge da “trappola” o “pozzo” per gli elettroni. Quindi l’anello tiazolico fornisce una struttura elettron deficiente o elettrofila nella quale gli elettroni del carbanione possono delocalizzarsi per risonanza.
Nell’ultimo passaggio si verifica la condensazione tra il gruppo idrossietilico e il ribosio-5-fosfato, l’aldeide accettore, attraverso l’attacco del carbanione sul carbonio aldeidico del ribosio-5-fosfato, a formare un addotto covalente legato alla tiamina pirofosfato.
La successiva scissione dell’addotto porta alla liberazione di sedoeptulosio-7-fosfato, rigenerando il carbanione della tiamina pirofosfato.
Transaldolasi
La transaldolasi, scoperta nel 1953 da Horecker e Smyrniotis nel lievito Saccharomyces cerevisiae, interviene nella settima tappa della via del pentoso fosfato, dove catalizza il trasferimento di una unità a tre atomi di carbonio da un substrato donatore, il sedoeptulosio-7-fosfato, a un substrato accettore, la gliceraldeide-3-fosfato, a dare fruttosio-6-fosfato ed eritrosio-4-fosfato.[18]
Da notare che, in analogia con quanto visto per le reazioni catalizzate dalla transchetolasi, anche in questo caso il donatore dell’unità carboniosa trasferita è un chetoso mentre l’accettore è un aldoso.
Nella reazione in direzione opposta il chetoso donatore sarà il fruttosio-6-fosfato mentre l’aldoso accettore l’eritrosio-4-fosfato.
Meccanismo catalitico della transaldolasi
A differenza della transchetolasi, la transaldolasi non necessita per la propria attività della presenza di cofattori.
La reazione si compone di una parte equivalente a una scissione aldolica e di una equivalente a una condensazione aldolica. Di seguito verrà analizzato il meccanismo di catalitico della transaldolasi B di E. coli, considerando la reazione nella direzione della formazione dell’eritrosio-4-fosfato e del fruttosio-6-fosfato.
Nel primo passaggio l’ε-amino gruppo della catena laterale di un residuo di lisina (Lys132) presente nel sito attivo, a seguito del trasferimento di un protone a un residuo di acido glutammico (Glu96), trasferimento mediato da una molecola d’acqua, porta un attacco nucleofilo al C-2 del sedoeptulosio-7-fosfato, il carbonio carbonilico. Il risultato è la formazione di una carbinolammina con il sedoeptulosio-7-fosfato.[5][14][18][22]
Meccanismo Catalitico della Transaldolasi
Segue la perdita di una molecola d’acqua dalla carbinolammina con formazione di una immina, o base di Schiff, legata all’enzima; anche in questo passaggio interviene il trasferimento di un protone da Glu96 alla molecola d’acqua “catalitica”.
Nota: l’intermedio covalente enzima-substrato che si è formato è simile a quello che quello che si forma nella reazione catalizzata dalla aldolasi (EC 4.1.2.13) nella quarta tappa della glicolisi.
Nel passaggio successivo, il gruppo carbossilico di un residuo di acido aspartico (Asp17) estrae un protone dal gruppo idrossilico legato sul C-4, con conseguente rottura del legame covalente tra C-3 e C-4, una scissione aldolica che porta alla liberazione dell’aldoso eritrosio-4-fosfato, il primo prodotto della reazione, mentre rimane legato all’enzima un carbanione a tre atomi di carbonio che è stabilizzato per risonanza, analogamente a quanto visto per la transchetolasi. Infatti, al pari dell’atomo di azoto dell’anello tiazolico della tiamina pirofosfato, anche l’atomo di azoto con carica positiva della base di Schiff agisce da trappola per gli elettroni stabilizzando la carica negativa del carbanione.
Una volta che il secondo substrato, la gliceraldeide-3-fosato, si trova in posizione nel sito attivo, il carbanione porta un attacco nucleofilo al carbonio carbonilico della gliceraldeide-3-fosfato a formare, mediante una condensazione aldolica, un nuovo legame C-C e un chetoso legato all’enzima.
Infine l’idrolisi della base di Schiff porta al rilascio del fruttosio-6-fosfato, un chetoso e il secondo prodotto della reazione, e permette l’inizio di un nuovo ciclo di reazione.
A questo punto, come visto in precedenza, nell’ottava tappa della via del pentoso fosfato, interviene nuovamente la transchetolasi che catalizza la formazione di fruttosio-6-fosfato e glicealdeide-3-fosfato a partire da eritrosio-4-fosfato e xilulosio-5-fosfato.
Fabbisogno di ribosio-5-fosfato, NADPH e ATP
Dal punto di vista molecolare il destino del glucosio-6-fosfato dipende in larga parte dalle attività relative degli enzimi che lo metabolizzano nella via glicolitica e nella via del pentoso fosfato, in particolare dalla fosfofruttochinasi-1 o PFK-1, acronimo dell’inglese phosphofructokinase-1 (EC 2.7.1.11) e la glucosio-6-fosfato deidrogenasi, enzimi la cui attività è altamente regolata.
La PFK-1 è inibita quando aumenta la concentrazione dell’ATP e/o di citrato, ossia quando la carica energetica della cellula è alta, mentre è attivata quando aumenta la concentrazione dell’AMP e/o del fruttosio-2,6-bisfosfato, ossia quando la carica energetica della cellula è bassa. Ne consegue che, quando la carica energetica della cellula è alta il flusso di carbonio, e quindi di glucosio-6-fosfato, attraverso la glicolisi si riduce.[20][21]
La glucosio-6-fosfato deidrogenasi è invece inibita dal NADPH e dagli acil-CoA, intermedi nella biosintesi degli acidi grassi.[15] Se quindi la concentrazione citosolica del NADPH aumenta il flusso di glucosio-6-fosfato attraverso la via del pentoso fosfato è inibito. Viceversa, se i livelli di NADPH cadono, l’inibizione viene meno, la via si “riaccende” e vengono sintetizzati NADPH e ribosio-5-fosfato.
Pertanto, in base alle necessità della cellula di NADPH, ribosio-5-fosfato e ATP, quello che accadrà è una “combinazione” di alcune reazioni della via glicolitica, della gluconeogenesi e della via del pentoso fosfato al fine di sintetizzare i metaboliti richiesti, sfruttando anche il fatto che la fase non ossidativa dello shunt dell’esoso monofosfato è controllata essenzialmente dalla disponibilità dei substrati.
Di seguito sono analizzate le quattro principali possibilità.[2]
Fabbisogno di NADPH molto maggiore di quello di ribosio-5-fosfato e ATP
Quando il fabbisogno di NADPH è molto maggiori di quello di ribosio-5-fosfato e la cellula non ha bisogno di ulteriore produzione di ATP, ossia la carica energetica della cellula è alta, il glucosio-6-fosfato entra nella via del pentoso fosfato e può essere completamente ossidato a CO2. Una condizione metabolica del genere si ritrova ad esempio nel tessuto adiposo nel corso di una intensa sintesi di acidi grassi.
Nella fase ossidativa della via, per ogni molecola di glucosio-6-fosfato ossidata a ribulosio-5-fosfato, sono prodotte due molecole di NADPH. Attraverso una combinazione delle reazione della fase non ossidativa e di alcune reazioni della gluconeogenesi, nello specifico quelle catalizzate dagli enzimi trioso fosfato isomerasi (EC 5.3.1.1), aldolasi (EC 4.1.2.13), fosfoglucosio isomerasi (EC 5.3.1.9), e fruttosio-1,6-bisfosfatasi (EC 3.1.3.11), sei molecole di ribulosio-5-fosfato vengono convertite in cinque di glucosio-6-fosfato. E’ quindi anche possibile affermare che le reazioni della fase non ossidativa permettono alle reazioni della fase ossidativa di procedere.
In questo processo è possibile individuare tre gruppi di reazioni che operano in sequenza
Nel primo gruppo si ritrovano le reazioni catalizzate dagli enzimi della fase ossidativa, e si ha la formazione di due molecole di NADPH e una di ribulosio-5-fosfato.
Al secondo gruppo appartengono le reazioni catalizzate dagli enzimi fosfopentoso epimerasi, ribosio-5-fosfato isomerasi, transchetolasi e transaldolasi, ossia quelli della fase non ossidativa della via, che portano alla conversione del ribulosio-5-fosfato in fruttosio-6-fosfato e gliceraldeide-3-fosfato.
Infine, attraverso le suddette reazioni della gluconeogenesi il fruttosio-6-fosfato e la gliceraldeide-3-fosfato sono “riciclati” a glucosio-6-fosfato, di modo che il ciclo possa ricominciare.
Sommando le ultime due reazioni si evince che sei molecole di ribulosio-5-fosfato sono convertite in cinque di glucosio-6-fosfato.
6 Ribulosio-5-fosfato + H2O → 5 Glucosio-6-fosfato + Pi
Dalla somma delle reazioni del primo, secondo e terzo gruppo si ottiene la reazione complessiva:
Glucosio-6-fosfato + 12 NADP+ + 7 H20 → 6 CO2 + 12 NADPH + 12 H+ + Pi
Dunque, una molecola di glucosio-6-fosfato, attraverso sei cicli della via del pentoso fosfato accoppiati con alcune reazioni della reazioni gluconeogenesi, sarà convertita in 6 molecole di CO2, con la concomitante produzione di 12 molecole di NADPH, ma nessuna produzione netta di ribosio-5-fosfato.
Fabbisogno di NADPH e ATP molto maggiore di quello di ribosio-5-fosfato
Quando il fabbisogno di NADPH è molto maggiore di quello di ribosio-5-fosfato e la carica energetica della cellula è bassa, ossia la cellula necessita di ATP, il ribulosio-5-fosfato prodotto nella fase ossidativa della via del pentoso fosfato viene convertito in fruttosio-6-fosfato e gliceraldeide-3-fosfato attraverso le reazioni della fase non ossidativa. Di seguito i due intermedi, attraverso le reazioni della glicolisi, sono ossidati a piruvato, la base coniugata dell’acido piruvico, con concomitante produzione di ATP.
La reazione complessiva è:
Se la cellula necessita di altro ATP, il piruvato prodotto potrà essere ossidato attraverso il ciclo dell’acido citrico. Se invece non è necessario produrre ulteriore ATP, il piruvato potrà essere utilizzato come precursore in diverse vie biosintetiche.
Da notare che anche in questo caso, come nel precedente, non si verifica alcuna produzione netta di ribosio-5-fosfato.
Fabbisogno di ribosio-5-fosfato molto maggiore di quello di NADPH
Quando il fabbisogno di ribosio-5-fosfato è molto maggiore rispetto a quello di NADPH, come nelle cellule che si dividono rapidamente, nelle quali è quindi in corso una rapida sintesi di nucleotidi precursori del DNA, le reazioni della fase ossidativa della via del pentoso fosfato non hanno luogo, e non vi è sintesi di NADPH. Al contrario, poiché le reazioni della fase non ossidativa sono facilmente reversibili, la riduzione della concentrazione del ribosio-5-fosfato, causata dal suo rapido utilizzo, avrà come effetto quello di stimolarne la sintesi.
Quello che accade è che, attraverso la via glicolitica, molto del glucosio-6-fosfato viene convertito in fruttosio-6-fosfato e gliceraldeide-3-fosfato. A questo punto la transaldolasi e la transchetolasi portano alla sintesi di ribosio-5-fosfato e xilulosio-5-fosfato. Quest’ultimo, attraverso le reazione catalizzate dalla fosfotopentoso epimerasi e di seguito dalla ribosio-5-fosfato isomerasi, verrà convertito in ribosio-5-fosfato.
La reazione complessiva è:
6 Glucosio-6-fosfato + ATP → 6-Ribosio-5-fosfato + ADP + H+
In questo caso quindi quello che si verifica è una combinazione di reazioni della glicolisi e della fase non ossidativa della via del fosfogluconato, con queste ultime nella direzione della sintesi del ribosio-5-fosfato.
Da notare che nessun metabolita torna alla glicolisi.
I fabbisogni di ribosio-5-fosfato e NADPH si equivalgono
Se una molecola di ribosio-5-fosfato e due molecole di NADPH per molecola di glucosio-6-fosfato metabolizzata sono sufficienti a soddisfare le necessità metaboliche della cellula, quello che si verifica sono le prime quattro reazioni della via del pentoso fosfato, ossia l’intera fase ossidativa, più la reazione catalizzata dalla ribosio-5-fosfato isomerasi.
La reazione complessiva è:
Anche in questa condizione metabolica nessun metabolita torna alla glicolisi.
Bibliografia
^ abc Au S.W.N., Gover S., Lam V.M.S., Adams M.J. Human glucose-6-phosphate dehydrogenase: the crystal structure reveals a structural NADP+ molecule and provides insights into enzyme deficiency. Structure 2000;8(3):293-303. doi:10.1016/S0969-2126(00)00104-0
^ abcd Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002
^ Brodie A.F., Lipmann F. Identification of a gluconolactonase. J Biol Chem 1955;212(2):677-85. doi:10.1016/S0021-9258(18)71006-5
^ Cosgrove M.S., Naylor C., Paludan S., Adams M.J., Richard Levy H. On the mechanism of the reaction catalyzed by glucose 6-phosphate dehydrogenase. Biochemistry 1998;37(9);2759-2767. doi:10.1021/bi972069y
^ Hanau S., Montin K., Cervellati C., Magnani M., Dallocchio F. 6-Phosphogluconate dehydrogenase mechanism: evidence for allosteric modulation by substrate. J Biol Chem 2010;285(28):21366-21371. doi:10.1074/jbc.M110.105601
^ ab Horecker B.L. The pentose phosphate pathway. J Biol Chem 2002;277(50):47965-47971. doi:10.1074/jbc.X200007200
^ Jelakovic S., Kopriva S., Süss K-H, Schulz G.E. Structure and catalytic mechanism of the cytosolic D-ribulose-5-phosphate 3-epimerase from rice. J Mol Biol 2003;326:127-135. doi:10.1016/S0022-2836(02)01374-8
^ Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003;100:5107-5112. doi:10.1073/pnas.0730817100
^ ab Kochetov G.A., Solovjeva O.N. Structure and functioning mechanism of transketolase. Biochim Biophys Acta 2014;1844(9):1608-1618. doi:10.1016/j.bbapap.2014.06.003
^ Levy H.R. Glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides. Biochem Soc Trans. 1989 Apr;17(2):313-5. doi:10.1042/bst0170313
^ abcd Michal G., Schomburg D. Biochemical pathways. An atlas of biochemistry and molecular biology. 2nd Edition. John Wiley J. & Sons, Inc. 2012
^ abcdef Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ abcd Patra K.C., Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci 2014;39(8):347-354. doi:10.1016/j.tibs.2014.06.005
^ abc Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009
^ Ruda G.F., Campbell G., Alibu V.P., Barrett M.P., Brenk R., Gilbert I.H. Virtual fragment screening for novel inhibitors of 6-phosphogluconate dehydrogenase. Bioorg Med Chem 2010;18(14):5056-62. doi:10.1016/j.bmc.2010.05.077
^ abcd Samland A.K., Sprenger G.A. Transaldolase: from biochemistry to human disease. Int J Biochem Cell Biol 2009;41(7):1482-1494. doi:10.1016/j.biocel.2009.02.001
^ Sprenger G.A. Genetics of pentose-phosphate pathway enzymes of Escherichia coli K-12. Arch Microbiol 1995;164(5):324-30. doi:10.1007/BF02529978
^ Van Schaftingen E., Hers H-G. Inhibition of fructose-1,6-bisphosphatase by fructose-2,6-bisphosphate. Proc Natl Acad Sci USA 1981;78(5):2861-2863. doi:10.1073/pnas.78.5.2861
^ Van Schaftingen E., Jett M-F., Hue L., Hers H-G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA 1981;78(6):3483-3486. doi:10.1073/pnas.78.6.3483
^ abcdefg Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011
^ Zhang R., Andersson C.E., Savchenko A., Skarina T., Evdokimova E., Beasley S., Arrowsmith C.H., Edwards A.M., Joachimiak A., Mowbray S.L. Structure of Escherichia coli ribose-5-phosphate isomerase: a ubiquitous enzyme of the pentose phosphate pathway and the Calvin cycle. Structure 2003;11(1):31-42. doi:10.1016/S0969-2126(02)00933-4
La glicolisi, dal greco glykys, dolce e lysis, sciogliere, può essere definita come la sequenza di reazioni enzimatiche che, nel citosol della cellula, anche in assenza di ossigeno, porta alla conversione di una molecola di glucosio, un composto a sei atomi di carbonio, in due di piruvato, un composto a tre atomi di carbonio, con la concomitante produzione di due molecole di ATP, la moneta di scambio energetico della cellula.[5]
La Via Glicolitica
La glicolisi, che si è evoluta prima che nell’atmosfera si accumulasse una quantità sostanziale di ossigeno, è la via metabolica con il maggior flusso di carbonio nella maggior parte degli esseri viventi. E’ presente in quasi tutti gli organismi.
Grazie al fatto di poter procedere in assenza di ossigeno, ha svolto un ruolo cruciale nei processi metabolici durante i primi due miliardi di anni di evoluzione della vita sulla terra. Probabilmente rappresenta il modo più antico che gli organismi hanno sviluppato per ottenere energia dalle molecole organiche quando la disponibilità del gas è scarsa. E’ inoltre una fonte di precursori per il catabolismo aerobico e per vari processi biosintetici.
Nota: la glicolisi è conosciuta anche come la via di Embden-Meyerhof, dal nome dei due ricercatori che delucidarono l’intero processo nel muscolo.[2][5]
Lo sviluppo della biochimica è andato di pari passo con la comprensione del metabolismo del glucosio e in particolare della glicolisi, la prima via metabolica a essere stata chiarita.[2][5]
Sebbene la delucidazione di questa via fu completata negli anni quaranta del secolo scorso, la scoperta chiave sul metabolismo del glucosio venne fatta in modo casuale nel 1897, a seguito di un problema sorto un anno prima quando un chimico tedesco, M. Hahn, nel tentativo di ottenere e conservare estratti proteici privi di cellule dal lievito, incontrò difficoltà nella loro conservazione. Un suo collega, Hans Buchner, ricordando quanto veniva, e viene tuttora fatto per la conservazione delle marmellate, suggerì di addizionare saccarosio all’estratto.
Eduard Buchner, fratello di Hans, mise in pratica l’idea di Hans, osservando però che dalla soluzione si sviluppavano bollicine. Questo indusse Eduard a concludere che fosse in corso una fermentazione, per l’epoca una scoperta sorprendente. Infatti sino ad allora la fermentazione, secondo quanto affermato da Pasteur nel 1860, era stata inestricabilmente legata alle cellule viventi, mentre ora veniva dimostrato che poteva avvenire anche al di fuori delle cellule stesse. In breve, questi due ricercatori demolirono il dogma vitalistico e diedero il via alla moderna biochimica.
Grazie a queste ricerche Eduard Buchner fu insignito del premio Nobel per la Chimica nel 1907. Buchner fu il primo di una serie di ricercatori che vinsero il premio grazie alle loro scoperte sulla glicolisi. In seguito fu dimostrato, lavorando con estratti muscolari, che molte delle reazioni della fermentazione lattica erano le stesse della fermentazione alcolica, il che evidenziò l’esistenza di una unità nella biochimica.
Come anticipato, la glicolisi fu poi chiarita in modo esaustivo nella prima metà del secolo scorso in gran parte grazie al lavoro di ricercatori quali Gerty and Carl Cori, Carl Neuberg, Arthur Harden, William Young, Jacob Parnas, Otto Warburg, Hans von Euler-Chelpin, Gustav Embden e Otto Meyerhof.
Perché è importante?
La glicolisi è una via metabolica essenziale sia dal punto di vista energetico che come fonte di precursori per diverse altre vie metaboliche. E la velocità del flusso di carbonio attraverso di essa, ossia la velocità con cui il glucosio viene convertito in piruvato, è regolata in modo da soddisfare queste due necessità fondamentali della cellula.[7]
Dal punto di vista energetico, sebbene sia un processo relativamente inefficiente, può avvenire in assenza di ossigeno, la condizione in cui si è sviluppata la vita sulla Terra e in cui tutt’oggi molte cellule, sia eucariote che procariote, si ritrovano. Di seguito alcuni esempi.
I muscoli di molti animali presentano una anaerobiosi dipendente dall’attività, ossia possono funzionare in assenza di ossigeno per brevi periodi. Ad esempio, quando gli animali, ma anche gli atleti, compiono esercizi molto intensi, il loro fabbisogno di ATP aumenta più rapidamente della capacità del corpo di rifornire di ossigeno il muscolo. In questa situazione il muscolo ricava l’energia metabolica necessaria al suo funzionamento in modo anaerobico, appunto attraverso la sola glicolisi.
Un altro esempio è la cornea dell’occhio, un tessuto scarsamente vascolarizzato.
Molti microorganismi, come quelli che vivono in ambienti quali il suolo, le profondità marine ma anche i pori della pelle, si ritrovano in zone in cui l’ossigeno è scarso o assente. Ed esistono microrganismi, definiti anaerobi obbligati, che non possono sopravvivere in presenza di ossigeno, una molecola altamente reattiva. Esempi sono il batterio responsabile della gangrena gassosa, Clostridium perfringens, del tetano, Clostridium tetani, e del botulismo, Clostridium botulinum.
Va inoltre sottolineato che la glicolisi gioca un ruolo centrale anche in tutte quelle cellule e tessuti nei quali il glucosio rappresenta la sola fonte di energia, come:
i globuli rossi, privi di mitocondri;
le cellule spermatiche;
il cervello, che solo in particolari condizioni può usare anche i corpi chetonici per produrre energia;
la midollare del surrene.
Una situazione simile si ritrova anche nel mondo vegetale dove molte piante acquatiche e alcuni tessuti vegetali specializzati nell’accumulo di amido, come i tuberi delle patate, utilizzano il glucosio come principale fonte di energia.
Nota: esistono organismi che sono anaerobi facoltativi, ossia possono sopravvivere in presenza e in assenza di ossigeno, come gli animali appartenenti al genere Mytilus, che quindi mostrano un funzionamento anaerobico dipendente dall’ambiente, o anaerobico habitat-dipendente, una situazione simile a quanto visto in precedenza nel muscolo sottoposto a lavori molto intesi.
Infine non va dimenticato che in condizioni aerobiche, nelle cellule dotate di mitocondri, la glicolisi rappresenta la parte iniziale della via metabolica che porta alla completa ossidazione del glucosio ad anidride carbonica (CO2) e acqua a scopi energetici.
Precursori Derivanti dalla Via Glicolitica
Diversi intermedi glicolitici, come il glucosio-6-fosfato (G-6-P), il fruttosio-6-fosfato (F-6-P,), il diidrossiacetone fosfato (DHAP) o il piruvato possono essere utilizzati come precursori biosintetici in vie metaboliche quali ad esempio quelle che portano alla sintesi del glicogeno, degli acidi grassi, dei trigliceridi, dei nucleotidi, di alcuni amminoacidi, o del 2,3-bisfosfoglicerato (2,3-BPG).
Tappe
Le 10 tappe che compongono la glicolisi possono essere suddivise in due fasi.
La prima, detta fase preparatoria, si compone di 5 reazioni e comporta dapprima la conversione di una molecola di glucosio in una di fruttosio-1,6-bisfosfato (F-1,6-BP), attraverso tre passaggi:
una prima fosforilazione sul C-1;
una isomerizzazione;
una seconda fosforilazione, stavolta sul C-6, con consumo complessivo di 2 ATP.
Segue la scissione, la lisi, del fruttosio-1,6-bisfosfato in due intermedi fosforilati a tre atomi di carbonio, la gliceraldeide-3-fosfato e il diidrossiacetone fosfato, e infine l’isomerizzazione di quest’ultimo in gliceraldeide-3-fosfato. Dunque, nella fase preparatoria una molecola di glucosio viene scissa in due di gliceraldeide-3-fosfato e si consumano due ATP.[2][5]
Nella seconda fase, detta fase di recupero energetico, composta dalle restanti 5 reazioni, le due molecole di gliceraldeide-3-fosfato sono convertite in altrettante molecole di piruvato, con concomitante produzione di 4 molecole di ATP. Quindi nella seconda fase si verifica l’estrazione e conservazione in forma di ATP di parte dell’energia chimica presente nella molecola del glucosio. Inoltre sono estratti anche equivalenti riducenti, immagazzinati nella molecola del NADH. Quello che sarà poi il destino del NADH dipende dal tipo di cellula e dalla presenza o meno di quantità adeguate di ossigeno.[2][5]
Nota: nella glicolisi è metabolizzato sia il glucosio di derivazione ematica, che entra nella cellula attraverso un trasporto mediato da specifici trasportatori proteici di membrana, che il glucosio-6-fosfato derivante dalla glicogenolisi.
Reazione 1
Nella prima tappa della via glicolitica il glucosio viene fosforilato a glucosio-6-fosfato a spese di una molecola di ATP.
Glucosio + ATP → Glucosio-6-fosfato + ADP + H+
Nella maggior parte delle cellule questa reazione è catalizzata dalla esochinasi (EC 2.7.1.1), enzima presente nelle cellule di tutti gli organismi, e negli esseri umani con quattro forme isoenzimatiche.
L’esochinasi e la piruvato chinasi (EC 2.7.1.40), l’altra chinasi della glicolisi, al pari di molte altre chinasi, richiede per il corretto funzionamento la presenza dello ione magnesio (Mg2+) o di un altro ione metallico bivalente come il manganese (Mn2+). Considerando lo ione magnesio, questi si va a legare all’ATP a formare il complesso MgATP2-, che è il vero substrato dell’enzima. E va sottolineato che l’attacco nucleofilo da parte di un gruppo ossidrilico (-OH) del glucosio all’atomo di fosforo terminale dell’ATP è facilitato proprio dall’azione degli ioni magnesio che vanno a interagire con le cariche negative dei gruppi fosforici del nucleoside trifosfato.
La formazione del legame fosfoestereo tra un gruppo fosforico e il gruppo ossidrilico sul C-6 è termodinamicamente sfavorita e richiede energia per procedere, energia che è fornita dall’ATP. Infatti, mentre la fosforilazione del glucosio sul C-6 a opera del solo fosfato inorganico ha un ΔG°’ pari a 3,3 kcal/mol (13,8 kJ/mol), ossia è una reazione endoergonica, l’idrolisi dell’ATP a dare ADP e Pi ha un ha un ΔG°’ pari -7,3 kcal/mol (30,5 kJ/mol), ossia è una reazione esoergonica. La reazione netta avrà un ΔG°’ = -7,3 + 3,3 = -4,0 kcal/mol (-30,5 + 13,8 = -16,7 kJ/mol). Nelle condizioni presenti nella cellula la reazione è ancora più favorevole, con un ΔG pari a -8,0 kcal/mol (-33,5 kJ/mol).
Dunque si tratta di una reazione essenzialmente irreversibile.
Nota: in biochimica le fosforilazioni sono reazioni fondamentali catalizzate da enzimi detti chinasi, una sottoclasse delle transferasi. Le chinasi catalizzano il trasferimento del gruppo fosforico terminale, o gruppo fosforico in posizione γ, di un nucleoside trifosfato a un nucleofilo accettore a formare un legame fosfoestereo. Nello specifico, le esochinasi catalizzano il trasferimento del gruppo fosforico in posizione γ dell’ATP a vari zuccheri a sei atomi di carbonio, o esosi, tra cui, oltre ovviamente al glucosio, anche al fruttosio e al mannosio.
Perché la fosforilazione del glucosio è importante?
La fosforilazione della molecola del glucosio è importante per almeno alcuni motivi.[2]
Il glucosio-6-fosfato grazie alla sua carica negativa e all’assenza sulla membrana plasmatica di trasportatori per gli zuccheri fosforilati non può diffondere attraverso di essa. Quindi, dopo la fosforilazione iniziale non è più necessario spendere energia per mantenere la molecola fosforilata all’interno della cellula, nonostante la grande differenza esistente tra le sue concentrazioni intra- ed extracellulari.
Considerazioni analoghe valgono per gli altri otto intermedi fosforilati che si ritrovano tra il glucosio-6-fosfato e il piruvato.
La rapida fosforilazione del glucosio mantiene la concentrazione intracellulare dell’esoso bassa, favorendo così il suo passaggio all’interno della cellula.
La fosforilazione comporta l’aumento del contenuto in energia della molecola, ossia inizia a destabilizzarla, facilitandone il successivo metabolismo.
Altri destini metabolici del glucosio-6-fosfato
Il glucosio-6-fosfato rappresenta un punto di ramificazione importante del metabolismo del glucosio. Infatti, oltre che proseguire nella via glicolitica, in condizioni anaboliche, può avere destini differenti. Di seguito alcuni esempi.
Può essere utilizzato per la sintesi di: glicogeno, immagazzinato principalmente nel fegato e nel muscolo;
polisaccaridi complessi presenti nella matrice extracellulare; galattosio;
glucosammina e altri zuccheri utilizzati per la glicosilazione delle proteine.
Può essere entrare nella via del pentoso fosfato, via attraverso la quale le cellule producono:
NADPH, necessario in generale per le biosintesi riduttive, come quella degli acidi grassi, del colesterolo, degli ormoni steroidei, e dei desossiribonucleotidi, ma anche indispensabile nella prevenzione del danno ossidativo in cellule come i globuli rossi;
riboso-5-fosfato, utilizzato nella sintesi dei nucleotidi ma anche del NADH, FADH2 e del coenzima A.
Reazione 2
Nella seconda tappa della glicolisi si verifica l’isomerizzazione del glucosio-6-fosfato, un aldoso, a fruttosio-6-fosfato, un chetoso, nella reazione catalizzata dalla fosfoglucosio isomerasi (EC 5.3.1.9).
Glucosio-6-fosfato ⇄ Fruttosio-6-fosfato
Glucosio-6-fosfato e fruttosio-6-fosfato sono un esempio di isomeria di gruppo funzionale
Come l’esochinasi, anche la fosfoglucosio isomerasi necessita della presenza di Mg2+.
Il ΔG°’ della reazione è pari a 0,4 kcal/mol (1,7 kJ/mol), mentre il ΔG è di -0,6 kcal/mol (-2,5 kJ/mol). Questo piccolo valore sta a indicare che la reazione è prossima all’equilibrio ed è facilmente reversibile. La reazione consiste essenzialmente nel passaggio del gruppo carbonilico presente sul C-1 della forma a catena aperta del glucosio-6-fosfato al C-2 della forma a catena aperta del fruttosio-6-fosfato.
La reazione enzimatica può essere suddivisa in almeno tre passaggi. Poiché entrambe gli esosi in soluzione acquosa sono presenti principalmente nella forma ciclica, l’enzima deve dapprima aprirne l’anello, catalizzare l’isomerizzazione, e infine richiudere l’anello stesso, portando alla formazione dell’anello a 5 atomi di carbonio del fruttosio-6-fosfato.
Reazione Catalizzata dalla Fosfoglucosio Isomerasi
Questa reazione di isomerizzazione è un passaggio cruciale per glicolisi in quanto prepara per le due tappe successive.
Perché?
La fosforilazione che si verifica nella reazione successiva, la terza, richiede la presenza di un gruppo alcolico sul C-1, e non di un gruppo carbonilico.
Nella quarta tappa si verifica la scissione del legame tra il C-3 e il C-4, scissione facilitata dalla presenza del gruppo carbonilico sul C-2.
Reazione 3
Nella terza tappa della via glicolitica si verifica una seconda reazione di fosforilazione nella quale il fruttosio-6-fosfato viene fosforilato sul C-1 a dare fruttosio-1,6-bisfosfato, a spese di una molecola di ATP. La reazione è catalizzata dalla fosfofruttochinasi-1 o PFK-1 (EC 2.7.1.11).
Fruttosio-6-fosfato + ATP → Fruttosio-1,6-bisfosfato + ADP + H+
La PFK-1 è così chiamata per distinguerla dall’enzima che catalizza la fosforilazione del fruttosio-6-fosfato a fruttosio-2,6-bisfosfato, ossia la fosfofruttochinasi-2 o PFK-2 (EC 2.7.1.105).
Al pari della reazione catalizzata dalla esochinasi/glucochinasi, anche questa fosforilazione è un passaggio essenzialmente irreversibile della glicolisi. E anche in questo caso l’irreversibilità è assicurata dall’accoppiamento, grazie alla PFK-1, con l’idrolisi dell’ATP. Infatti la fosforilazione del fruttosio-6-fosfato a opera del solo fosfato inorganico è una reazione endoergonica, con un ΔG°’ pari a 3,9 kcal/mol (16,3 kJ/mol), mentre, quando la reazione è accoppiata all’idrolisi dell’ATP, la reazione complessiva diviene esoergonica, con un ΔG°’ = 7,3-3,9 = -3,4 kcal/mol (-30,5 + 16,3 = -14,2 kJ/mol) e un ΔG di -5,3 kcal/mol (-22,2 kJ/mol).
Mentre la esochinasi permette alla cellula di “intrappolare” il glucosio al suo interno, la fosfofruttochinasi-1 evita che lo scheletro carbonioso del monosaccaride sia utilizzato per la sintesi del glicogeno o per la produzione di altri zuccheri, ma venga invece metabolizzato nella via glicolitica. Infatti, a differenza del glucosio-6-fosfato, il fruttosio-1,6-bisfosfato è un metabolita esclusivo della glicolisi/gluconeogenesi. In altri termini, la fosfofruttochinasi-1 catalizza la prima tappa di comando della via glicolitica. Reazioni di questo tipo sono di solito sono catalizzate da enzimi regolati per via allosterica, la cui regolazione impedisce l’accumulo sia dei prodotti intermedi che di quelli finali. La PFK-1 non fa eccezione. La chinasi è infatti soggetta a una fine regolazione allosterica da parte di numerosi metaboliti che segnalano il livello di energia e lo stato ormonale dell’organismo.
Alcuni batteri e protisti, e forse tutte le piante, posseggono una fosfofruttochinasi che utilizza, nella sintesi del fruttosio-1,6-bisfosfato, come donatore del gruppo fosforico non l’ATP ma il pirofosfato, una reazione con un ΔG°’ pari a -12,1 kcal/mol (-2,9 kJ/mol).
Fruttosio-6-fosfato + PPi → Fruttosio-1,6-bisfosfato + Pi
Nota: il prefisso bis– in bisfosfato, come fruttosio-1,6-bisfosfato, sta a indicare la presenza di due gruppi fosforici separati, mentre il prefisso di– in difosfato, come in adenosina difosfato, indica che sono presenti due gruppi fosforici connessi da un legame anidridico a dare un gruppo pirofosforico. Regole simili si applicano anche per la denominazione di molecole che presentano tre gruppi fosforici separati, come l’inositolo 1,4,5-trisfosfato, o connessi da legame anidridico come l’ATP o la guanosina trifosfato o GTP.
Reazione 4
Nella quarta tappa della glicolisi si verifica la scissione reversibile del fruttosio-1,6-bisfosfato in gliceraldeide-3-fosfato, un aldoso, e diidrossiacetone fosfato, un chetoso. La reazione è catalizzata dalla fruttosio-1,6-bisfosfato aldolasi o semplicemente aldolasi (EC 4.1.2.13), che catalizza la scissione del legame tra il C-3 e il C-4.
Tutti gli intermedi glicolitici a valle di questa reazione sono molecole a tre atomi di carbonio anziché a 6 come le precedenti.
Il ΔG°’ della reazione nella direzione della produzione di gliceraldeide-3-fosfato e diidrossiacetone fosfato è positivo, pari a 5,7 kcal/mol (23,8 kJ/mol), mentre la Km è pari a circa 10-4 M, valori che indicherebbero che la reazione non procede da sinistra verso destra. In realtà nelle normali condizioni cellulari, a causa delle basse concentrazioni dei reagenti presenti nella cellula, il ΔG è pari a -0,3 kcal/mol (-1,3 kJ/mol), un valore molto piccolo, per cui la reazione è facilmente reversibile, ossia essenzialmente all’equilibrio.
Nota: l’enzima aldolasi deriva il nome proprio nome dalla natura della reazione inversa, ossia una condensazione aldolica.
Reazione 5
Dei due prodotti della reazione catalizzata dalla aldolasi, la gliceraldeide-3-fosfato si trova sulla via diretta della glicolisi, al contrario del diidrossiacetone fosfato che come tale non può proseguire. Per farlo deve essere convertito, isomerizzato, a gliceraldeide-3-fosfato. Suddetta isomerizzazione avviene nella reazione catalizzata dalla trioso fosfato isomerasi (EC 5.3.1.1).
La trioso fosfato isomerasi, nel convertire il diidrossiacetone fosfato in gliceraldeide-3-fosfato, catalizza il trasferimento di un atomo d’idrogeno dal C-1 a C-2, ossia una ossidoriduzione intramolecolare. In pratica l’enzima fa si che i carboni C-1, C-2 e C-3 della molecola di glucosio di partenza divengano equivalenti rispettivamente ai carboni C-6, C-5 e C-4.
Il risultato netto dell’azione della aldolasi e della trisofosfato isomerasi è dunque la produzione di due molecole di gliceraldeide-3-fosfato.
Il ΔG°’ della reazione è pari a 1,8 kcal/mol (7,5 kJ/mol), mentre il ΔG è pari a 0,6 kcal/mol (2,5 kJ/mol). All’equilibrio, circa il 96% del triosofosfato presente sarebbe rappresentato dal diidrossiacetone fosfato. Tuttavia ciò non accade, e la reazione procede rapidamente verso la formazione della gliceraldeide-3-fosfato grazie alla tappa successiva della glicolisi che rimuove la gliceraldeide-3-fosfato prodotta.
Una delle caratteristiche distintive della trioso fosfato isomerasi è la sua grande capacità catalitica. L’enzima è infatti considerato cineticamente perfetto. Perché? Se si considera la velocità con cui avviene l’isomerizzazione in presenza di un catalizzatore basico come può essere l’acetone, l’enzima è in grado di accelerarla di un fattore 1010. Infatti, analizzando il rapporto Kcat/KM questi è pari a 2×108 M-1s-1, valore prossimo alla limite controllato dalla diffusione. Quindi, il passaggio limitante nella catalisi è l’incontro tra enzima e substrato controllato appunto dalla diffusione.
Considerando dal punto di vista energetico gli ultimi due passaggi della glicolisi, questi sono sfavorevoli, con ΔG°’ pari a 7,5 kcal/mol (31,3 kJ/mol), mentre il ΔG°’complessivo dei primi 5 passaggi è pari a 0,5 kcal/mol (2,1 kJ/mol), con una Keq complessiva di circa 0,43. Ed è l’energia libera derivante dall’idrolisi delle due molecole di ATP che, in condizioni standard, sposta la costante di equilibrio complessiva vicino a uno. Se invece si considera il ΔG questi è ampiamente negativo, -13,6 kcal/mol (-56,8 kJ/mol).
Nota: il diidrossiacetone fosfato può anche essere ridotto a glicerolo-3-fosfato nella reazione catalizzata dalla glicerolo-3-fosfafo deidrogenasi (EC 1.1.1.8).
L’enzima fa da ponte tra il metabolismo glucidico e lipidico in quanto il glicerolo-3-fosfato prodotto è utilizzato nella sintesi di lipidi come i trigliceridi.
Questa via è particolarmente importante nel tessuto adiposo e nell’intestino tenue.
Reazione 6
Nella sesta tappa della via glicolitica, la prima della seconda fase, si verifica l’ossidazione della gliceraldeide-3-fosfato a dare 1,3-bisfosfoglicerato (1,3-BPG) e la concomitante riduzione del NAD+ a NADH. La reazione è catalizzata dalla gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12).
Questa reazione è la prima delle due reazione della glicolisi in cui viene resa disponibile l’energia chimica necessaria per la successiva sintesi dell’ATP; l’altra è quella catalizzata dalla enolasi (EC 4.2.1.11). Perché?
La reazione è la somma di due processi.
Nella prima reazione si verifica l’ossidazione del gruppo aldeidico a gruppo carbossilico, passaggio nel quale il NAD+ funge da agente ossidante. La reazione è fortemente esoergonica, con un ∆G’° di -10,3 kcal/mol (-43 kJ/mol).
Nella seconda reazione si verifica la formazione del legame tra l’ortofosfato e il gruppo carbossilico sul C-1 dell’1,3-bisfosfoglicerato a dare un’anidride mista, detta acil fosfato. Questa reazione è fortemente endoergonica, con ∆G’° di 11,8 23kcal/mol (49,3 kJ/mol).
Affinché possa verificarsi la formazione dell’acil fosfato, le due reazioni precedenti non debbono avvenire in successione ma debbono essere accoppiate. In questo modo l’ossidazione dell’aldeide viene utilizzata per guidare la formazione dell’anidride mista, con un ΔG°’ complessivo leggermente endoergonico, 1,5 kcal/mol (6,3 kJ/mol), mentre il valore del ΔG è 0,6 kcal/mol (2,5 kJ/mol).
Dunque la maggior parte dell’energia libera derivante dall’ossidazione del gruppo aldeidico anziché dissiparsi in calore viene conservata grazie alla formazione dell’acil fosfato.
Nota: la riduzione, reversibile dell’anello nicotinamidico del NAD+ o del NADP+ è conseguente alla perdita di due atomi d’idrogeno da parte di un’altra molecola, in questo caso il gruppo aldeidico della gliceraldeide-3-fosfato, che quindi si ossida, e al successivo trasferimento di uno ione idruro, ossia due elettroni e un protone, all’anello suddetto. L’H+ rimanente è rilasciato nel mezzo acquoso. Di seguito le semireazioni per entrambe i coenzimi.
NAD+ + 2 e– + 2 H+ → NADH + H+
NADP+ + 2 e– + 2 H+ → NADPH + H+
Reazione 7
Nella settima tappa della glicolisi si verifica il trasferimento del gruppo fosforico ad alta energia dall’acil fosfato dell’1,3-bisfosfoglicerato all’ADP con formazione di ATP e 3-fosfoglicerato. La reazione è catalizzata dalla fosfoglicerato chinasi (EC 2.7.2.3).
1,3-Bisfosfoglicerato + ADP + H+ ⇄ 3-Fosfoglicerato + ATP
Il ΔG°’ della reazione è pari a -4,4 kcal/mol (-18,5 kJ/mol), quindi si tratta di una reazione esoergonica. Il ΔG è pari a 0,3 kcal/mol (1,3 kJ/mol).
L’elevato potenziale di trasferimento del gruppo fosforico dell’acil fosfato viene sfruttato per la fosforilazione dell’ADP, reazione definita fosforilazione a livello del substrato. In altri termini, parte dell’energia rilasciata nel corso dell’ossidazione del gruppo aldeidico nel passaggio precedente viene ora conservata attraverso la formazione di ATP dall’ADP e Pi.
La reazione catalizzata dalla fosfoglicerato chinasi è la prima delle due reazione della glicolisi in cui parte dell’energia chimica presente nella molecola del glucosio viene conservata in forma di ATP. E, poiché dall’azione dell’aldolasi e della trioso fosfato isomerasi, reazione 4 e 5, si formano due molecole di gliceraldeide-3-fosfato per molecola di glucosio, in questo passaggio sono prodotte due molecole di ATP che vanno a “pareggiare” le due molecole di ATP consumate nella fase preparatoria.
Nota: la fosfoglicerato chinasi prende il nome dalla reazione che procede nella direzione opposta rispetto a quella appena descritta, ossia la fosforilazione del 3-fosfoglicerato a dare 1,3-bisfosfoglicerato a spese di una molecola di ATP. L’enzima infatti, al pari di tutti gli altri enzimi, è in grado di catalizzare la reazione in entrambe le direzioni. E la direzione che porta alla sintesi dell’1,3-bisfosfoglicerato è seguita durante la fissazione fotosintetica della CO2 e la gluconeogenesi.
La reazione sei e la sette nel loro insieme rappresentano un processo di accoppiamento energetico in cui l’intermedio comune è rappresentato dall’1,3-bisfosfoglicerato. Mentre la reazione che porta alla sintesi dell’1,3-bisfosfoglicerato è endoergonica, con un ΔG°’ = 1,5 kcal/mol (6,3 kJ/mol), la seconda è esoergonica, ΔG°’ = -4,4 kcal/mol (-18,5 kJ/mol). Il ΔG°’ complessivo è pari a (-4,4 + 1,5) = 2,9 kcal/mol (-18,5 + 6,3 = 12,2 kJ/mol), ossia la razione catalizzata dalla fosfoglicerato chinasi è sufficientemente esoergonica da “trascinare” anche la precedente, rendendo la reazione complessiva esoergonica.
Gliceraldeide-3-fosfato + ADP + Pi + NAD+ ⇄ 3-Fosfoglicerato + ATP + NADH + H+
In verità, la reazione della fosfoglicerato chinasi è sufficientemente esoergonica da trascinare anche le reazioni catalizzate dalla aldolasi e dalla trioso fosfato isomerasi.
Poiché l’energia libera standard d’idrolisi del gruppo fosforico sul C-3 del 3-fosfoglicerato è pari a -3 kcal/mol (-12,5 kJ/mol), non è possibile alcuna sintesi di ATP dall’ADP e dal 3-fosfoglicerato stesso. Le due reazioni successive della glicolisi porteranno alla conversione del 3-fosfoglicerato in fosfoenolpiruvato, una molecola con un potenziale di trasferimento del gruppo fosforico sufficientemente alto da permettere la sintesi di ATP.
Reazione 8
Nell’ottava tappa della glicolisi il 3-fosfoglicerato viene convertito in 2-fosfoglicerato, in una reazione reversibile catalizzata dalla fosfoglicerato mutasi (EC 5.4.2.1). La reazione richiede la presenza di Mg2+, ha un ΔG molto basso, pari a circa 0,2 kcal/mol (0,8 kJ/mol) e un ΔG°’ pari a 1,1 kcal/mol (4,4 kJ/mol).
L’enzima è una mutasi, enzimi che catalizzano lo spostamento intramolecolare di un gruppo chimico, in questo caso lo spostamento di un gruppo fosforico dal C-3 al C-2 del 3-fosfoglicerato. A loro volta le mutasi sono una sottoclasse delle isomerasi.
Il meccanismo con cui si svolge questa reazione dipende dal tipo di organismo considerato. Ad esempio, nel lievito o nel muscolo di coniglio la reazione avviene in due passaggi e comporta la formazione di un intermedio fosfo-enzima. Nella prima parte della reazione un gruppo fosforico legato covalentemente a un residuo di istidina presente nel sito attivo viene trasferito all’ossidrile presente sul C-2 del 3-fosfoglicerato a dare il 2,3-bisfosfoglicerato. Nel passaggio successivo l’enzima opera come una fosfatasi convertendo il 2,3-bisfosfoglicerato in 2-fosfoglicerato; tuttavia il gruppo fosforico presente sul C-3 non viene rilasciato ma si lega al residuo d’istidina del sito attivo a rigenerare l’intermedio enzima-His-fosfato. Schematicamente:
Vale la pena notare che il gruppo fosforico del 2-fosfoglicerato prodotto non è lo stesso di quello del substrato 3-fosfoglicerato.
All’incirca una volta ogni 100 passaggi catalitici il 2,3-bisfosfoglicerato si dissocia dal sito attivo dall’enzima, lasciandolo privo dell’istidina fosforilata, e quindi in forma inattiva. L’enzima in forma inattiva può essere riattivato dal 2,3-BPG, che è quindi necessario sia sempre presente in piccole quantità. E il 2,3-bisfosfoglicerato è in effetti presente in piccole quantità nella maggior parte delle cellule. Fanno eccezione i globuli rossi, dove è prodotto attraverso lo shunt di Rapoport-Luebering e agisce anche come inibitore allosterico riducendo l’affinità dell’emoglobina per l’ossigeno, e dove la sua concentrazione è di circa 4-5 mM.
Nota: il 3-fosfoglicerato, oltre che proseguire nella via glicolitica, può essere utilizzato anche per la sintesi della serina, da cui derivano glicina e cisteina. La sintesi della serina ha inizio con la reazione catalizzata dalla fosfoglicerato deidrogenasi (EC 1.1.1.95), in cui si verifica l’ossidazione del 3-fosfoglicerato a 3-fosfoidrossipiruvato e la concomitante riduzione del NAD+ a NADH. Questa reazione costituisce anche il passaggio limitante dell’intera via biosintetica, in quanto la serina inibisce l’attività della deidrogenasi.
Reazione 9
Nella nona tappa della glicolisi si verifica la deidratazione, la rimozione di una molecola di acqua, del 2-fosfoglicerato a dare fosfoenolpiruvato, un enolo. La reazione, reversibile, è catalizzata dalla enolasi.
2-Fosfoglicerato ⇄ Fosfoenolpiruvato + H2O
La reazione richiede la presenza di Mg2+ che va a stabilizzare l’intermedio enolico che si forma nel corso del processo.
Il ΔG°’ della reazione è pari a 1,8 kcal/mol (7,5 kJ/mol), mentre il ΔG è pari a -0,8 kcal/mol (-3,3 kJ/mol).
Come anticipato fosfoenolpiruvato e 1,3-bisfosfoglicerato sono gli unici due intermedi della glicolisi a possedere un potenziale di trasferimento del gruppo fosforico sufficiente a permettere la sintesi dell’ATP. Da dove deriva l’elevata energia libera standard d’idrolisi del gruppo fosforico del fosfoenolpiruvato. Sebbene fosfoenolpiruvato e 2-fosfoglicerato contengano all’incirca la stessa quantità totale di energia, rispetto alla decomposizione a CO2 e H2O e Pi, la deidratazione del 2-fosfoglicerato porta a una diversa ridistribuzione della stessa, tale che l’energia libera standard d’idrolisi dei due gruppi fosforici corrisponde a:
-4,2 kcal/mol (-17,6 kJ/mol) per il 2-fosfoglicerato, un estere fosforico;
-14,8 kcal/mol (-61,9 kJ/mol) per il fosfoenolpiruvato, un enol fosfato.
Quello che accade è che il gruppo fosforico del fosfoenolpiruvato intrappola la molecola nella sua forma enolica, instabile. Quando, nell’ultima reazione della glicolisi, il fosfoenolpiruvato cede il gruppo fosforico all’ADP si formano ATP e la forma enolica del piruvato, instabile, che rapidamente e non enzimaticamente tautomerizza alla più stabile forma chetonica, prevalente a pH 7. Quindi, alla base dell’elevato potenziale di trasferimento del gruppo fosforico del fosfoenolpiruvato c’è la successiva tautomerizzazione enolo-chetone del piruvato.
Reazione 10
Nell’ultimo passaggio della via glicolitica si verifica il trasferimento del gruppo fosforico dal fosfoenolpiruvato all’ADP con produzione di una molecola di ATP e una di piruvato.
Fosfoenolpiruvato + ADP + H+ → Piruvato + ATP
La reazione, la seconda fosforilazione a livello del substrato della glicolisi, è catalizzata dalla piruvato chinasi, enzima tetramerico la cui attività, al pari della PFK-1, è altamente regolata. Infatti l’enzima possiede siti di legame per numerosi effettori allosterici. Inoltre nei vertebrati sono presenti tre forme isoenzimatiche, di cui una prevalente nel fegato, indicata come forma L, e un’altra, detta forma M, prevalente nel muscolo e nel cervello. I diversi isoenzimi hanno molte proprietà in comune, ma differiscono, oltre che nella distribuzione tissutale, anche nella risposta a ormoni quali glucagone, epinefrina e insulina.
L’attività dell’enzima è stimolata dallo ione potassio, K+, e da alcuni altri cationi monovalenti.
La reazione è essenzialmente irreversibile, con un ΔG°’ pari a -7,5 kcal/mol (-31,4 kJ/mol), e un ΔG pari a -4,0 kcal/mol (-16,7 kJ/mol), irreversibilità in gran parte dovuta, come anticipato nel paragrafo precedente, alla rapida tautomerizzazione spontanea del piruvato dalla forma enolica a quella chetonica.
Tautomeri, Enolico e Chetonico, del Piruvato
E, delle 14,8 kcal/mol (-61,9 kJ/mol) derivanti dall’idrolisi dell’enol fosfato del fosfoenolpiruvato, circa la metà viene conservata grazie alla formazione del legame fosfoanidridico tra Pi e ADP, il cui ΔG°’ d’idrolisi è pari a -7,3 kcal/mol (-30,5 kJ/mol). La restante energia, -7,5 kcal/mol (-31,4 kJ/mol), è la forza trainante che spinge la reazione verso la produzione di ATP.
Se, con la reazione catalizzata dalla fosfoglicerato chinasi venivano pareggiate le due molecole di ATP spese nella fase preparatoria della glicolisi, con la reazione catalizzata dalla piruvato chinasi si ha un guadagno netto di due molecole di ATP per molecola di glucosio.
Destino del piruvato e NADH
I prodotti della glicolisi sono tre: ATP, piruvato e NADH.
Il NADH deve essere riossidato a NAD+ per permettere alla glicolisi di procedere.[2]
Il NAD+, un coenzima derivante dalla vitamina niacina, è presente nella cellula in quantità limitata, ≤ 10-5M, valore ben inferiore a quello del glucosio metabolizzato in pochi minuti, e deve essere quindi continuamente rigenerato. E il passaggio finale della via glicolitica è proprio la sua rigenerazione dal NADH attraverso vie metaboliche aerobiche o anaerobiche, ognuna delle quali comporta un ulteriore metabolismo del piruvato. Queste vie permettono quindi il mantenimento del bilancio redox della cellula.
Il piruvato è una molecola assai versatile che può entrare in diverse vie metaboliche, sia anaboliche che cataboliche, a seconda del tipo di cellula, dello stato energetico della cellula e della disponibilità di ossigeno.[5]
Catabolismo del Piruvato
Con l’esclusione di alcune “variazioni” che si incontrano nel mondo dei batteri, sfruttate anche dall’industria alimentare per la produzione di vari alimenti tra cui molti formaggi, sono essenzialmente tre le vie cataboliche che possono essere imboccate dal piruvato:
la riduzione ad acido lattico, attraverso la fermentazione lattica;
la riduzione ad alcol etilico, attraverso la fermentazione alcolica;
l’ossidazione aerobica.
Questo permette alla glicolisi di procedere sia in condizioni anaerobiche che aerobiche.
E’ quindi anche possibile affermare, ampliando la visuale, che il destino catabolico dello scheletro carbonioso del glucosio è influenzato dal tipo di cellula, dal suo stato energetico e dalla disponibilità di ossigeno.
Fermentazione lattica
Negli animali, con poche eccezioni, e in molti microrganismi quando la disponibilità di ossigeno non è sufficiente a soddisfare le richieste energetiche della cellula, o se la cellula è priva di mitocondri, il piruvato prodotto dalla glicolisi viene ridotto a lattato nella reazione catalizzata dall’enzima citosolico lattato deidrogenasi (EC 1.1.1.27).
Piruvato + NADH + H+ ⇄ Lattato + NAD+
Nella reazione il NADH fornisce gli equivalenti riducenti e si ossida a NAD+. L’equilibrio complessivo della reazione favorisce fortemente la formazione del lattato, come testimoniato anche del valore del ΔG°’ pari a -6 kcal/mol (-25,1 kJ/mol).
La conversione del glucosio in acido lattico viene definita fermentazione lattica. L’equazione complessiva del processo è:
Glucosio + 2 Pi + 2 ADP + 2 H+ → 2 Lattato + 2 ATP + 2 H2O
Si noti che la fermentazione, scoperta da Louis Pasteur che la definì “la vie sans l’air”, è un processo che:
estrae energia dalla molecola del glucosio immagazzinandola in forma di ATP;
non consuma ossigeno;
non modifica la concentrazione del NAD+ o del NADH.
Riguardo ai coenzimi si noti che nell’equazione complessiva della fermentazione lattica non compaiono ne NAD+ ne NADH, sebbene entrambe cruciali nel processo. Quindi non si verifica alcuna ossidoriduzione netta. In altri termini, nel passaggio da glucosio C6H12O6, a lattato, C3H6O3, il rapporto H:C non varia.
Dal punto di vista energetico va sottolineato che la fermentazione permette di estrarre una ridotta quantità dell’energia chimica contenuta nella molecola del glucosio.
Nell’uomo molto del lattato prodotto entra nel ciclo di Cori, per essere riutilizzato nella gluconeogenesi. In definitiva si può anche affermare che la produzione di lattato sposta parte del carico metabolico dalla periferia, ad esempio dal muscolo scheletrico di un atleta impegnato in un esercizio molto inteso, come uno sprint, quando la velocità della glicolisi può quasi istantaneamente aumentare anche di 2000 volte, al fegato.
Al contrario del muscolo scheletrico che rilascia in circolo lattato, il muscolo cardiaco è in grado di assumere lattato e utilizzarlo come carburante per produrre ATP. Questo è possibile grazie alle proprietà dell’isoenzima cardiaco della lattato deidrogenasi, indicato come H4, e al suo metabolismo completamente aerobico. Quindi, parte del lattato rilasciato dal muscolo scheletrico sottoposto a un lavoro inteso sarà utilizzato dal muscolo cardiaco come carburante.
Nota: il lattato prodotto dai microorganismi durante la fermentazione lattica è responsabile sia del profumo che del gusto dei crauti, ossia dei cavoli fermentati, come anche del gusto del latte acido.
Fermentazione alcolica
In microorganismi come il lievito di birra e il lievito da panificazione, ma anche in certi tessuti vegetali, e in alcuni invertebrati e protisti, il piruvato, in condizioni ipossiche o anerobiche, può essere ridotto in due passaggi ad alcol etilico o etanolo, con liberazione di CO2.
Il primo passaggio comporta la decarbossilazione non ossidativa del piruvato a dare acetaldeide, una reazione essenzialmente irreversibile. La reazione è catalizzata dalla piruvato decarbossilasi (EC 4.1.1.1), enzima che richiede la presenza di Mg2+ e tiamina pirofosfato, un coenzima derivante dalla vitamina tiamina o vitamina B1. L’enzima è assente nei tessuti dei vertebrati e negli altri organismi che operano la fermentazione lattica.
Nel secondo passaggio si verifica la riduzione dell’acetaldeide a etanolo nella reazione catalizzata dalla alcol deidrogenasi (EC 1.1.1.1), enzima che ha un atomo di zinco legato nel sito attivo. Nella reazione il NADH fornisce gli equivalenti riducenti e si ossida a NAD+. A pH neutro, l’equilibrio della reazione è fortemente spostato verso la formazione dell’alcol etilico.
La conversione del glucosio in etanolo e CO2 viene definita fermentazione alcolica. La reazione complessiva è:
Glucosio + 2 Pi + 2 ADP + 2 H+ → 2 Etanolo + 2 CO2 + 2 ATP + 2 H2O
E, al pari della fermentazione lattica, anche nella fermentazione alcolica non si verifica alcuna ossido-riduzione netta.
La fermentazione alcolica è alla base della produzione della birra e del vino. Da notare che la CO2 prodotta dal lievito di birra è responsabile delle caratteristiche bollicine della birra, dello champagne e dello spumante, mentre quella prodotta dal lievito usato nella panificazione porta alla lievitazione dell’impasto.
Destino del piruvato e NADH in condizioni aerobiche
Nelle cellule dotate di mitocondri e in condizioni aerobiche, la situazione più comune negli organismi multicellulari e in molti unicellulari, l’ossidazione del NADH e il catabolismo del piruvato seguono vie distinte.
Il piruvato viene dapprima convertito in acetil-CoA nelle reazioni catalizzate dal complesso della piruvato deidrogenasi, uno dei complessi multienzimatici mitocondriali. Nella reazione, una decarbossilazione ossidativa, la molecola perde un atomo di carbonio in forma di CO2, e l’unità a due atomi di carbonio rimanente è legata al coenzima A a dare acetil-coenzima A o semplicemente acetil-CoA.
Piruvato + NAD+ + CoA → Acetil-CoA + CO2 + NADH + H+
Il gruppo acetilico dell’acetil-CoA è quindi completamente ossidato a CO2 attraverso le reazioni del ciclo dell’acido citrico, con produzione di ulteriore NADH, e anche di FADH2. Il complesso della piruvato deidrogenasi rappresenta quindi un ponte tra la glicolisi, che si verifica nel citosol, e il ciclo dell’acido citrico, una via metabolica mitocondriale.
Gli elettroni derivati dalle ossidazioni che si verificano durante la glicolisi sono trasportati nel mitocondrio a seguito della riduzione di intermedi metabolici citosolici. In questo modo nel citosol viene rigenerato NAD+ dal NADH, mentre l’intermedio ridotto, una volta nella matrice mitocondriale, è riossidato grazie al trasferimento dei suoi equivalenti riducenti al Complesso I della catena di trasporto degli elettroni mitocondriale. In questa sede gli elettroni passano all’ossigeno a dare H2O, trasferimento che fornisce l’energia necessaria per la sintesi dell’ATP attraverso il processo della fosforilazione ossidativa.
Ovviamente anche gli elettroni trasportati dal NADH prodotto nelle reazioni catalizzate dal complesso della piruvato deidrogenasi e nel ciclo dell’acido citrico, e quelli del FADH2, seguono un analogo destino.
Nota: il FADH2 cede i suoi equivalenti riducenti non al Complesso I ma al Complesso II.
Destino anabolico del piruvato
In condizioni anaboliche lo scheletro carbonioso del piruvato può avere destini diversi dalla completa ossidazione a CO2, o dalla conversione in lattato. Infatti, previa conversione in acetil-CoA, potrà essere utilizzato ad esempio per la sintesi degli acidi grassi, o dell’aminoacido alanina.
Glicolisi e produzione di ATP
Nella glicolisi una molecola di glucosio viene convertita in due di piruvato.
Durante la prima fase, la fase preparatoria, sono consumate due molecole di ATP nelle reazioni catalizzate dalla esochinasi e dalla PFK-1. Nella seconda fase, la fase di recupero energetico, sono prodotte 4 molecole di ATP attraverso fosforilazioni a livello del substrato nelle reazioni catalizzate dalla fosfoglicerato chinasi e dalla piruvato chinasi. Quindi si ha un guadagno netto di due molecole di ATP per molecola di glucosio metabolizzata. In più, nella reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi, per ogni molecola di glucosio sono prodotte due molecole di NADH.[5]
ΔG°’ e ΔG delle Reazioni della Glicolisi
Il ΔG°’ dell’intero processo è pari a -20,3 kcal/mol (-85 kJ/mol), valore che deriva dalla differenza tra il ΔG°’ della conversione del glucosio in due molecole di piruvato, pari a -34,9 kcal/mol (146 kJ/mol), e il ΔG°’ della formazione dell’ATP da ADP e Pi, pari a 2 x 7,3 kcal/mol = 14,6 kcal/mol (2 x 30,5 kJ/mol = 61 kJ/mol). Di seguito le due reazioni.
Glucosio + 2 NAD+ → 2 Piruvato + 2 NADH + 2 H+
2 ADP + 2 Pi → 2 ATP + 2 H2O
La somma delle due reazioni da la reazione complessiva della glicolisi.
Eliminando i termini comuni si ottiene l’equazione complessiva mostrata sopra.
Produzione di ATP in condizioni anaerobiche
In condizioni anaerobiche, a prescindere da quello che sarà il destino metabolico del piruvato, conversione in lattato, etanolo o altre molecole, non sono prodotte altre molecole di ATP a valle della glicolisi.
Quindi in queste condizioni la glicolisi permette di estrarre solamente una frazione dell’energia chimica contenuta nella molecola del glucosio, pari a 679 kcal/mol (2840 kJ/mol) rilasciate a seguito della sua ossidazione a CO2 e H2O. Infatti, la conversione del glucosio in due molecole di piruvato porta al rilascio di 34,9 kcal/mol, il che significa che solamente il 5%, [(34,9/679) x 100], dell’energia chimica contenuta nella molecola del glucosio è rilascia a seguito della sua conversione in piruvato. Quindi il piruvato contiene ancora la maggior parte dell’energia chimica del glucosio.
Analogamente, neppure i 4 elettroni, in forma di ioni idruro, trasportati dalle due molecole di NADH potranno essere utilizzati per la produzione di ATP.
Considerando la fermentazione lattica, il ΔG°’ associato alla conversione di una molecola di glucosio in due di lattosio è pari a -43,9 kcal/mol (-183,6 kJ/mol) e la percentuale dell’energia libera rilasciata e immagazzinata in forma di ATP sarà pari a: (14,6/43,9) x 100 = 33,2%, mentre se si considera la sola glicolisi la percentuale è del 41,8%. Da notare che nelle condizioni intracellulari la quantità di energia libera necessaria per la sintesi dell’ATP da ADP e Pi è molto più elevata di quella in condizioni standard, per cui la percentuale dell’energia libera disponibile immagazzinata è maggiore, pari a circa il 50% del totale.
Produzione di ATP in condizioni aerobiche
In condizioni aerobiche, nelle cellule dotate di mitocondri, la quantità di energia chimica che può essere estratta dalla molecola del glucosio e immagazzinata in forma di ATP è molto maggiore rispetto a quanto accade in assenza di ossigeno. Se si considerano solo le due molecole di NADH prodotte durante la glicolisi, il trasferimento dei loro 4 equivalenti riducenti lungo la catena di trasporto degli elettroni mitocondriale permette la produzione di 2-3 molecole di ATP per coppia di elettroni attraverso la fosforilazione ossidativa. In questo caso si avrà una produzione netta di ATP compresa tra 6 e 8 molecole per ogni molecola di glucosio ossidata a due di piruvato, dalla glicolisi e 4-6 dalla fosforilazione ossidativa.
Nota: la quantità complessiva di ATP prodotta dagli equivalenti riducenti del NADH dipende dal sistema attraverso cui gli equivalenti riducenti del coenzima ridotto entrano nel mitocondrio.
Se invece si analizza l’azione concertata della glicolisi, del complesso della piruvato deidrogenasi, del ciclo dell’acido citrico, della catena di trasporto degli elettroni mitocondriale e della fosforilazione ossidativa, viene estratta e immagazzinata in forma di ATP molta altra dell’energia chimica disponibile presente nel monosaccaride. In questo caso, secondo quanto riportato dal Lehninger sono prodotti 30-32 ATP/molecola di glucosio, sebbene recenti stime suggeriscano una produzione netta pari a 29,85 ATP/molecola di glucosio, o 29,38 ATP/molecola di glucosio se anche l’ATP derivato dal GTP, prodotto dal ciclo dell’acido citrico, viene esportato. Considerando entrambe le stime, la produzione di ATP è circa 15 volte maggiore rispetto a quanto accade in assenza di ossigeno.
Vie di alimentazione
Oltre al glucosio, altri carboidrati, sia semplici che complessi, possono essere catabolizzati attraverso la glicolisi, previa conversione enzimatica in uno degli intermedi della via metabolica stessa.[2][7]
Tra i più importanti si ritrovano:
due polisaccaridi di deposito, il glicogeno e l’amido;
i monosaccaridi galattosio e fruttosio; un terzo monosaccaride è il mannosio, meno comune rispetto ai precedenti.
Vie di Alimentazione della Glicolisi
A livello intestinale, amido e disaccaridi assunti con l’alimentazione incontrano gli enzimi responsabili della digestione dei carboidrati. Segue l’assorbimento dei monosaccaridi liberati e derivanti dall’alimentazione. Una volta nel circolo venoso i monosaccaridi raggiungono, attraverso la vena porta, il fegato, ed è principalmente in questa sede che sono metabolizzati.
Glicogeno e amido
Per quanto riguarda il catabolismo fosforolitico dell’amido e del glicogeno endogeno si rimanda ai rispettivi articoli.
Fruttosio
In condizioni fisiologiche, il fegato rimuove dal circolo ematico molto del fruttosio ingerito, prima che lo stesso possa raggiungere i tessuti extraepatici.
La via metabolica epatica che porta alla conversione del monosaccaride in intermedi della glicolisi si compone di alcuni passaggi.
Nel primo il fruttosio viene fosforilato a fruttosio-1-fosfato nella reazione catalizzata dalla fruttochinasi (EC 2.7.1.4), con consumo di una molecola di ATP.
Fruttosio + ATP → Fruttosio-1-fosfato + ADP + H+
La reazione richiede la presenza di ioni magnesio.
Come per il glucosio, anche per il fruttosio la fosforilazione ha come effetto quello di intrappolare la molecola all’interno della cellula.
Nel secondo passaggio il fruttosio-1-fosfato viene scisso a diidrossiacetone fosfato e gliceraldeide, nella reazione catalizzata dalla fruttosio-1-fosfato aldolasi.
Il diidrossiacetone fosfato è un intermedio della glicolisi e prosegue lungo la via previa conversione in gliceraldeide-3-fosfato. Invece, la gliceraldeide, che non è un intermedio della glicolisi, viene fosforilata a gliceraldeide-3-fosfato nella reazione catalizzata dalla trioso chinasi (EC 2.7.1.28), con consumo di una seconda molecola di ATP.
Gliceraldeide + ATP → Gliceraldeide-3-fosfato + ADP + H+
La reazione richiede la presenza di ioni Mg2+.
Quindi nell’epatocita, una molecola di fruttosio viene convertita in due di gliceraldeide-3-fosfato, con consumo di due molecole di ATP, come per il glucosio.
In sedi extraepatiche come il muscolo scheletrico, il rene o il tessuto adiposo non è presente la fruttochinasi, e il fruttosio entra nella via glicolitica in forma di fruttosio-6-fosfato. Infatti tra i substrati della esochinasi c’è anche il fruttosio, che viene fosforilato in posizione C-6.
Fruttosio + ATP → Fruttosio-6-fosfato + ADP + H+
L’affinità dell’enzima per il fruttosio è però circa 20 volte minore rispetto a quella per il glucosio, per cui nell’epatocita, dove il glucosio è molto più abbondante del fruttosio, o nel muscolo scheletrico in condizioni di scarsa disponibilità di ossigeno, quando cioè il glucosio diviene il carburante preferito, si forma poco fruttosio-6-fosfato. Al contrario, nel tessuto adiposo il fruttosio è più abbondante del glucosio per cui la sua fosforilazione a opera della esochinasi non è inibita competitivamente in modo significativo dal glucosio. In questo tessuto quindi la sintesi del fruttosio-6-fosfato rappresenta il modo principale per incanalare il monosaccaride nella via glicolitica.
Ai fini degli effetti metabolici del fruttosio è importante sottolineare che nel fegato il monosaccaride, essendo fosforilato in posizione C-1, entra nella glicolisi a livello dei trioso fosfati, dunque a valle della reazione catalizzata dalla PFK-1, enzima che gioca un ruolo chiave nella regolazione del flusso del carbonio attraverso la glicolisi stessa. Al contrario, quando il monosaccaride viene fosforilato sul C-6, entra nella via glicolitica a monte della reazione catalizzata dalla PFK-1.
Sorbitolo
Il fruttosio rappresenta il punto d’ingresso nella via glicolitica anche per il sorbitolo. La molecola è presente in molti frutti e vegetali e utilizzata anche come uno dei dolcificanti e stabilizzanti. Nel fegato è convertita in fruttosio nella reazione catalizzata dall’enzima citosolico sorbitolo deidrogenasi (EC 1.1.99.21), enzima che fa parte anche della via dei polioli, per la conversione del glucosio a fruttosio.
Sorbitolo + NAD+ → Fruttosio + NADH + H+
La reazione richiede la presenza di ioni Zn2+.
Galattosio
Il galattosio, che per la maggior parte deriva dalla digestione intestinale del lattosio, raggiunto il fegato viene convertito, attraverso la via di Leloir, in glucosio-1-fosfato. Il destino del glucosio-1-fosfato dipende dallo stato energetico della cellula. In condizioni che favoriscono il deposito di glucosio, la molecola può essere incanalata nella via di sintesi del glicogeno. In condizioni che favoriscono l’utilizzo del glucosio per la produzione di energia, il glucosio-1-fosfato viene isomerizzato in glucosio-6-fosfato nella reazione reversibile catalizzata dalla fosfoglucomutasi (EC 5.4.2.2).
Glucosio-1-fosfato ⇄ Glucosio-6-fosfato
A sua volta il glucosio-6-fosfato prodotto potrà essere incanalato nella via glicolitica per essere utilizzato per la produzione di energia, o essere defosforilato a glucosio nella reazione catalizzata dal complesso proteico della glucosio-6-fosfatasi ed essere quindi rilasciato nel circolo ematico.
Mannosio
Il mannosio è presente in vari polisaccaridi, glicolipidi e glicoproteine della dieta. Una volta liberato da queste fonti, viene assorbito, e raggiunto il fegato è fosforilato in posizione C-6 a dare mannosio-6-fosfato, nella reazione catalizzata dalla esochinasi.
Mannosio + ATP → Mannosio-6-fosfato + ADP + H+
Il mannosio-6-fosfato è quindi isomerizzato a fruttosio-6-fosfato nella reazione catalizzata dalla mannosio-6-fosfato isomerasi (EC 5.3.1.8.).
Mannosio-6-fosfato ⇄ Fruttosio-6-fosfato
Regolazione della glicolisi
Il flusso di carbonio attraverso la glicolisi viene regolato in base alle condizioni metaboliche all’interno e all’esterno della cellula, rispondendo essenzialmente a due bisogni:
la produzione di ATP;
la produzione di precursori per molte vie biosintetiche.
Inoltre, nel fegato, per evitare uno spreco di energia, glicolisi e gluconeogenesi sono finemente regolate in modo che quando una via procede l’altra rallenta. Nel corso dell’evoluzione ciò è stato ottenuto selezionando enzimi differenti per catalizzare le reazioni essenzialmente irreversibili delle due vie, enzimi la cui attività è regolata separatamente. Se infatti queste reazioni procedessero simultaneamente a elevata velocità creerebbero un ciclo futile o ciclo del substrato.[7] Una regolazione così fine non potrebbe essere ottenuta se uno stesso enzima catalizzasse la reazione nelle due direzioni.
Il controllo del flusso di carbonio attraverso la glicolisi coinvolge principalmente le reazioni catalizzate dagli enzimi esochinasi, PFK-1 e piruvato chinasi, la cui attività è regolata mediante:
meccanismi allosterici, che si svolgono in un arco temporale di millisecondi e sono istantaneamente reversibili;
modificazioni covalenti, ossia fosforilazioni e defosforilazioni, che si svolgono in secondi;
modificazioni nella concentrazione degli enzimi coinvolti, conseguenti a modifiche nella loro velocità di sintesi/degradazione, che avvengono in ore.
Nota: per la gluconeogenesi i principali punti di regolazione sono le reazioni catalizzate dagli enzimi piruvato carbossilasi (EC 6.4.1.1) e fruttosio-1,6-bisfosfatasi (EC 3.1.3.11).
Esochinasi
Nell’uomo la esochinasi è presente con quattro forme isoenzimatiche tessuto specifiche, designate da I a IV, e codificate da altrettanti geni differenti.[2][5]
La esochinasi I è l’isoenzima prevalente nel cervello, mentre nel muscolo scheletrico si ritrova sia l’esochinasi I, che costituisce il 70-75% del totale, che la esochinasi II, che rappresenta il restante 25-30%.
L’esochinasi IV, detta anche glucochinasi (EC 2.7.1.2) è presente prevalentemente negli epatociti e nelle cellule β del pancreas, dove è il principale isoenzima. Nel fegato, con la glucosio-6-fosfatasi, catalizza il ciclo del substrato tra glucosio e glucosio-6-fosfato.
La glucochinasi differisce dalle altre isoforme della esochinasi per quanto riguarda la cinetica e le proprietà regolatorie.[1]
Nota: gli isoenzimi o isozimi sono proteine differenti che catalizzano la stessa reazione, e che in genere si differenziano per le proprietà cinetiche e regolatorie, distribuzione subcellulare, o per i cofattori utilizzati. Possono essere presenti contemporaneamente in una stessa specie, tessuto o anche cellula.
Proprietà cinetiche delle esochinasi
Le esochinasi I, II e III hanno proprietà cinetiche simili. La esochinasi I e la II hanno una Km per il glucosio, ossia la concentrazione del glucosio alla quale l’enzima è per metà saturato, rispettivamente pari a 0,03 mM e 0,1 mM. Pertanto questi due isoenzimi lavorano in modo molto efficiente ai normali valori di glicemia, che sono pari a circa 4-5 mM.[7]
Invece la glucochinasi ha una Km per il glucosio assai più alta, pari a circa 10 mM; questo significa che l’attività dell’enzima diviene importante solo quando i valori della glicemia sono alti, come dopo un pasto ricco di carboidrati ad alto indice glicemico.
Regolazione delle esochinasi I-III
Le esochinasi I-III sono inibite allostericamente dal glucosio-6-fosfato, il prodotto della loro reazione. Questo assicura che il glucosio-6-fosfato non si accumuli nella cellula quando non è richiesto ulteriore glucosio per la produzione di energia, la sintesi del glicogeno, la via del pentoso fosfato, o come fonte di intermedi per la sintesi di altre molecole, e al contempo permette di ridurre l’assunzione dal circolo di altro glucosio che rimane quindi disponibile per altri organi e tessuti. Ad esempio, quando la PFK-1 è inibita, si accumula fruttosio-6-fosfato e, grazie alla reazione catalizzata dalla fosfoglucosio isomerasi, glucosio-6-fosfato. Dunque, l’inibizione della PFK-1 porta alla inibizione delle esochinasi I-III.
Nel muscolo scheletrico l’attività della esochinasi I e II è coordinata con quella di GLUT4, un trasportatore del glucosio con una bassa Km per il monosaccaride (5 mM), la cui traslocazione sulla membrana plasmatica è indotta sia dall’insulina che dall’attività fisica. L’azione combinata delle esochinasi e di GLUT4 mantiene un equilibrio tra l’ingresso del glucosio nella cellula e la sua fosforilazione. Considerando che la concentrazione ematica del monosaccaride è compresa tra 4 e 5 mmol/L, il suo ingresso nel miocita per mezzo di GLUT4 può portare la sua concentrazione a valori sufficientemente elevati da saturare o quasi l’enzima, che dunque può lavora vicino o addirittura alla sua Vmax.
Regolazione della glucochinasi epatica
La glucochinasi differisce per almeno tre aspetti dalle esochinasi I-III, ed è particolarmente idonea per il ruolo che il fegato svolge nella regolazione della glicemia.[1] Perché?
Come detto in precedenza, la glucochinasi ha una Km per il glucosio pari a circa 10 mM, molto più elevata rispetto alla Km per il glucosio delle esochinasi I-III, e più alta anche del valore della glicemia a digiuno, pari a circa 4-5 mM.[7] Nel fegato, dove rappresenta l’isoenzima prevalente, il suo ruolo è quello di fornire glucosio-6-fosfato per la sintesi del glicogeno e degli acidi grassi. L’enzima lavora in modo coordinato con il trasportatore del glucosio GLUT2, il principale carrier per il glucosio nell’epatocita, la cui Km per lo zucchero è circa 10 mM.
Quindi GLUT2 è molto attivo quando la glicemia è elevata, equilibrando rapidamente la concentrazione del glucosio nel citosol dell’epatocita con quella presente nel sangue. In queste condizioni la glucochinasi è attiva e catalizza la conversione del glucosio in glucosio-6-fosfato, e, grazie alla elevata Km per il glucosio la sua attività continua ad aumentare anche quando la concentrazione intracellulare dello zucchero raggiunge o supera le 10 mM. Quindi la velocità con cui il monosaccaride entra nella cellula e di seguito viene fosforilato è determinata dal valore della stessa glicemia.
Quando invece la disponibilità del glucosio è scarsa, la sua concentrazione nel citosol dell’epatocita è altrettanto bassa, ben più bassa del valore della Km della glucochinasi. In questa condizione il glucosio prodotto attraverso la gluconeogenesi e/o la glicogenolisi non viene fosforilato e può lasciare la cellula. Una situazione simile si verifica anche nelle cellule β del pancreas, dove il sistema GLUT2/glucochinasi fa si che la concentrazione intracellulare del glucosio-6-fosfato equipari quella del glucosio nel sangue, permettendo alla cellula di rilevare e rispondere a elevate glicemie.
A differenza delle esochinasi I-III, la glucochinasi non è inibita dal glucosio-6-fosfato, per cui continua a catalizzarne la sintesi anche quando questi si accumula.
La glucochinasi viene inibita a seguito del legame reversibile a una specifica proteina regolatrice detta anche GKRP, acronimo dell’inglese glucokinase regulatory protein.[4] Il meccanismo con cui la proteina regolatrice agisce implica l’ancoraggio della glucochinasi all’interno del nucleo, dove rimane separata dagli altri enzimi della glicolisi.
Regolazione della Glucochinasi Epatica
Il legame tra glucochinasi e GKRP è molto più forte in presenza del fruttosio-6-fosfato mentre è indebolito dal glucosio e dal fruttosio-1-fosfato.
In assenza di glucosio la glucochinasi si trova nella sua conformazione “super-aperta” che è dotata di bassa attività. L’aumento nei livelli citosolici di glucosio determina una transizione concentrazione-dipendente dell’enzima verso la sua conformazione chiusa, la conformazione attiva che non è accessibile alla GKRP. Ne consegue che la glucochinasi è attiva e non più inibita.
Da notare che il fruttosio-1-fosfato è presente nell’epatocita solamente quando viene metabolizzato il fruttosio, che quindi, quando disponibile fa venire meno l’inibizione della glucochinasi da parte di GKRP.
Esempio
Quando dopo un pasto ricco di carboidrati la glicemia sale, il glucosio tramite il GLUT2 entra nella cellula epatica, e quindi attraverso i pori nucleari nel suo nucleo dove determina la transizione della glucochinasi verso la sua conformazione chiusa, attiva e non accessibile a GKRP, permettendo così all’enzima di diffondere nel citosol dove potrà fosforilare il glucosio.
Viceversa, quando il glucosio è scarso, come nel digiuno quando la glicemia può scendere sotto il valore di 4 mM, la concentrazione del glucosio nell’epatocita è bassa, e il fruttosio-6-fosfato associandosi a GKRP permette a quest’ultima di legarsi in modo più saldo alla glucochinasi. Da ciò ne risulta una inibizione molto efficace dell’enzima. Questo concorre a evitare che il fegato, in condizioni di bassa glicemia, competa con gli altri organi, in primis il cervello, per il poco glucosio disponibile.
Nella cellula il fruttosio-6-fosfato è presente in equilibrio con il glucosio-6-fosfato grazie alla reversibilità della reazione catalizzata dalla fosfoglucosio isomerasi. Tramite la sua associazione con GKRP, il fruttosio-6-fosfato opera quindi un feedback negativo segnalando alla cellula che non è necessario produrre altro glucosio-6-fosfato, ossia che l’attività della glucochinasi può ridursi per prevenire l’accumulo di intermedi.
Riassumendo si può dire che quando i valori della glicemia sono normali, il glucosio viene fosforilato prevalentemente dalle esochinasi I-III, mentre quando la glicemia è elevata l’esoso è fosforilato dalla glucochinasi.
Regolazione della fosfofruttochinasi-1
La fosfofruttochinasi-1 è il principale punto di controllo del flusso di carbonio attraverso la glicolisi.[7]
L’enzima, oltre ai siti di legame per i substrati, presenta molti altri siti su cui vanno a legarsi effettori allosterici sia inibitori che attivatori. Tra gli effettori allosterici negativi si ha l’ATP, che è anche un substrato dell’enzima e un prodotto finale della glicolisi, il citrato e gli ioni idrogeno.
Tra gli effettori allosterici positivi si ritrovano l’AMP, il Pi e il fruttosio-2,6-bisfosfato.[8][9]
Nota: la PFK-1 presenta due siti di legame distinti per l’ATP, uno nel sito attivo e dotato di grande affinità per la molecola, e l’altro, regolatorio, a bassa affinità.
Che cosa segnalano i diversi effettori allosterici?
ATP, AMP e Pi segnalano lo stato energetico della cellula. L’attività della fosfofruttochinasi-1 aumenta quando la cellula necessita di energia, ossia quando c’è necessità di ATP, mentre si riduce quando lo stato energetico della cellula è alto, ossia quando la cellula è ricca di ATP.
In che modo?
Quando la concentrazione di ATP è elevata, quando cioè il nucleotide viene prodotto a una velocità superiore a quella con cui è consumato, lo stesso legandosi allo specifico sito allosterico va a inibire la fosfofruttochinasi-1, riducendone l’affinità per il fruttosio-6-fosfato. Dal punto di vista cinetico l’aumento della concentrazione dell’ATP determina una cambiamento della rapporto tra la velocità di reazione e la concentrazione del fruttosio-6-fosfato che diviene sigmoide da iperbolico, e quindi si ha un aumento della Km dell’enzima per il fruttosio-6-fosfato stesso.
Tuttavia nella maggior parte delle condizioni cellulari la concentrazione dell’ATP non varia più di tanto. Ad esempio, nel muscolo durante un esercizio vigoroso si possono avere riduzioni della concentrazione di ATP di circa un 10% rispetto alla situazione di riposo mentre la velocità della glicolisi varia molto di più rispetto a quanto ci si aspetterebbe da tale riduzione. Quando la velocità di produzione dell’ATP è inferiore a quella con cui viene consumato, si verifica l’aumento della concentrazione dell’AMP e dell’ADP, e in particolare dell’AMP, grazie all’azione dell’enzima adenilato chinasi (EC 2.7.4.3 ), secondo la reazione di seguito descritta.
ADP + ADP ⇄ ATP + AMP
La costante di equilibrio Keq della reazione è:
Keq = [ATP][AMP]/[ADP]2= 0,44
Nelle normali condizioni cellulari la concentrazione dell’ADP e dell’AMP corrispondono rispettivamente a circa il 10% e a meno dell’1% di quella dell’ATP. Pertanto, considerando che il pool dell’adenilato è costante nel breve periodo, anche una piccola variazione nella concentrazione dell’ATP porterà, grazie all’attività della adenilato chinasi, a una variazione relativa molto maggiore della concentrazione dell’AMP. A sua volta l’AMP agisce rimuovendo l’inibizione dovuta all’ATP.
Quindi, l’attività della fosfofruttochinasi-1 dipende dallo stato energetico della cellula:
quando l’ATP è abbondante l’attività dell’enzima si riduce;
quando i livelli di AMP aumentano l’attività dell’enzima aumenta.
Perché l’ADP non è un effettore allosterico positivo della fosfofruttochinasi-1? Ci sono due ragioni.
Quando la carica energetica della cellula si riduce, l’ADP è utilizzato per riformare l’ATP, nella reazione catalizzata dalla adenilato chinasi. Inoltre, come detto in precedenza, una piccola riduzione nei livelli di ATP porta una più elevata variazione percentuale nei livelli dell’ADP e soprattutto dell’AMP.
Gli ioni idrogeno inibiscono la fosfofruttochinasi-1. Tale inibizione previene, controllando la velocità della glicolisi, l’eccessivo accumulo di lattato e la conseguente caduta del pH ematico.
Il citrato è un inibitore allosterico della fosfofruttochinasi-1 che agisce aumentando l’effetto inibitorio dell’ATP. Il citrato è il prodotto del primo passaggio del ciclo dell’acido citrico, una via metabolica che fornisce intermedi metabolici per le vie biosintetiche e indirizza gli elettroni verso la catena di trasporto degli elettroni mitocondriale per la sintesi dell’ATP via fosforilazione ossidativa. Elevati livelli citosolici di citrato indicano che nel mitocondrio si sta verificando la produzione di un eccesso di precursori e che le richieste energetiche della cellula sono state soddisfatte, ossia, il ciclo dell’acido citrico ha raggiunto la saturazione; pertanto la glicolisi, che rifornisce il ciclo in condizioni aerobiche, può rallentare, risparmiando glucosio.
Quindi, va sottolineato che la PFK-1 accoppia la glicolisi e il ciclo dell’acido citrico.
Nel fegato il punto centrale per la regolazione sia della glicolisi che della gluconeogenesi è rappresentato dal ciclo del substrato tra fruttosio-6-fosfato e fruttosio-1,6-bisfosfato, catalizzato dagli enzimi fosfofruttochinasi-1 e fruttosio-1,6-bisfosfatasi.
Il fegato gioca un ruolo cruciale nel mantenimento della glicemia entro valori normali.
Se la glicemia scende, il glucagone a livello epatico stimola la sintesi, via glicogenolisi e gluconeogenesi, di glucosio, e al contempo segnala all’organo di non consumare l’esoso per soddisfare i propri fabbisogni.
Quando invece la glicemia è elevata, l’insulina induce il fegato a utilizzare il glucosio per produrre energia, sintetizzare glicogeno e trigliceridi. In quest’ottica, la regolazione della glicolisi e della gluconeogenesi è mediata dal fruttosio-2,6-bisfosfato, una molecola che permette all’organo di svolgere un ruolo di primo piano nella regolazione della glicemia segnalando il rapporto tra insulina e glucagone.
A seguito del legame allo specifico sito allosterico sulla fosfofruttochinasi-1, il fruttosio-2,6-bisfosfato aumenta l’affinità dell’enzima per il fruttosio-6-fosfato, il suo substrato, mentre diminuisce quella per gli inibitori allosterici citrato e ATP. Ed è notevole sottolineare che in presenza di concentrazioni fisiologiche dei substrati e degli effettori allosterici sia positivi che negativi l’enzima, in assenza di fruttosio-2,6-bisfosfato, è praticamente inattivo.
Viceversa, il legame del fruttosio-2,6-bisfosfato alla fruttosio-1,6-bisfosfatasi ne comporta l’inibizione, anche in assenza di AMP, un altro inibitore allosterico dell’enzima.
Grazie a queste azioni la molecola aumenta il flusso netto di glucosio attraverso la glicolisi.
Altro metabolita importante per il controllo del flusso del carbonio attraverso la glicolisi e la gluconeogenesi è lo xilulosio-5-fosfato.[3]. Si tratta di un intermedio della via del pentoso fosfato, la cui concentrazione nell’epatocita aumenta a seguito dell’ingestione di un pasto ricco di carboidrati. La molecola, attivando la protein fosfatasi 2A porta infine a un aumento della concentrazione del fruttosio-2,6-bisfosfato e quindi a un incremento del flusso del carbonio attraverso la glicolisi e alla riduzione di quello attraverso la gluconeogenesi.
Regolazione della piruvato chinasi
Un ulteriore punto di regolazione del flusso di carbonio attraverso la glicolisi e la gluconeogenesi è rappresentato dal ciclo del substrato tra il fosfoenolpiruvato e il piruvato. Suddetto ciclo è catalizzato dalla piruvato chinasi, per la glicolisi, e dall’azione combinata della piruvato carbossilasi e della fosfoenolpiruvato carbossichinasi (EC 4.1.1.32) per la gluconeogenesi.[5][7]
Tutte le isoforme della piruvato chinasi sono allostericamente inibite da elevate concentrazioni di ATP, acetil-CoA e acidi grassi a catena lunga, segnali che la cellula si trova in uno stato energetico ottimale. Anche l’alanina, che può essere sintetizzata dal piruvato attraverso una reazione di transaminazione, è un inibitore allosterico dell’enzima. L’accumulo di alanina segnala l’abbondanza di precursori per le vie biosintetiche.
Regolazione della Piruvato Chinasi Epatica
Di contro la piruvato chinasi è allostericamente attivata dal fruttosio-1,6-bisfosfato, il prodotto della prima reazione esclusiva della glicolisi. Questo permette all’enzima di tenere il passo con il flusso di intermedi in arrivo. E va sottolineato il fatto che, in una situazione di concentrazioni fisiologiche di substrato, ossia il fosfoenolpiruvato, e degli inibitori ATP e alanina, la piruvato chinasi sarebbe completamente inibita senza l’effetto stimolatorio del fruttosio-1,6-bisfosfato.
L’isoenzima epatico, ma non quello muscolare, è soggetto anche a regolazione attraverso fosforilazione a opera della:
protein chinasi A, attivata a seguito del legame del glucagone allo specifico recettore o dell’epinefrina ai recettori β-adrenergici;
protein chinasi calcio/calmodulina dipendente, attivata dal legame dell’epinefrina ai recettori α1-adrenergici.
La fosforilazione dell’enzima ne riduce l’attività a seguito di un aumento della sua Km per il fosfoenolpiruvato, e rallenta la glicolisi.
Ad esempio, a seguito di una riduzione della glicemia, la fosforilazione indotta dal glucagone riduce l’attività dell’enzima. Nella forma fosforilata l’enzima è anche meno facilmente stimolato dal fruttosio-1,6-bisfosfato ma più facilmente inibito dall’alanina e dall’ATP.
Di contro, l’enzima in forma defosforilata è più sensibile al fruttosio-1,6-bisfosfato, e meno agli inibitori allosterici ATP e alanina. In questo modo quando la glicemia è bassa, il fegato rallenta l’ossidazione del glucosio a scopi energetici, e lo zucchero rimane quindi disponibile per altri tessuti e organi, come il cervello. Va comunque notato che la piruvato chinasi non subisce la fosforilazione indotta dal glucagone in presenza del fruttosio 1,6-bisfosfato.
Viceversa, un aumento nel rapporto insulina/glucagone porta infine alla defosforilazione dell’enzima e quindi alla sua attivazione. Nella forma defosforilata l’enzima è meno sensibile agli inibitori allosterici alanina e ATP, ma più sensibile al suo attivatore allosterico, il fruttosio-1,6-bisfosfato.
Bibliografia
ab de la Iglesia N., Mukhtar M., Seoane J., Guinovart J.J., & Agius L. The role of the regulatory protein of glucokinase in the glucose sensory mechanism of the hepatocyte. J Biol Chem 2000;275(14):10597-10603. doi: 10.1074/jbc.275.14.10597
^ Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003;100:5107-5112. doi:10.1073/pnas.0730817100
^ Kaminski M.T., Schultz J., Waterstradt R., Tiedge M., Lenzen S., Baltrusch S. Glucose-induced dissociation of glucokinase from its regulatory protein in the nucleus of hepatocytes prior to nuclear export. BBA – Molecular Cell Research 2014;1843(3):554-564. doi:10.1016/j.bbamcr.2013.12.002
^ abcdefghi Nelson D.L., M. M. Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ Oslund R.C., Su X., Haugbro M., Kee J-M., Esposito M., David Y., Wang B., Ge E., Perlman D.H., Kang Y., Muir T.W., & Rabinowitz J.D. Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat Chem Biol 2017;13:1081-1087. doi:10.1038/nchembio.2453
^ abcdefg Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
^ Van Schaftingen E., and Hers H-G. Inhibition of fructose-1,6-bisphosphatase by fructose-2,6-bisphosphate. Proc Natl Acad Sci USA 1981;78(5):2861-2863. doi:10.1073/pnas.78.5.2861
^ Van Schaftingen E., Jett M-F., Hue L., and Hers H-G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA 1981;78(6):3483-3486. doi:10.1073/pnas.78.6.3483
La gluconeogenesi è la via metabolica che permette, anche agli organismi non fotosintetizzanti, di produrre glucosio a partire dal piruvato, la base coniugata dell’acido piruvico, e da altri precursori non glucidici.[1][6]
E’ presente in tutti gli animali, piante, funghi e microorganismi, con essenzialmente le stesse reazioni, che portano, da due molecole di piruvato, alla sintesi di una molecola di glucosio. Dunque è essenzialmente l’inverso della glicolisi, che invece procede dal glucosio fino al piruvato, e con essa condivide molti enzimi.[5]
Gluconeogenesi e Glicolisi
La glicogenolisi è distinta dalla gluconeogenesi, non corrispondendo a una sintesi de novo del monosaccaride, come si può facilmente evidenziare osservando la sua reazione complessiva:
Glicogeno o (glucosio)n → n molecole di glucosio
La discussione successiva verterà sulla gluconeogenesi che avviene negli animali superiori, e in particolare nel fegato dei mammiferi.
La gluconeogenesi è una via metabolica di grande importanza per almeno due motivi.
Assicura il mantenimento di una adeguata concentrazione ematica di glucosio quando le riserve epatiche di glicogeno sono prossime all’esaurimento e lo zucchero non viene assunto con l’alimentazione.[6]
Il mantenimento della glicemia entro il range di normalità, 3,3-5,5 mmol/L (60 e i 99 mg/dL), è essenziale in quanto molte cellule e tessuti dipendono largamente o totalmente dal glucosio per soddisfare le loro richieste di ATP; tra questi i globuli rossi, i neuroni, il muscolo scheletrico quando lavora in anaerobiosi, la midollare del rene, i testicoli, la lente e la cornea dell’occhio, e i tessuti embrionali. Considerando ad esempio il cervello, il suo fabbisogno giornaliero di glucosio è di circa 120 g, una quantità pari a:
oltre il 50% delle riserve corporee totali del monosaccaride, circa 210 g, di cui 190 g immagazzinato come glicogeno muscolare ed epatico, e 20 g in forma libera nei fluidi corporei;
circa il 75% del fabbisogno giornaliero di glucosio dell’intero organismo, quindi sui 160 g.[1]
Nel digiuno breve, come nell’intervallo tra i pasti o durante la notte, la glicemia è mantenuta entro il range di normalità grazie alla glicogenolisi epatica e al rilascio di acidi grassi dal tessuto adiposo e corpi chetonici dal fegato. Acidi grassi e corpi chetonici, utilizzati di preferenza dal muscolo scheletrico, consentono un risparmio di glucosio che sarà disponibile per le cellule e i tessuti che da esso dipendono. Tuttavia, dopo circa 18 ore di digiuno, le riserve di glicogeno sono prossime all’esaurimento, riserve che possono divenire insufficienti anche durante l’attività fisica intensa e prolungata. Ed è a questo punto che, se non sono assunti carboidrati con l’alimentazione, la gluconeogenesi diviene essenziale.
E, a sottolineare ulteriormente l’importanza della sintesi de novo del glucosio il fatto che se i valori della glicemia scendono al di sotto di 2 mmol/L si verifica la perdita di coscienza.
L’eventuale escrezione del piruvato comporterebbe la perdita della possibilità di produrre ATP attraverso la sua ossidazione aerobica, ossia più di 10 molecole di ATP per ogni piruvato ossidato.
Dove avviene?
Negli animali superiori la gluconeogenesi avviene nel fegato, nella corticale del rene e nelle cellule epiteliali dell’intestino tenue, gli enterociti.[5]
Quantitativamente il fegato rappresenta la sede principale, producendo circa il 90% di tutto il glucosio sintetizzato, seguito dalla corticale del rene, con circa il 10%.[1] La maggiore capacità di sintesi del fegato rispetto alla corticale del rene è dovuta solo alle sue maggiori dimensioni; se infatti si considera il rapporto peso/sintesi, la corticale del rene produce più glucosio del fegato.
A livello della corticale del rene, le cellule che portano a termine la gluconeogenesi sono quelle del tubulo prossimale, la porzione del nefrone successiva al glomerulo. Molto del glucosio prodotto nel rene viene utilizzato dalla midollare del rene stesso, mentre l’azione dell’organo sul mantenimento della omeostasi glicemica diviene più importante durante il digiuno prolungato e nell’insufficienza epatica. Va tuttavia sottolineato che il rene, essendo privo di depositi di glicogeno, può contribuire alla regolazione della glicemia solo attraverso la gluconeogenesi e non anche attraverso la glicogenolisi, come invece può fare il fegato.
Parte della via gluconeogenetica può verificarsi anche nel muscolo scheletrico e cardiaco e nel cervello, anche se a velocità estremamente ridotta. Nell’adulto il muscolo ha circa 18 volte la massa del fegato, per cui la sua sintesi di glucosio potrebbe avere una qualche importanza quantitativa.
Tuttavia in questi tessuti la sintesi de novo del glucosio non porta alla sua liberazione in circolo, essendo assente la glucosio-6-fosfatasi (EC 3.1.3.9), l’enzima che catalizza l’ultima tappa della via.[6] Quindi, l’eventuale produzione di glucosio-6-fosfato, compreso quello derivante dalla glicogenolisi, non contribuirà al mantenimento della glicemia ma aiuterà solo a ripristinare le scorte del glicogeno, per la verità nel cervello piccole e limitate per lo più agli astrociti. L’unico contributo diretto al mantenimento della glicemia operato da questi tessuti, e in particolare dal muscolo scheletrico, vista la sua grande massa, deriva dalla piccola quota di glucosio rilasciata dall’enzima deramificante (EC 3.2.1.33) della glicogenolisi.
Per quello che riguarda la localizzazione cellulare, la maggior parte delle reazioni della gluconeogenesi avvengono nel citosol, alcune nel mitocondrio, e la tappa finale all’interno delle cisterne del reticolo endoplasmatico.
Tappe irreversibili
Come detto, glicolisi e gluconeogenesi sono essenzialmente una l’inverso dell’altra. E, delle dieci reazioni che costituiscono la gluconeogenesi, ben 7 sono in comune con la glicolisi. Si tratta di reazioni caratterizzate da un ΔG prossimo allo zero, per cui facilmente reversibili. Ma nelle normali condizioni cellulari, il ΔG complessivo della glicolisi è pari a circa -63 kJ/mole (-15 kcal/mole) e quello della gluconeogenesi a circa -16 kJ/mole (-3,83 kcal/mole), ossia si tratta di processi irreversibili.[5]
Nel caso della glicolisi l’irreversibilità è conseguenza di tre reazioni fortemente esoergoniche, che non potranno essere utilizzate nella gluconeogenesi, e di seguito elencate.
La fosforilazione del glucosio a glucosio-6 fosfato, catalizzata dalla esochinasi (EC 2.7.1.1) o dalla glucochinasi (EC 2.7.1.2).
ΔG = -33,4 kJ/mole (-8 kcal/mole)
ΔG°’ = -16,7 kJ/mole (-4 kcal/mole)
La fosforilazione del fruttosio-6-fosfato a fruttosio-1,6-bisfosfato, catalizzata dalla fosfofruttochinasi-1 o PFK-1 (EC 2.7.1.11).
ΔG = -22,2 kJ/mole (-5,3 kcal/mole)
ΔG°’ = -14,2 kJ/mole (-3,4 kcal/mole)
La conversione del fosfoenolpiruvato o PEP, acronimo dell’inglese phosphoenolpyruvate, in piruvato, catalizzata dalla piruvato chinasi (EC 2.7.1.40).
ΔG = -16,7 kJ/mole (-4,0 kcal/mole)
ΔG°’ = -31,4 kJ/mole (-7,5 kcal/mole)
Nella gluconeogenesi queste tre tappe unidirezionali sono superate grazie a enzimi specifici che catalizzano passaggi irreversibili nella direzione della sintesi del glucosio.[6] In questo modo è assicurata l’irreversibilità dell’intera via metabolica.
Di seguito sono analizzate tali reazioni.
Conversione del piruvato in fosfoenolpiruvato
Il primo passaggio della gluconeogenesi che by-passa una tappa irreversibile della glicolisi, nello specifico quella catalizzata dalla piruvato chinasi, è la conversione del piruvato in fosfoenolpiruvato.[8]
La sintesi del fosfoenolpiruvato è ottenuta attraverso una sequenza di due reazioni catalizzate nell’ordine dagli enzimi:
piruvato carbossilasi (EC 6.4.1.1);
fosfoenolpiruvato carbossichinasi o PEP carbossichinasi (EC 4.1.1.32).
Piruvato → Ossalacetato → Fosfoenolpiruvato
La piruvato carbossilasi catalizza la carbossilazione del piruvato in ossalacetato, con consumo di una molecola di ATP. L’enzima richiede la presenza di ioni manganese o magnesio.
Piruvato + HCO3– + ATP → Ossalacetato + ADP + Pi
L’enzima, scoperto nel 1960 da Merton Utter, è una proteina mitocondriale formata da quattro subunità identiche, ognuna dotata di attività catalitica. Le subunità hanno come coenzima la biotina, legata attraverso legame ammidico al gruppo amminico ε di un residuo di lisina, e la cui funzione è quella di trasportatore di CO2 attivata nel corso della reazione enzimatica. In ogni subunità è presente anche un sito di legame per l’acetil-CoA.
Va notato che la reazione catalizzata dalla piruvato carbossilasi, portando alla produzione di ossalacetato, fornisce intermedi anche al ciclo dell’acido citrico o di Krebs.
La fosfoenolpiruvato carbossichinasi è presente, all’incirca nelle stesse quantità, sia nel mitocondrio che nel citosol dell’epatocita. Le due forme isoenzimatiche sono codificate da distinti geni nucleari.
L’enzima catalizza la decarbossilazione e fosforilazione dell’ossalacetato a dare fosfoenolpiruvato, in una reazione in cui il GTP funge da donatore di un gruppo fosfato, e richiede la presenza sia di ioni manganese che magnesio. La reazione nelle normali condizioni cellulari è reversibile.
Ossalacetato + GTP ⇄ PEP + CO2 + GDP
Nella reazione la CO2 aggiunta nella tappa catalizzata dalla piruvato carbossilasi viene rimossa. La sequenza di carbossilazione e decarbossilazione è un modo per “attivare” il piruvato, poiché la decarbossilazione dell’ossalacetato facilita, rende termodinamicamente possibile la formazione del fosfoenolpiruvato.
Più in generale le sequenze carbossilazione-decarbossilazione sono utilizzate per favorire reazioni che altrimenti sarebbero fortemente endoergoniche, e sono utilizzate anche nel ciclo dell’acido citrico, nella via del pentoso fosfato, detta via dell’esoso monofosfato, e nella sintesi degli acidi grassi.
Prima della nascita i livelli di PEP carbossichinasi sono molto bassi, mentre, poche ore dopo il parto la sua attività aumenta di diverse volte. Questo è il motivo per cui la gluconeogenesi è attiva solo dopo la nascita.
La somma delle reazioni catalizzate dalla piruvato carbossilasi e dalla fosfoenolpiruvato carbossichinasi è:
Piruvato + ATP + GTP + HCO3– → PEP + ADP + GDP + Pi + CO2
Il ΔG°’ della reazione è pari a 0,9 kJ/mole (0,2 kcal/mole), mentre la variazione di energia libera standard associata alla formazione di piruvato dal fosfoenolpiruvato per semplice inversione della reazione catalizzata dalla piruvato chinasi è di + 31,4 kJ/mole (7,5 kcal/mole).
Sebbene il ΔG’° dei due passaggi che portano alla formazione di fosfoenolpiruvato dal piruvato sia leggermente positivo, il ΔG calcolato dalle concentrazioni intracellulari degli intermedi è molto negativo, -25 kJ/mole (-6 kcal/mole), grazie al rapido consumo del fosfoenolpiruvato in altre reazioni, il che mantiene la sua concentrazione molto bassa. Quindi, nelle condizioni esistenti nella cellula la suddetta sintesi del PEP dal piruvato è un processo irreversibile.[5]
Particolarità della sintesi del fosfoenolpiruvato dal piruvato è che la via seguita dipende dal precursore prevalente: piruvato o alanina, oppure il lattato.
Substrato prevalente piruvato o alanina
I passaggi di seguito descritti prevalgono quando i substrati gluconeogenetici prevalenti sono piruvato o alanina.
La piruvato carbossilasi è un enzima mitocondriale, per cui il piruvato dovrà essere trasportato dal citosol nell’organello. Ciò avviene grazie a due trasportatori presenti sulla membrana mitocondriale interna, indicati come MPC1 and MPC2, che, associandosi, formano un eteropolimero che facilita il passaggio della molecola.[4]
Il piruvato può anche essere prodotto direttamente all’interno del mitocondrio dall’alanina per transaminazione, nella reazione catalizzata dalla alanina aminotransferasi o ALT (EC 2.6.1.2), mentre il gruppo amminico sarà infine convertito in urea attraverso il ciclo dell’urea.
Da Piruvato e Alanina a PEP
Poiché gli enzimi che intervengono nelle tappe successive della gluconeogenesi, fino alla formazione del glucosio-6-fosfato, sono citosolici, l’ossalacetato prodotto nei mitocondri dovrà essere trasportato nel citosol. La membrana mitocondriale interna è però priva di trasportatori per l’ossalacetato. Il suo passaggio nel citosol avviene a seguito della sua riduzione a malato, che al contrario può attraversare la membrana mitocondriale interna. La reazione è catalizzata dalla malato deidrogenasi mitocondriale (EC 1.1.1.37), un enzima che interviene anche nel ciclo dell’acido citrico dove però il flusso dei metaboliti procede in direzione opposta. Nella reazione il NADH viene ossidato a NAD+.
Ossalacetato + NADH + H+ ⇄ Malato + NAD+
Sebbene ΔG°’ della reazione sia piuttosto elevato, il ΔG calcolato sulla base della concentrazione intracellulare dell’ossalacetato, molto bassa, è prossimo allo zero per cui la reazione è facilmente reversibile.
Il malato prodotto attraversa la membrana mitocondriale interna grazie a un componente dello shuttle del malato-aspartato, il trasportatore malato-α-chetoglutarato. Una volta nel citosol il malato è riossidato a ossalacetato nella reazione catalizzata dalla malato deidrogenasi citosolica, con produzione di NADH.
Malato + NAD+ → Ossalacetato + NADH + H+
Nota: lo shuttle del malato-aspartato è il più attivo nel trasporto degli equivalenti riducenti del NADH dal citosol all’interno del mitocondrio, ed è presente nei mitocondri del fegato, rene, e cuore.
Grazie a questa reazione equivalenti riducenti mitocondriali, in forma di NADH, sono trasferiti nel citosol.[8] Tale trasferimento è necessario per il proseguimento della gluconeogenesi, in quanto nel citosol il NADH, utilizzato nella reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12), è presente in bassa concentrazione, con un rapporto [NADH]/[NAD+] pari a 8 x 10-4, circa 100000 volte più basso di quello osservato nei mitocondri.[5]
Infine l’ossalacetato viene convertito in fosfoenolpiruvato nella reazione catalizzata dalla PEP carbossichinasi.
Substrato prevalente lattato
Il lattato è un importante precursore gluconeogenico. Esempi di cellule e tessuti che lo producono in grande quantità sono i globuli rossi, che, completamente dipendenti dalla glicolisi anaerobica, lo producono continuamente, e il muscolo scheletrico in forte attività, quando la velocità della glicolisi supera la velocità del ciclo dell’acido citrico e della fosforilazione ossidativa.[5]
Quando il lattato è il precursore gluconeogenico prevalente la sintesi del PEP segue una via differente rispetto a quanto visto in precedenza.
Nel citosol dell’epatocita, dove come detto la concentrazione del NAD+ è elevata, il lattato viene ridotto a piruvato nella reazione catalizzata dall’isoenzima epatico della lattato deidrogenasi (EC 1.1.1.27). Nella reazione il NAD+ viene ridotto a NADH.
Lattato + NAD+ → Piruvato + NADH + H+
La produzione citosolica di NADH rende non necessaria l’esportazione di equivalenti riducenti dal mitocondrio.
Il piruvato passa nella matrice mitocondriale per essere convertito in ossalacetato nella reazione catalizzata dalla piruvato carbossilasi. Nel mitocondrio l’ossalacetato è convertito in fosfoenolpiruvato nella reazione catalizzata dall’isoenzima mitocondriale della piruvato carbossilasi, che, a mezzo di un trasportatore anionico della membrana mitocondriale interna, esce dal mitocondrio per continuare nella via gluconeogenetica.
Nota: la sintesi del glucosio dal lattato può essere considerata anche come una parte del “ramo epatico” del ciclo di Cori.
Conversione del fruttosio-1,6-bisfosfato in fruttosio-6-fosfato
Il secondo passaggio della gluconeogenesi che supera una reazione irreversibile della via glicolitica, quella catalizzata dalla PFK-1, è la defosforilazione del fruttosio-1,6-bisfosfato a fruttosio-6-fosfato.
La reazione, catalizzata dalla fruttosio-1,6-bisfosfatasi o FBPasi-1 (EC 3.1.3.11), enzima citosolico e Mg2+ dipendente, comporta l’idrolisi del fosfato sul C-1, senza alcuna produzione di ATP.
Fruttosio-1,6-bisfosfato + H2O → Fruttosio-6-fosfato + Pi
Il ΔG°’ della reazione è pari a -16,3 kJ/mol (-3,9 kcal/mol), dunque una reazione irreversibile.
Conversione del glucosio-6-fosfato a glucosio
Il terzo passaggio esclusivo della gluconeogenesi permette di superare la reazione della glicolisi catalizzata dalla esochinasi o dalla glucochinasi. Nello specifico si tratta della defosforilazione del glucosio-6-fosfato a glucosio catalizzata dall’unità catalitica della glucosio-6-fosfatasi, un complesso proteico presente nella membrana del reticolo endoplasmatico degli epatociti, enterociti e cellule del tubulo prossimale del rene.[8] La glucosio-6-fosfatasi è composta da una subunità catalitica dotata di attività fosfatasica e un trasportatore bidirezionale specifico per il glucosio-6-fosfato detto glucosio-6-fosfato traslocasi o T1.
La subunità catalitica della glucosio-6-fosfatasi presenta il sito attivo rivolto verso la superficie luminale dell’organello: quindi l’enzima catalizza una idrolisi intraluminale del substrato.
Il glucosio-6-fosfato, sia quello derivante dalla gluconeogenesi, rilasciato dalla reazione catalizzata dalla glucosio-6-fosfato isomerasi o fosfoglucosio isomerasi (EC 5.3.1.9), che dalla glicogenolisi, rilasciato dalla reazione catalizzata dalla fosfoglucomutasi (EC 5.4.2.2), è prodotto nel citosol, e dovrà entrare nel lume del reticolo endoplasmatico per essere defosforilato. Il suo passaggio è operato dalla glucosio-6-fosfato traslocasi.
La subunità catalitica della glucosio-6-fosfatasi, un enzima Mg2+-dipendente, catalizza la reazione corrisponde all’ultima tappa sia della gluconeogenesi che della glicogenolisi. E, al pari della reazione che porta alla sintesi del fruttosio-6-fosfato, anche questa è una semplice idrolisi di un estere fosforico.
Glucosio-6-fosfato + H2O → Glucosio + Pi
Va inoltre sottolineato che, grazie all’orientamento del sito attivo, la cellula separa questa reazione dal citosol, e dunque dalla glicolisi, che verrebbe bloccata dall’azione dell’enzima sul glucosio-6-fosfato.
Il ΔG°’ della reazione è pari a -13,8 kJ/mol (-3,3 kcal/mol), dunque una reazione irreversibile. Se invece la reazione fosse l’inverso di quella catalizzata dalla esochinasi/glucochinasi, comporterebbe il trasferimento di un gruppo fosforico all’ADP dal glucosio-6-fosfato, una reazione con ΔG pari a +33,4 kJ/mole (+8 kcal/mol), quindi fortemente endoergonica. Analoghe considerazioni possono essere estese alla reazione catalizzata dalla FBPasi-1.
Sembra che il glucosio e il gruppo Pi prodotti siano trasportati nel citosol da due distinti trasportatori, indicati rispettivamente come T2 e T3, quest’ultimo un trasportatore anionico.
Infine, grazie al trasportatore di membrana GLUT2, il glucosio potrà lasciare l’epatocita ed entrare in circolo per essere trasportato ai tessuti che lo richiedano. Come accennato in precedenza invece, il glucosio prodotto nel rene, in condizioni di normale efficienza epatica, viene utilizzato per la maggior parte dalla midollare del rene stesso.
La gluconeogenesi è energeticamente costosa
Al pari di quanto accade nella glicolisi, sono le tappe irreversibili della gluconeogenesi le responsabili del consumo della maggior parte dell’energia necessaria al processo.[5]
Sono consumati sei legami fosforici ad alta energia, due forniti dal GTP e quattro dall’ATP, cui vanno aggiunte due molecole di NADH per la riduzione di altrettante molecole di 1,3-bisfosfoglicerato nella reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi. Il consumo dei due NADH comporta la mancata produzione di 5 molecole di ATP che sarebbero potute essere sintetizzate nel caso in cui gli elettroni del coenzima ridotto fossero stati utilizzati per la sintesi di ATP nel mitocondrio attraverso la fosforilazione ossidativa.
Anche da queste considerazioni prettamente energetiche emerge che la gluconeogenesi non è semplicemente l’inverso della glicolisi, nel qual caso necessiterebbe del consumo di sole due molecole di ATP, come si evince dalla equazione glicolitica complessiva.
Almeno nel fegato, l’ATP necessario a sostenere la gluconeogenesi deriva di solito dall’ossidazione degli acidi grassi, o dall’ossidazione degli scheletri carboniosi degli amminoacidi, a seconda del “carburante” disponibile.
Regolazione coordinata della glicolisi e gluconeogenesi
Se la glicolisi e la gluconeogenesi procedessero simultaneamente ad alta velocità nella stessa cellula, ne risulterebbe solo il consumo di ATP e la produzione di calore, in particolare in corrispondenza delle tappe irreversibili dei due processi, e nulla di più.
Ad esempio, considerando PFK-1 e FBPasi-1:
ATP + Fruttosio-6-fosfato → ADP + Fruttosio-1,6-bisfosfato
Fruttosio 1,6-bisfosfato + H2O → Fruttosio 6-fosfato + Pi
E, dalla somma delle due reazioni:
ATP + H2O → ADP + Pi + Calore
Quando reazioni contrapposte di questo tipo procedono simultaneamente si parla di ciclo futile o ciclo del substrato.
La contemporanea attività ad alta velocità di enzimi che catalizzano reazioni contrapposte è evitata grazie al controllo dell’attività degli enzimi stessi, che può avvenire mediante:
meccanismi allosterici;
modificazioni covalenti, in particolare fosforilazioni e defosforilazioni;
modificazioni nella concentrazione degli enzimi coinvolti, conseguenti a variazioni del rapporto tra la loro sintesi e degradazione.
I meccanismi allosterici sono molto rapidi e istantaneamente reversibili, avvenendo in un arco temporale di millisecondi. Gli altri, innescati da segnali che provengono dall’esterno della cellula e trasportati da ormoni quali insulina, glucagone, o adrenalina, richiedono tempi più lunghi, dai secondi ai minuti per le modificazioni covalenti, e fino a ore per le modificazioni della concentrazione degli enzimi.
Grazie a questi meccanismi regolatori è possibile ottenere una regolazione coordinata delle due vie, tale da assicurare che quando il flusso di piruvato procede attraverso la gluconeogenesi, il flusso del glucosio attraverso la glicolisi rallenta, e viceversa.
Regolazione della gluconeogenesi
La regolazione della gluconeogenesi e della glicolisi avviene attraverso controlli esercitati sugli enzimi specifici delle singole vie, e non su quelli comuni.[1]
Mentre i principali punti di regolazione della glicolisi sono le reazioni catalizzate dagli enzimi PFK-1 e piruvato chinasi, i principali punti di regolazione della via gluconeogenetica sono le reazioni catalizzate dagli enzimi fruttosio-1,6-bisfosfatasi e piruvato carbossilasi. Anche gli altri due enzimi esclusivi della via gluconeogenetica, glucosio-6-fosfatasi e PEP carbossichinasi, sono soggetti a regolazione ma solo a livello trascrizionale.
Piruvato carbossilasi
Nel mitocondrio il piruvato può essere convertito in:
ossalacetato, nella reazione catalizzata dalla privato carbossilasi, l’enzima che catalizza la prima tappa della gluconeogenesi.
Il destino metabolico del piruvato dipende dai livelli dell’acetil-CoA e dunque dalla disponibilità di acidi grassi nel mitocondrio.
Quando gli acidi grassi sono disponibili, la loro beta-ossidazione porta alla liberazione di molecole di acetil-CoA, che entrano nel ciclo di Krebs per dar luogo alla formazione di GTP e NADH. Una volta che i fabbisogni energetici della cellula sono soddisfatti, la fosforilazione ossidativa rallenta, il rapporto NADH/NAD+ cresce, il NADH inibisce il ciclo dell’acido citrico e si verifica un accumulo di acetil-CoA nella matrice mitocondriale.[1][5]
L’acetil-CoA è un effettore allosterico positivo della piruvato carbossilasi e un effettore allosterico negativo della piruvato chinasi. Inoltre inibisce il complesso della piruvato deidrogenasi sia attraverso una inibizione da prodotto finale che mediante la sua fosforilazione, a seguito dell’attivazione di una specifica chinasi.
Destini del Piruvato
Dunque, quando la carica energetica della cellula è alta, quello che accade è che la formazione di acetil-CoA dal piruvato è rallentata, mentre viene stimolata la conversione del piruvato in glucosio. L’acetil-CoA è quindi un segnale metabolico che indica che un’ulteriore ossidazione del glucosio a scopo energetico non è necessaria e che i metaboliti carboniosi possono essere utilizzati per la sintesi e il deposito di glucosio.
Viceversa, quando i livelli di acetil-CoA si riducono, l’attività della piruvato chinasi e del complesso della piruvato deidrogenasi aumentano, e quindi anche il flusso di metaboliti attraverso il ciclo di Krebs, il tutto per rifornire di energia la cellula.
Si potrebbe riassumere dicendo che la piruvato carbossilasi è attiva quando la carica energetica della cellula è alta e che il primo punto di controllo della gluconeogenesi determina quello che sarà il destino del piruvato nel mitocondrio.
Fruttosio-1,6-bisfosfatasi
Il secondo punto principale di controllo della gluconeogenesi è rappresentato dalla reazione catalizzata dalla fruttosio-1,6-bisfosfatasi. L’enzima è inibito allostericamente dall’AMP. Quindi quando la concentrazione dell’AMP è alta, e di conseguenza quella dell’ATP è bassa, la gluconeogenesi rallenta. Ossia, come visto in precedenza, l’enzima è attivo quando la carica energetica della cellula è adeguata a sostenere la sintesi de novo del glucosio.
Di contro la PFK-1, l’enzima glicolitico corrispondente, è stimolata allostericamente dall’AMP e dall’ADP e inibita dall’ATP e dal citrato, quest’ultimo derivante dalla condensazione tra l’acetil-CoA e l’ossalacetato. Quindi riassumendo:
quando la concentrazione dell’AMP è alta la gluconeogenesi rallenta, mentre la glicolisi accelera;
quando la concentrazione dell’ATP è elevata, e quindi è bassa quella di ADP e AMP, o quando l’acetil-CoA o il citrato sono presenti in concentrazioni adeguate, viene promossa la gluconeogenesi mentre rallenta la glicolisi.
L’aumento della concentrazione del citrato segnala che l’attività del ciclo dell’acido citrico può rallentare; in questo modo il piruvato potrà essere utilizzato per la sintesi del glucosio.
Fruttosio-1,6-bisfosfatasi, PFK-1 e fruttosio-2,6-bisfosfato
Il fegato ha un ruolo centrale nel mantenimento della glicemia a valori costanti: questo richiede la presenza di meccanismi regolatori che coordinino il consumo e la produzione del glucosio. Due sono gli ormoni principalmente coinvolti: il glucagone e l’insulina.
Il glucagone, rilasciato in circolo quando la glicemia scende, segnala al fegato di ridurre il suo consumo di glucosio e di aumentarne la produzione de novo e il rilascio dal glicogeno.[6]
L’azione degli ormoni suddetti è mediata da un effettore allosterico della PFK-1 e della fruttosio-1,6-bisfosfatasi: il fruttosio-2,6-bisfosfato, una molecola strutturalmente correlata al fruttosio-1,6-bisfosfato ma che non è ne un intermedio della glicolisi nel della gluconeogenesi.[1]
La molecola fu scoperta nel 1980 da Emile Van Schaftingen and Henri-Gery Hers come un potente stimolatore della PFK-1; l’anno successive gli stessi ricercatori dimostrarono che è anche un potente inibitore dalla fruttosio-1,6-bisfosfatasi.[9][10]
A seguito del legame allo specifico sito allosterico sulla PFK-1, il fruttosio-2,6-bisfosfato svolge un duplice effetto: riduce l’affinità della proteina per i suoi inibitori allosterici ATP e citrato e ne aumenta l’affinità per il fruttosio-6-fosfato, il suo substrato. La PFK-1, in assenza di fruttosio-2,6-bisfosfato, e in presenza di concentrazioni fisiologiche di ATP, fruttosio-6-fosfato, e dei suoi effettori allosterici AMP, ATP e citrato, è praticamente inattiva. La presenza di fruttosio-2,6-bisfosfato ha invece l’effetto di attivare l’enzima quindi stimolare la glicolisi nell’epatocita. Nel contempo la molecola rallenta la gluconeogenesi, inibendo la fruttosio-1,6-bisfosfatasi, anche in assenza di AMP. Tuttavia gli effetti di AMP e fruttosio-2,6-bisfosfato sull’inibizione di FBPasi-1 sono sinergici.
F2,6BP e Regolazione della Glicolisi e Gluconeogenesi
La concentrazione di fruttosio-2,6-bisfosfato è regolata dalle velocità relative della sua sintesi e degradazione. Viene sintetizzato a partire dal fruttosio-6-fosfato nella reazione catalizzata dalla fosfofruttochinasi 2 o PFK-2 (EC 2.7.1.105), e idrolizzato a fruttosio-6-fosfato nella reazione catalizzata dalla fruttosio-2,6-bisfosfatasi o FBPasi-2 (EC 3.1.3.46). Le due attività enzimatiche sono presenti su una stessa proteina, che dunque è un enzima bifunzionale, anche detto enzima tandem, e nel fegato sono regolate dall’insulina e dal glucagone, nel modo di seguito descritto.
Il glucagone, a seguito del legame allo specifico recettore di membrana, stimola la adenilato ciclasi (EC 4.6.1.1) presente sulla membrana plasmatica a produrre 3’-5’ AMP ciclico o cAMP, che, a seguito del legame alla protein chinasi cAMP-dipendente o protein chinasi A o PKA (EC 2.7.11.11), la attiva. La chinasi attivata catalizza la fosforilazione, a spese di una molecola di ATP, di uno specifico residuo di serina (Ser32) di PFK-2/FBPasi-2. La fosforilazione comporta un aumento dell’attività fosfatasica a spese di quella chinasica, che si riduce in conseguenza di un aumento della Km per il fruttosio-6-fosfato. Tutto ciò porta a una riduzione dei livelli di fruttosio-2,6-bisfosfato, con conseguente stimolazione della gluconeogenesi e inibizione della glicolisi. Quindi, in risposta al segnale trasportato dal glucagone, aumenta la produzione epatica di glucosio attraverso la gluconeogenesi, il che rende l’organo capace di contrastare la riduzione della glicemia segnalata dall’ormone.
Nota: il glucagone, al pari dell’adrenalina, stimola la gluconeogenesi in parte anche aumentando la disponibilità di substrati quali il glicerolo e gli amminoacidi.
L’insulina, a seguito del legame agli specifici recettori di membrana dell’epatocita, va ad attivare una protein fosfatasi, la fosfoprotein fosfatasi 2A che catalizza la rimozione del gruppo fosforico dalla PFK-2/FBPasi-2, attivando così la PFK-2 e riducendo l’attività della FBPasi-2. (Nel contempo stimola anche una cAMP fosfodiesterasi che idrolizza il cAMP ad AMP). Il risultato è l’aumento dei livelli intracellulari di fruttosio-2,6-bisfosfato e la conseguente inibizione della gluconeogenesi e attivazione della glicolisi.
Inoltre il fruttosio-6-fosfato inibisce allostericamente la FBPasi-2 mentre attiva la PFK-2. Riguardo l’attività dell’enzima bifunzionale PFK-2/FBPasi-2 va sottolineato che entrambe le attività sono inibite dai rispettivi prodotti di reazione, e tuttavia i fattori predominanti sono la concentrazione del fruttosio-6-fosfato e lo stato di fosforilazione dell’enzima stesso.
Glucosio-6-fosfatasi
A differenza della piruvato carbossilasi e della fruttosio-1,6-bisfosfatasi, la subunità catalitica della glucosio-6-fosfatasi non è soggetta a regolazione allosterica o covalente, mentre viene regolata a livello trascrizionale.[7] La bassa glicemia e il glucagone, dunque fattori che determinano una maggiore produzione di glucosio, e i glucocorticoidi ne stimolano la sintesi, che al contrario è inibita dall’insulina.
Inoltre la sua Km per il glucosio-6-fosfato è decisamente più alta rispetto al normale intervallo di concentrazione della molecola stessa. Ne risulta che l’attività dell’enzima mostra una dipendenza quasi lineare rispetto alla concentrazione del substrato. Per questo si dice che l’enzima è sotto controllo da parte della concentrazione del substrato.
PEP carbossichinasi
La regolazione dell’enzima avviene principalmente a livello della sua sintesi e demolizione. Ad esempio elevati livelli di glucagone o il digiuno ne aumentano la produzione, a seguito della stabilizzazione del suo mRNA e dell’aumento della sua velocità di trascrizione. Glicemie elevate o l’insulina hanno effetto opposto.
Xilulosio-5-fosfato
Anche un altro meccanismo regolatorio scoperto di recente, attraverso l’azione dello xilulosio-5-fosfato, un intermedio della via del pentoso fosfato, stimola la glicolisi e inibisce la gluconeogenesi, intervenendo nel controllo della concentrazione del fruttosio-2,6-bisfosfato nell’epatocita.
Quando la concentrazione ematica del glucosio aumenta, come dopo un pasto ricco di carboidrati, nel fegato si verifica l’attivazione della glicolisi e della via dell’esoso monofosfato. In quest’ultima via metabolica viene prodotto anche xilulosio-5-fosfato che è in grado di attivare la protein fosfatasi 2A. Ciò porta alla defosforilazione della PFK-2/FBPasi-2, inibendo così la FBPasi-2 e stimolando la PFK-2. Ne risulta un aumento della concentrazione del fruttosio-2,6-bisfosfato, e quindi l’inibizione della gluconeogenesi e la stimolazione della glicolisi, con conseguente aumento della produzione di acetil-CoA, il principale substrato per la sintesi dei lipidi.[2]
Il concomitante aumento del flusso attraverso la via dell’esoso monofosfato produce NADPH, fonte di elettroni per la sintesi dei lipidi. Infine, la protein fosfatasi 2A defosforila anche ChREBP, un fattore di trascrizione che attiva l’espressione dei geni epatici per la sintesi dei lipidi. Quindi, in risposta a un aumento della glicemia, a livello epatico sarà stimolata la sintesi dei lipidi.
Risulta dunque evidente che lo xilulosio-5-fosfato è un regolatore chiave del metabolismo sia dei carboidrati che dei grassi.
Precursori
Oltre al piruvato, i principali precursori gluconeogenici sono il lattato, di cui si è parlato in precedenza, il glicerolo, la maggior parte degli amminoacidi, e comunque qualunque composto che possa essere convertito in piruvato od ossalacetato.[1]
Glicerolo
Il glicerolo deriva dalla lipolisi nel tessuto adiposo. Con l’esclusione del propionil-CoA, è l’unica parte delle molecole dei lipidi che negli animali possa essere utilizzata per la sintesi del glucosio.
Il suo punto di ingresso nella gluconeogenesi, o nella glicolisi, a seconda delle condizioni energetiche in cui si trova la cellula, è rappresentato dal diidrossiacetone fosfato, la cui sintesi avviene in due passaggi.
Nel primo il glicerolo è fosforilato a glicerolo-3-fosfato, nella reazione catalizzata dalla glicerolo chinasi (EC 2.7.1.30). La reazione consuma una molecola di ATP.
Glicerolo + ATP → Glicerolo-3-fosfato + ADP + Pi
L’enzima assente negli adipociti, ma presente nel fegato. Ciò significa che il glicerolo dovrà raggiungere il fegato prima di essere ulteriormente metabolizzato.
Il glicerolo-3-fosfato viene quindi ossidato a diidrossiacetone fosfato, nella reazione catalizzata dalla glicerolo-3-fosfato deidrogenasi (EC 1.1.1.8). Nella reazione il NAD+ viene ridotto a NADH.
Durante il digiuno prolungato il glicerolo è il principale precursore gluconeogenetico, essendo responsabile della produzione di circa il 20% del glucosio.[3]
Amminoacidi glucogenici
Piruvato e ossalacetato rappresentano i punti di ingresso per gli amminoacidi glucogenici, ossia quelli il cui scheletro carbonioso o parte di esso può essere utilizzato per la sintesi del glucosio.
Gli amminoacidi derivano dalla demolizione delle proteine, sia di origine alimentare che endogena, come quelle del muscolo scheletrico nel corso del digiuno o dell’attività fisica intensa e prolungata.
I processi catabolici a carico di ognuno dei venti amminoacidi che compongono le proteine convergono verso la sintesi di sette prodotti principali: acetil-CoA, acetoacetil-CoA, α-chetoglutarato, succinil-CoA, fumarato, ossalacetato e piruvato.
Con l’esclusione di acetil-CoA e acetoacetil-CoA, le altre cinque molecole possono essere utilizzate per la sintesi del glucosio; quindi gli amminoacidi glucogenici possono essere definiti anche come quelli il cui scheletro carbonioso, in parte o in toto, può essere convertito in una o più delle suddette molecole.
Solo leucina e lisina sono esclusivamente chetogenici, essendo il loro scheletro carbonioso catabolizzato ad acetil-CoA e/o acetoacetil-CoA.
Di seguito sono elencati gli amminoacidi glucogenici con i rispettivi punti di ingresso.
Piruvato: alanina, cisteina, glicina, serina, treonina e triptofano.
Ossalacetato: aspartato e asparagina.
α-Chetoglutarato: glutammato, arginina, glutammina, istidina e prolina.
Succinil-CoA: isoleucina, metionina, treonina e valina.
Fumarato: fenilalanina e tirosina.
α-Chetoglutarato, succinil-CoA e fumarato, tutti intermedi del ciclo dell’acido citrico, entrano nella via gluconeogenetica previa conversione in ossalacetato.
Propionato
Il propionato, che fa parte del gruppo degli acidi grassi a catena corta, in forma di propionil-CoA è un precursore gluconeogenetico in quanto può essere convertito in succinil-CoA.
Di seguito sono analizzate le diverse fonti di propionato.
Può derivare dalla beta-ossidazione di acidi grassi a catena dispari, come ad esempio l’acido margarico, acido grasso saturo con 17 atomi di carbonio. Tali acidi grassi sono molto rari rispetto a quelli a catena pari, e presenti in quantità significative nei lipidi di alcuni organismi marini, delle piante, e nel grasso dei ruminanti. Nell’ultimo passaggio del loro ciclo di ossidazione, il substrato è non a 4 ma a 5 atomi di carbonio, per cui una volta ossidato e scisso in due frammenti, darà origine a un acetil-CoA e un propionil-CoA.
Altra fonte è la ossidazione degli acidi grassi a catena ramificata, con ramificazioni costituite da gruppi alchilici con un numero dispari di atomi di carbonio. Un esempio è l’acido fitanico, prodotto nei ruminanti dall’ossidazione del fitolo, un derivato della degradazione della clorofilla.
Nei ruminanti, è prodotto anche a partire dal glucosio liberato dall’idrolisi della cellulosa grazie all’azione di batteri presenti nel rumine, una delle quattro camere che compongono lo stomaco di questi animali. Sempre nel rumine, gli stessi batteri convertono, attraverso fermentazione, il glucosio in propionato, che potrà, una volta assorbito, essere utilizzato per la gluconeogenesi, essere ossidato per la produzione di energia, o essere utilizzato per la sintesi degli acidi grassi.[5]
Nei ruminanti, dove la gluconeogenesi tende a essere un processo continuo, il propionato è il più importante precursore gluconeogenetico.
Il propionato può derivare anche catabolismo della valina, leucina e isoleucina.
Nel colon, il propionato viene prodotto dai batteri del microbiota intestinale attraverso la fermentazione anaerobica di carboidrati non digeribili, come l’amido resistente e le fibre. Assorbito dagli enterociti, per la maggior parte non è ivi metabolizzato, passa nel circolo portale e raggiunge il fegato, dove può entrare nella gluconeogenesi.
L’ossidazione del propionil-CoA a succinil-CoA avviene attraverso tre reazioni che si verificano nel fegato e in altri tessuti.
Nella prima reazione il propionil-CoA è carbossilato a dare D-metilmalonil-CoA, nella reazione catalizzata dalla propionil-CoA carbossilasi (EC 6.4.1.3), enzima che ha come cofattore la biotina. La reazione consuma un ATP.
Propionil-CoA + HCO3– + ATP → D-Metilmalonil-CoA+ ADP + Pi
Nella reazione successiva il D-metilmalonil-CoA viene epimerizzato nello stereoisomero L. La reazione è catalizzata dalla metilmalonil-CoA epimerasi (EC 5.1.99.1).
D-Metilmalonil-CoA ⇄ L-Metilmalonil-CoA
Infine l’L-metilmalonil-CoA, nella reazione catalizzata dalla metilmalonil-CoA mutasi (EC 5.4.99.2), enzima che richiede come coenzima la 5-deossiadenosilcobalamina, un derivato della cobalamina o vitamina B12, subisce un riarrangiamento intramolecolare a dare succinil-CoA.
^ Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003;100:5107-5112. doi:10.1073/pnas.0730817100
^ Kuriyama H. et all. Coordinated regulation of fat-specific and liver-specific glycerol channels, aquaporin adipose and aquaporin 9. Diabetes 2002;51(10):2915-2921. 10.2337/diabetes.51.10.2915
^ McCommis K.S. and Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J 2015;466(3):443-454. doi:10.1042/BJ20141171
^ abcdefghi Nelson D.L., M. M. Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ abcde Rosenthal M.D., Glew R.H. Medical biochemistry – Human metabolism in health and disease. John Wiley J. & Sons, Inc., Publication, 2009
^ Soty M., Chilloux J., Delalande F., Zitoun C., Bertile F., Mithieux G., and Gautier-Stein A. Post-Translational regulation of the glucose-6-phosphatase complex by cyclic adenosine monophosphate is a crucial determinant of endogenous glucose production and is controlled by the glucose-6-phosphate transporter. J Proteome Res 2016;15(4):1342-1349. doi:10.1021/acs.jproteome.6b00110
^ abc Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
^ Van Schaftingen E., and Hers H-G. Inhibition of fructose-1,6-bisphosphatase by fructose-2,6-bisphosphate. Proc Natl Acad Sci USA 1981;78(5):2861-2863. doi:10.1073/pnas.78.5.2861
^ Van Schaftingen E., Jett M-F., Hue L., and Hers H-G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA 1981;78(6):3483-3486. doi:10.1073/pnas.78.6.3483
Il ciclo glucosio-alanina, anche detto ciclo di Cahill, proposto per la prima volta tra il 1969 e il 1970 da Mallette, Exton e Park, e Felig e collaboratori, consiste in una serie di reazioni attraverso le quali i tessuti extraepatici, come ad esempio il muscolo scheletrico, esportano al fegato piruvato, la base coniugata dell’acido piruvico, e gruppi amminici in forma di alanina, e ricevono, attraverso il circolo sanguigno, glucosio prodotto nel fegato.[1][4]
Di seguito ne sono riassunte le tappe.[5][6]
Quando nei tessuti extraepatici gli amminoacidi sono utilizzati a fini energetici, il piruvato, prodotto dal glucosio attraverso la glicolisi, funge da accettore del loro gruppo amminico alfa, formando alanina, un aminoacido non essenziale.
L’alanina diffonde nel circolo sanguigno, grazie al quale raggiunge il fegato.
Nel fegato, il gruppo amminico dell’alanina viene trasferito all’alfa-chetoglutarato a dare rispettivamente piruvato e glutammato.
Il glutammato cede per la maggior parte il gruppo amminico al ciclo dell’urea, mentre in parte funge da donatore di azoto in molti processi biosintetici.
Il piruvato entra nella gluconeogenesi e viene utilizzato per la produzione di glucosio.
Il glucosio neoformato diffonde dall’epatocita nel circolo sanguigno e raggiunge i tessuti periferici dove, grazie alla glicolisi, può essere convertito in piruvato, di nuovo disponibile per accettare il gruppo amminico α degli amminoacidi liberi, chiudendo così il ciclo.
Il ciclo glucosio-alanina fornisce quindi un collegamento tra il metabolismo dei carboidrati e quello degli amminoacidi.[6]
In breve:
Il ciclo glucosio-alanina esiste non solo tra il muscolo scheletrico, il primo tra i tessuti in cui fu osservato, e il fegato, ma coinvolge anche altre cellule e tessuti extraepatici tra cui le cellule del sistema immunitario, ad esempio gli organi linfoidi.
L’analisi successiva verrà fatta considerando il ciclo tra muscolo scheletrico e fegato.
Le proteine, sia intracellulari che extracellulari, sono continuamente idrolizzate nei loro amminoacidi costituenti e risintetizzate; e la velocità con cui avvengono i due processi è tale da evitare una perdita netta di massa magra dall’organismo.[3]
Tuttavia, in condizioni cataboliche, come nel digiuno o nell’esercizio intenso e prolungato, la velocità con cui avviene l’idrolisi delle proteine muscolari supera quella della loro sintesi de novo. Questo porterà alla liberazione di amminoacidi, alcuni dei quali sono utilizzati a fini energetici, altri a fini glucogenetici. Infatti, l’ossidazione dello scheletro carbonioso degli amminoacidi, in particolare di quelli a catena ramificata, valina, leucina, e isoleucina, rappresenta una significativa fonte di energia per il muscolo. Ad esempio, dopo circa 90 minuti dall’inizio di un esercizio fisico intenso, l’ossidazione intramuscolare degli amminoacidi fornisce il 10-15% dell’energia necessaria alla contrazione.
L’utilizzazione degli scheletri carboniosi degli amminoacidi a fini energetici implica la rimozione del loro gruppo amminico α, e quindi il successivo smaltimento di tale azoto in una forma non tossica.[5][6]
La rimozione del gruppo amminico α avviene attraverso reazioni di transaminazione che possono essere schematizzate come segue:
alfa-Chetoacido + Aminoacido ⇄ Nuovo aminoacido + Nuovo alfa-chetoacido
Tali reazioni, catalizzate da enzimi detti amminotransferasi o transaminasi (EC 2.6.1-) sono liberamente reversibili.
Gli amminoacidi ramificati, ad esempio, trasferiscono il gruppo amminico α all’α-chetoglutarato o acido 2-ossoglutarico, a dare glutammato e l’α-chetoacido derivato dall’amminoacido stesso, in una reazione catalizzata dalla transaminasi specifica per tale gruppo di amminoacidi o BCAT (EC 2.6.1.42), acronimo dell’inglese branched chain aminotransferases.
Ciclo glucosio-alanina nel muscolo scheletrico
Nel muscolo scheletrico, il glutammato prodotto potrà accettare un altro gruppo amminico a dare glutammina, per molti tessuti e organi, come ad esempio il cervello, la principale forma di trasporto interorgano dell’azoto.[7] La reazione è catalizzata dall’enzima citosolico glutammina sintetasi (EC 6.3.1.2) e consuma un ATP.
Glutammato + NH4+ + ATP → Glutammina + ADP + Pi
Ciò tuttavia comporterebbe l’uscita dal ciclo glucosio-alanina.
In alternativa, e a differenza di quanto accade nella maggior parte degli altri tessuti, il glutammato prodotto potrà partecipare a una reazione di transaminazione catalizzata dalla alanina aminotransferasi o ALT (EC 2.6.1.2), enzima presente nella maggior parte dei tessuti animali e vegetali. In tale reazione il glutammato dona il gruppo amminico alfa al piruvato, derivante dalla glicolisi, a dare alanina e alfa-chetoglutarato:
L’alanina prodotta e quella derivante dalla degradazione delle proteine, e le proteine muscolari ne sono piuttosto ricche, può lasciare la cellula ed essere veicolata dal circolo ematico al fegato, trasportandovi quindi il gruppo amminico. La velocità con cui l’alanina formata per transaminazione dal piruvato viene trasferita in circolo è proporzionale alla produzione intracellulare del piruvato.
Nota: alanina e glutammina sono le principali fonti di azoto e carbonio nel metabolismo interorgano degli amminoacidi.
Ciclo glucosio-alanina nel fegato
Una volta nel fegato si verifica una transaminazione catalizzata dalla alanina aminotransferasi epatica, in cui l’alanina, il principale amminoacido gluconeogenico, funge da donatore del gruppo amminico alfa, e l’alfa-chetoglutarato da chetoacido accettore.[6] I prodotti della reazione sono il piruvato, ossia lo scheletro carbonioso dell’alanina, e il glutammato.
Il glutammato, nella reazione catalizzata dalla glutammato deidrogenasi (EC 1.4.1.2), enzima presente nella matrice mitocondriale, rilascia ione ammonio, che entra nel ciclo dell’urea, e una molecola di alfa-chetoglutarato, che può entrare nel ciclo dell’acido citrico. Questa reazione rappresenta processo anaplerotico che lega il metabolismo degli amminoacidi con il ciclo dell’acido citrico.[6]
Tuttavia il glutammato potrà entrare anche nella reazioni di transaminazione, catalizzata dalla aspartato amminotransferasi (EC 2.6.1.1), con l’ossalacetato a dare aspartato e alfa-chetoglutarato. L’aspartato è uno degli amminoacidi coinvolti nella produzione di urea attraverso il ciclo dell’urea, ma può essere utilizzato pure nella sintesi delle purine e pirimidine.
Anche il piruvato prodotto potrà seguire destini metabolici differenti: essere ossidato per la produzione di ATP, e quindi uscire dal ciclo glucosio-alanina, o entrare nella via gluconeogenetica, e dunque proseguire nel ciclo glucosio-alanina.
Il glucosio prodotto verrà rilasciato dall’epatocita e attraverso il circolo ematico distribuito ai vari tessuti che lo richiedono, tra cui il muscolo scheletrico, dove viene utilizzato per la produzione di piruvato, di nuovo disponibile per accettare il gruppo amminico α del glutammato, chiudendo così il ciclo.
Transaminasi
Come detto in precedenza, la rimozione del gruppo amminico α degli amminoacidi avviene in reazioni di transaminazione, catalizzate da enzimi detti amminotransferasi o transaminasi.[6]
Sono enzimi citosolici, presenti in tutte le cellule e particolarmente abbondanti nel fegato, rene, intestino e muscolo, la maggior parte dei quali richiede come coenzima il piridossal fosfato o PLP, acronimo dell’inglese pyridoxal phosphate, la forma attiva della vitamina B6 o piridossina. Il coenzima è legato strettamente al sito attivo dell’enzima.
Nelle reazioni di transaminazione il gruppo amminico α degli amminoacidi liberi, con l’esclusione della treonina e lisina, è incanalato verso un numero ristretto di chetoacidi, in particolare piruvato, ossalacetato e alfa-chetoglutarato.
Le cellule contengono diversi tipi di amminotransferasi: molte sono specifiche per l’alfa-chetoglutarato come alfa-chetoacido, ma differiscono nella specificità per l’amminoacido, da cui prendono parte del nome. Esempi sono le già citate alanina aminotransferasi, anche detta alanina transaminasi e glutammico piruvico transferasi (GPT), e l’aspartato aminotransferasi (AST) o glutammico ossalacetico transaminasi (GOT) (EC 2.6.1.1).
Va sottolineato che nelle reazioni di transaminazione non si verifica alcuna deaminazione netta, nessuna perdita di gruppi amminici, in quanto l’alfa-chetoacido accettore viene amminato e l’amminoacido deaminato.
Trasporta azoto in una forma non tossica dai tessuti periferici al fegato.
Trasporta al fegato piruvato, un substrato gluconeogenico.
Rimuove piruvato dai tessuti periferici nei quali è così possibile ottenere una maggior produzione di ATP dal glucosio. Infatti il NADH prodotto durante la glicolisi può entrare nei mitocondri ed essere ossidato attraverso la fosforilazione ossidativa.
Permette di mantenere nell’epatocita una concentrazione relativamente alta di alanina, tale da inibire la degradazione delle proteine.
Può avere un ruolo nella difesa dell’ospite nei confronti delle malattie infettive.
Infine è importante sottolineare che nel ciclo glucosio-alanina non c’è sintesi netta di glucosio.
Costo energetico
Al pari del ciclo di Cori, anche il ciclo glucosio-alanina ha un costo energetico netto, che corrisponde a 3-5 molecole di ATP.
La parte del ciclo che si svolge nei tessuti periferici comporta la produzione di 5-7 molecole di ATP per molecola di glucosio:
2 ATP sono prodotti dalla glicolisi;
3-5 ATP derivano dal trasferimento degli elettroni dal NADH/FADH2 alla catena di trasporto degli elettroni.
Nel fegato invece la gluconeogenesi e il ciclo dell’urea consumano 10 ATP:
6 ATP sono consumati nel corso della gluconeogenesi;
4 ATP sono necessari per il ciclo dell’urea per ogni molecola di urea prodotta.
Il ciclo glucosio-alanina, al pari del ciclo di Cori, sposta parte del carico metabolico dai tessuti extraepatici al fegato.[6] Tuttavia il prezzo pagato dal fegato è ampiamente giustificato dai vantaggi che il ciclo apporta all’intero organismo in quanto consente, in particolari condizioni, un efficiente catabolismo delle proteine nei tessuti extraepatici, il che a sua volta permette di ottenere substrati per la gluconeogenesi come anche l’utilizzazione a fini energetici degli aminoacidi nei tessuti extraepatici.
Analogie e differenze con il ciclo di Cori
Tra i due cicli esistono alcune analogie di seguito elencate.[6][7]
Il ciclo di Cahill in parte si sovrappone al ciclo di Cori quando il piruvato viene convertito in glucosio e lo stesso trasportato ai tessuti extraepatici, dove attraverso la via glicolitica rigenera piruvato.
L’ingresso nella gluconeogenesi epatica è simile per i due cicli: sia l’alanina che il lattato sono infatti convertiti in piruvato.
Al pari del ciclo di Cori, anche il ciclo glucosio-alanina si estende attraverso tipi cellulari differenti, al contrario di quanto accade con vie metaboliche come la glicolisi, il ciclo dell’acido citrico o la gluconeogenesi che sono confinate all’interno di singole cellule.
Ciclo di Cori vs Ciclo Glucosio-Alanina
Di seguito, alcune differenze tra i due cicli.
La principale riguarda l’intermedio a tre atomi di carbonio che dai tessuti periferici raggiunge il fegato: il lattato nel il ciclo di Cori e l’alanina nel ciclo glucosio-alanina.
Un’altra differenza riguarda il destino del NADH prodotto dalla glicolisi nei tessuti periferici.
Nel ciclo di Cori il coenzima funge da donatore di agenti riducenti nella riduzione del piruvato a lattato, nella reazione catalizzata dalla lattato deidrogenasi (EC 1.1.1.27).
Nel ciclo glucosio-alanina tale riduzione non si verifica e gli elettroni del NADH potranno essere trasportati all’interno del mitocondrio dai sistemi navetta del malato-aspartato o del glicerolo-3-fosfato, generando NADH la prima navetta e FADH2 l’altra, da cui si otterranno rispettivamente 2,5 e 1,5 molecole di ATP.
Infine, dal punto precedente emerge che, a differenza del ciclo di Cori, per il ciclo glucosio-alanina è richiesta nei tessuti periferici anche la presenza di ossigeno e mitocondri.
Bibliografia
^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
^ Gropper S.S., Smith J.L., Groff J.L. Advanced nutrition and human metabolism. Cengage Learning, 2009
^ Lecker S.H., Goldberg A.L. and Mitch W.E. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J Am Soc Nephrol 2006;17(7):1807-1819.doi:10.1681/ASN.2006010083
^ Mallette L. E., Exton J.H., and Park C.R. Control of gluconeogenesis from amino acids in the perfused rat liver. J Biol Chem 1969;244(20):5713-5723. doi:10.1016/S0021-9258(18)63618-X
^ abc Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
^ abcdefgh Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ abc Wu G. Amino acids: biochemistry and nutrition. CRC Press, 2010
Il ciclo di Cori fu scoperto negli anni 30 e 40 del secolo scorso dai coniugi Carl e Gerty Cori, i quali scoprirono l’esistenza di una cooperazione metabolica tra il muscolo scheletrico che lavora in condizioni di limitata disponibilità di ossigeno e il fegato.[1]
Il ciclo permette la conversione del lattato, la forma in cui l’acido lattico è quasi esclusivamente presente a pH fisiologico, in glucosio, assicurando così un continuo apporto del monosaccaride ai tessuti periferici. La sua importanza è testimoniata anche dal fatto che è responsabile di circa il 40% del turn over del glucosio plasmatico.[7][8]
Dal punto di vista biochimico, il ciclo di Cori connette la glicolisi anaerobica con la gluconeogenesi, utilizzando tessuti differenti per compartimentalizzare processi opposti.[10]
Questa cooperazione metabolica è stata dimostrata esistere anche tra il fegato e tessuti extraepatici diversi dal muscolo scheletrico. Si può infatti affermare che, come per il ciclo glucosio-alanina, al ciclo di Cori possono partecipare i tessuti che non ossidino completamente il glucosio a CO2 e H2O.[11]
Di seguito sono riassunte le tappe del ciclo di Cori, considerando il lattato prodotto dalle fibre muscolari o dal globulo rosso.[12]
Il ciclo ha inizio con la conversione del glucosio a lattato, attraverso la glicolisi anaerobica e la successiva azione della lattato deidrogenasi (EC 1.1.1.27), che catalizza la riduzione del piruvato, la base coniugata dell’acido piruvico.
Segue la diffusione del lattato dalla cellula al circolo sanguigno, grazie al quale raggiunge, tra gli altri, il fegato, che è il suo principale utilizzatore, e la corteccia renale, in particolare i tubuli prossimali, essendo questi un altro sito dove avviene la gluconeogenesi.
Nel fegato e nella corteccia renale si verifica l’ossidazione, catalizzata dalla lattato deidrogenasi, del lattato a piruvato, che è quindi convertito a glucosio a mezzo della via gluconeogenica.[8]
Infine il glucosio prodotto diffonde nel circolo sanguigno, grazie al quale potrà raggiungere i globuli rossi o le fibre muscolari, chiudendo il ciclo.[10][11]
Globulo rosso
I globuli rossi, essendo privi di nucleo, ribosomi e mitocondri, sono più piccoli rispetto a molte altre cellule, e le ridotte dimensioni permettono loro il passaggio attraverso gli stretti capillari. Tuttavia la mancanza dei mitocondri li rende completamente dipendenti dalla glicolisi anaerobica per la produzione di ATP, il che significa che queste cellule producono continuamente acido lattico.[8][10]
La possibilità di procedere della glicolisi, come la sua velocità, dipendono anche dalla disponibilità di NAD+. Il coenzima nella sua forma ossidata è infatti necessario per l’ossidazione della gliceraldeide-3-fosfato a 1,3-bisfosfoglicerato nella reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12).
L’accumulo di NADH è evitato dalla riduzione del piruvato a lattato, in una reazione catalizzata dalla lattato deidrogenasi nella quale il NADH funge da donatore di agenti riducenti, ossidandosi a NAD+.[4]
Fibre muscolari
Le fibre muscolari a contrazione rapida contengono un numero ridotto di mitocondri e, in condizioni di limitata disponibilità di ossigeno, come durante un lavoro intenso, producono notevoli quantità di acido lattico.[8] In queste condizioni infatti:
la velocità di produzione del piruvato attraverso la via glicolitica eccede la capacità del ciclo dell’acido citrico di ossidarlo, tanto che meno del 10% del piruvato prodotto entra nel ciclo stesso;
la velocità alla quale l’ossigeno è assunto dalle cellule non è sufficiente ad assicurare l’ossidazione aerobica di tutto il NADH formato.[3]
In tali condizioni la glicolisi anaerobica porta alla produzione di 2 molecole di ATP per molecola di glucosio, 3 se il glucosio proviene dal glicogeno muscolare, una quantità decisamente inferiore rispetto alle 29-30 molecole di ATP prodotte dalla completa ossidazione del glucosio.[9] Tuttavia la velocità con cui l’ATP è prodotto dalla glicolisi anaerobica è molto maggiore rispetto a quella ottenibile dalla completa ossidazione del glucosio.[10]
Infine, come nel globulo rosso, la reazione catalizzata dalla lattato deidrogenasi, rigenerando NAD+, permette alla glicolisi di procedere, ma produce lattato.[4]
Lattato
L’acido lattico è un prodotto finale del metabolismo, e per essere utilizzato dalla cellula deve essere convertito in piruvato.[3]
La membrana plasmatica della maggior parte delle cellule è liberamente permeabile sia al piruvato che al lattato, che possono quindi raggiungere il circolo ematico.[3] Considerando ad esempio la fibra muscolare, la quantità di lattato che lascia la cellula è maggiore rispetto a quella del piruvato grazie all’elevato rapporto NADH/NAD+ intracellulare e alle proprietà catalitiche dell’isoenzima muscolare della lattato deidrogenasi.[5]
Una volta in circolo il lattato raggiunge, tra gli altri, il fegato e la corteccia renale, dove viene ossidato a piruvato, nella reazione catalizzata dagli specifici isoenzimi della lattato deidrogenasi.
Nell’epatocita questa ossidazione è favorita dal basso rapporto NADH/NAD+ presente nel citosol. Il piruvato è quindi disponibile per entrare nella tappa successiva del ciclo di Cori, la gluconeogenesi.[3]
Il glucosio prodotto, tramite il circolo ematico raggiunge il muscolo e il globulo rosso, chiudendo così il ciclo. Ovviamente il monosaccaride raggiungerà anche tutti gli altri tessuti e cellule che lo richiedono.
Costo energetico
Il ciclo di Cori comporta un consumo netto di 4 molecole di ATP.
La parte del ciclo che comprende la gluconeogenesi consuma 6 equivalenti di ATP, nello specifico 4 ATP e 2 GTP, nelle reazioni catalizzate dagli enzimi:
piruvato carbossilasi (EC 6.4.1.1): un ATP;
fosfoenolpiruvato carbossichinasi (EC 4.1.1.32): un GTP;
gliceraldeide-3-fosfato deidrogenasi: un ATP.
Poiché sono utilizzate due molecole di lattato per la sintesi di ogni molecola di glucosio, il costo totale è di 2 x 3 = 6 legami ad alta energia per molecola di glucosio.[2]
Di contro, la parte del ciclo che comprende la glicolisi anaerobica ne produce 2.
In definitiva, è richiesta più energia per produrre glucosio dall’acido lattico nel fegato e nella corteccia renale rispetto a quella ottenuta dall’ossidazione anaerobica del glucosio nei tessuti extraepatici.
Il ciclo di Cori è un ciclo futile?
La demolizione e risintesi del glucosio caratteristica del ciclo di Cori può sembrare uno spreco di energia. In realtà questo ciclo permette l’efficace funzionamento di numerose cellule extraepatiche a spese del fegato e della corteccia renale, ed è quindi forse più corretto definirlo ciclo del substrato piuttosto che ciclo futile.[10] Di seguito alcuni esempi.
Il ciclo di Cori permette di smaltire parte dell’acido lattico che i globuli rossi producono.[8]
Se si considera il muscolo scheletrico, la glicolisi anaerobica rappresenta una efficiente sorgente di ATP nel corso di un esercizio intenso. Ma questo potrebbe portare a un accumulo intracellulare di lattato, e a una pericolosa diminuzione del pH intracellulare. Ovviamente tale accumulo non si verifica, grazie anche al ciclo di Cori, che scarica parte del lattato muscolare e del costo energetico per il suo smaltimento sui tessuti gluconeogenici.[3] Ad esempio, il debito di ossigeno, il “fiatone”, che sempre si presenta dopo un’attività fisica sostenuta, è in gran parte dovuto all’aumentata richiesta di ossigeno da parte degli epatociti per sostenere l’ossidazione degli acidi grassi, il loro principale carburante, ossidazione che porterà alla produzione dell’ATP necessario per la gluconeogenesi.[6][12]
Nel corso di traumi, sepsi, ustioni, o dopo grossi interventi chirurgici, si verifica un’intensa proliferazione cellulare nelle ferite, che sono tessuti ipossici, e nel midollo osseo. Questo a sua volta risulta in una maggiore produzione di acido lattico e un aumento del flusso attraverso il ciclo di Cori e, a livello epatico, in un consumo di ATP. Una situazione simile sembra presentarsi anche in quei pazienti oncologici che vanno incontro a una progressiva perdita di peso.[2]
Il ciclo di Cori è fondamentale anche nel digiuno.[6]
Ciclo di Cori e ciclo glucosio-alanina
Tra i due cicli esistono analogie e differenze.
Il ciclo glucosio-alanina e quello di Cori sono vie metaboliche che si estendono attraverso tipi cellulari differenti e contribuiscono ad assicurare un continuo rifornimento di glucosio ai tessuti.[12] In entrambe l’ingresso nella gluconeogenesi comporta la conversione del lattato e dell’alanina in piruvato. Infine, in entrambe, il glucosio prodotto è quindi trasportato ai tessuti periferici dove la via glicolitica rigenera piruvato.[13]
Invece, la loro principale differenza consiste nell’intermedio a tre atomi di carbonio che viene riciclato: nel ciclo di Cori il carbonio torna al fegato in forma di piruvato, mentre nel ciclo glucosio-alanina in forma di alanina.[8] I cicli differiscono anche per il destino metabolico del NADH: nel ciclo di Cori funge da agente riducente nella reazione catalizzata dalla lattato deidrogenasi, mentre nel ciclo glucosio-alanina gli elettroni del coenzima sono utilizzati per la sintesi di ATP nel mitocondrio. Pertanto, un’altra differenza è che il ciclo glucosio-alanina richiede la presenza di ossigeno, mentre non la richiede il ciclo di Cori.[8]
^ Gleeson T.T. Post-exercise lactate metabolism: a comparative review of sites, pathways, and regulation. Annu Rev Physiol 1996;58:565-81. doi:10.1146/annurev.ph.58.030196.003025
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
Gli acidi e i sali biliari sono derivati polari del colesterolo, e rappresentano la principale via per l’eliminazione dello steroide dal corpo.
Sono un gruppo di specie molecolari con struttura chimica simile ma non identica, proprietà fisiche differenti e caratteristiche biologiche anche più divergenti.
Sono sintetizzati nel fegato, immagazzinati nella cistifellea, secreti nel duodeno, e infine per la maggior parte riassorbiti nell’ileo.
Poiché a pH fisiologico sono presenti in forma di anioni, i termini acido biliare e sale biliare saranno di seguito utilizzati come sinonimi.
I sali biliari presentano analogia e differenze con la molecola del colesterolo.
Al pari dello steroide posseggono un nucleo formato da 4 anelli fusi, tre a sei atomi di carbonio, indicati come A, B e C, e uno, indicato come D, a 5; tale struttura è il ciclopentanoperidrofenantrene, più comunemente noto come nucleo steroideo.
Acidi BiIiari e Loro Coniugati
Negli vertebrati superiori sono formati da 24 atomi di carbonio, poiché hanno una coda idrocarburica più corta di tre atomi di carbonio rispetto a quella del colesterolo. Nei vertebrati inferiori sono formati a 25, 26 o 27 atomi di carbonio. La coda idrocarburica termina con un gruppo carbossilico, spesso ionizzato a pH 7, che può essere legato all’aminoacido glicina o taurina.
Oltre al gruppo ossidrilico in posizione 3, possono avere gruppi ossidrilici in posizione 7 e/o 12.
Tutto ciò rende queste molecole molto più polari del colesterolo.
Poiché gli anelli A e B sono fusi in configurazione cis, la struttura del nucleo steroideo risulta curva, ed è possibile individuarvi:
un lato concavo, verso cui sono orientati i gruppi idrossilici e il gruppo carbossilico della catena laterale, con o senza l’aminoacido a esso legato, e che risulta idrofilico;
un lato convesso, verso cui sono orientati i gruppi metilici presenti in posizione 18 e 19, che risulta idrofobico.
Rappresentazione dell’Acido Colico
Dunque, possedendo sia gruppi polari che non polari sono molecole anfifiliche e ottimi surfattanti. Tuttavia la loro struttura chimica li rende molto diversi rispetto ai surfattanti tradizionali, dove spesso si individua una testa polare e una lunga coda non polare.
Acidi e sali biliari primari, coniugati e secondari
Sono definiti acidi biliari primari le molecole sintetizzate negli epatociti direttamente dal colesterolo. Nell’uomo i principali sono l’acido colico e l’acido chenodesossicolico, che da soli formano fino all’80% di tutti gli acidi biliari.
Prima di essere secreti nell’albero biliare sono coniugati quasi per intero, sino al 98%, con gli amminoacidi glicina o taurina, a dare rispettivamente glicoconiugati e tauroconiugati. In particolare circa il 75% dell’acido colico e chenodesossicolico sono coniugati con la glicina, a dare acido glicocolico e acido glicochenodesossicolico, il restante 25% con la taurina, a dare acido taurocolico e acido taurochenodesossicolico.
Acidi Biliari Coniugati
Gli acidi biliari coniugati sono molecole dotate di gruppi maggiormente idrofilici rispetto a quelle di origine, dunque sono emulsionanti molto più efficaci. La coniugazione ha infatti l’effetto di ridurre il loro pKa facendo si che rimangano ionizzate in un intervallo più ampio di pH. Se infatti il pKa tipico dei non coniugati è di circa 6, si passa a circa 4 con l’acido glicocolico, e circa 2 con l’acido taurocolico.
L’idrofilicità dei comuni acidi e sali biliari decresce secondo il seguente ordine: coniugati della glicina < coniugati della taurina < acido litocolico < acido desossicolico < acido chenodesossicolico < acido colico < acido ursodesossicolico.
Infine la coniugazione ha anche l’effetto di ridurre la citotossicità delle molecole di origine.
Gli acidi biliari secondari derivano dai primari che non sono stati riassorbiti durante il loro passaggio nell’intestino tenue. Una volta raggiunto il colon, gli acidi biliari primari possono infatti subire diverse modificazioni ad opera del microbiota intestinale, che è parte del più ampio microbiota umano, a dare appunto gli acidi biliari secondari. Con gli acidi grassi a catena corta sono sono i due principali tipi di metaboliti batterici prodotto nel colon, e formano il il restante 20% del pool corporeo totale degli acidi biliari.
Un altro modo di suddividere i sali biliari fa riferimento alla loro coniugazione con aminoacidi e al grado di idrossilazione. Su queste base si individuano tre categorie.
I coniugati triidrossilati, quali l’acido taurocolico e il glicocolico.
I coniugati diidrossilati, come l’acido glicodesossicolico, glicochenodesossicolico, taurochenodesossicolico e taurodesossicolico. Nella bile rappresentano circa il 60% del totale dei sali biliari.
Forme non coniugate come l’acido colico, desossicolico, chenodesossicolico, e litocolico.
Funzione dei sali biliari
Premessa: tutte le funzioni fisiologiche sono portate a termine dai sali biliari in forma coniugata.
Rappresentano la via principale per l’eliminazione del colesterolo. Nell’uomo infatti non esiste il corredo enzimatico necessario per rompere nessuno dei quattro anelli del nucleo steroideo, ne per ossidare il colesterolo ad anidride carbonica e acqua.
L’altro modo per eliminare il colesterolo è con la bile, in forma libera.
I sali biliari sono potenti surfattanti. E in particolare, i coniugati di- e triidrossilati sono i surfattanti migliori, molto più efficaci rispetto ai corrispettivi non coniugati, avendo un numero maggiore di gruppi polari.
Una volta a contatto con i lipidi apolari nel lume del piccolo intestino, il lato convesso apolare va a interagire con i lipidi idrofobici, quali trigliceridi, esteri del colesterolo e delle vitamine liposolubili, mentre il lato concavo polare prende contatto con il mezzo acquoso circostante. Ciò incrementa la dispersione nel mezzo acquoso dei lipidi apolari, favorendo la formazione di minuscole goccioline lipidiche, che subiranno l’attacco delle lipasi, in particolare della lipasi pancreatica, nella cui attivazione i sali biliari hanno un ruolo diretto, e delle esterasi intestinali. In seguito facilitano l’assorbimento dei lipidi prodotti, nonché delle vitamine liposolubili, ad opera della mucosa intestinale grazie alla formazione di micelle miste.
Una funzione simile è svolta nella cistifellea dove, formando micelle miste con i fosfolipidi, prevengono la precipitazione del colesterolo.
Nota: grazie alla disposizione dei gruppi polari e non polari, una volta in soluzione acquosa, i sali biliari tendono a formare micelle, di solito composte da meno di 10 monomeri, purché la loro concentrazione sia superiore alla cosiddetta concentrazione micellare critica.
A livello intestinale modulano la secrezione degli enzimi pancreatici e della colecistochinina.
Sia nell’intestino tenue che nel colon hanno una potente attività antimicrobica, in primis l’acido desossicolico, in particolare contro i batteri Gram-positivi. Questa attività potrebbe essere dovuta a danno ossidativo al DNA e/o al danno alle membrane cellulari batteriche. Sono dunque importanti nella prevenzione della sovracrescita batterica, ma sembra abbiano anche un ruolo nella regolazione della composizione del microbiota intestinale.
Negli ultimi anni è divenuto evidente il loro ruolo regolatorio sul controllo del metabolismo energetico, in particolare per la “movimentazione” epatica del glucosio.
Circolo enteroepatico
A seguito del consumo di lipidi con la dieta, le cellule enteroendocrine del duodeno secernono in circolo la colecistochinina. Il successivo legame dell’ormone alle cellule muscolari lisce della parete della cistifellea ne promuove la contrazione; inoltre l’ormone causa anche il rilascio dello sfintere di Oddi. Da tutto ciò risulta la secrezione pulsatile della bile, e degli acidi biliari in essa contenuti, nel duodeno.
In condizioni fisiologiche il pool corporeo di acidi biliari è costante, e pari a circa 3-5 g; ciò è reso possibile da due processi:
il loro riassorbimento a livello intestinale;
la loro sintesi de novo.
Fino al 95% dei sali biliari secreti viene riassorbito a livello intestinale, non assieme ai prodotti della digestione dei lipidi, ma attraverso un processo definito circolo enteroepatico.
Si tratta di un sistema di recupero estremamente efficiente, che sembra avvenire almeno due volte per ogni pasto, cui partecipano il fegato, l’albero biliare, il duodeno, il colon, e il circolo portale attraverso cui le molecole riassorbite tornano al fegato. Tale ricircolo è reso necessario dal fatto che la capacità dell’epatocita di produrre acidi biliari è limitata e insufficiente a soddisfare le necessità fisiologiche intestinali se gli stessi sali andassero perduti in elevate quantità.
La maggior parte dei sali biliari è riassorbita una volta raggiunto l’ileo distale, la parte più bassa dell’intestino tenue, a mezzo di un trasportatore sodio-dipendente presente nell’orletto a spazzola degli enterociti, detto ASBT, acronimo dell’inglese apical sodium-dependent bile acid transporter, che opera un cotrasporto di due ioni sodio e un acido biliare.
Una volta nell’enterocita si ritiene che, a mezzo della proteina IBABP, acronimo dell’inglese ileal bile acid-binding protein, siano trasportati attraverso il citosol alla membrana basolaterale, che attraversano grazie all’intervento del trasportatore OSTalfa/OSTbeta, acronimo dell’inglese organic solute transporter alpha and beta. Tramite il circolo portale, veicolati dall’albumina, raggiungono il fegato.
Da notare che una piccola parte di acidi biliari raggiunge il fegato attraverso l’arteria epatica.
A livello epatico la loro estrazione dal circolo è molto efficiente, tanto che dal 50 al 90% sono rimossi al primo passaggio, percentuale che varia in funzione della struttura molecolare. Gli acidi biliari coniugati sono in gran parte rimossi attraverso un meccanismo di trasporto attivo sodio-dipendente, a mezzo del trasportatore NTCP, acronimo dell’inglese Na+-dependent taurocholate co-transport polipeptide. Tuttavia può avvenire anche un trasporto sodio-indipendente ad opera di proteine della famiglia OATP, acronimo dell’inglese organic anion transporting polypeptides, principalmente le isoforme OATP1B1 e OATP1B3.
Nel circolo enteroepatico il passaggio limitante è rappresentato dalla loro secrezione nei canalicoli biliari, in gran parte ad opera di BSEP, acronimo dell’inglese bile salt export pump, in un processo ATP-dipendente. Questa pompa trasporta gli acidi biliari monoanionici, che sono la maggior parte. Gli acidi biliari solforati o glucuronati, dianionici, sono secreti a mezzo di trasportatori differenti, quali MRP2 e BCRP.
Nota: il livello sierico degli acidi biliari varia sulla base della velocità di riassorbimento e quindi è più alto durante i pasti, quando il circolo enteroepatico è più attivo.
Metabolismo intestinale
Gli acidi biliari che sfuggono al riassorbimento ileale passano nel colon dove in parte subiscono l’azione di enzimi della flora batterica e sono trasformati in acidi biliari secondari.
Di seguito sono elencate le principali reazioni.
Deconiugazione
A livello della catena laterale si può verificare l’idrolisi del legame con l’aminoacido coniugato in posizione 24, con liberazione di acidi biliari non coniugati e glicina o taurina. Le reazioni sono catalizzate da idrolasi batteriche presenti sia nell’intestino tenue che nel colon.
7alfa-Deidrossilazione
E’ la reazione quantitativamente più importante, portata a termine da deidratasi batteriche coloniche che rimuovono il gruppo ossidrilico in posizione 7 dando origine ad un 7-deossi acido biliare. In particolare, dall’acido colico si formerà l’acido desossicolico, mentre dal chenodesossicolico il litocolico, due acidi biliari secondari tossici.
Da notare che la 7alfa-deidrossilazione, a differenza di quanto accade con l’ossidazione e l’epimerizzazione, può avvenire solamente sugli acidi biliari non coniugati, per cui la deconiugazione è un prerequisito essenziale.
Ossidazione ed epimerizzazione
Sono reazioni che interessano i gruppi idrossilici in posizione 3, 7 e 12, catalizzate da idrossisterolo deidrogenasi batteriche. Ad esempio, l’epimerizzazione dell’acido chenodesossicolico da origine all’acido ursodesossicolico.
Metabolismo Intestinale degli Acidi Biliari
Parte degli acidi biliari secondari sono poi riassorbiti e tornano al fegato, per essere riconiugati, se necessario, e secreti nuovamente. Quelli che invece sfuggono al riassorbimento saranno perduti con le feci.
Mentre le reazioni di deconiugazione e ossidazione sono portate a termine da un ampio spettro di batteri anaerobi, le 7alfa-deidrossilazioni sono effettuate da un numero ristretto di anaerobi.
La 7alfa-deidrossilazione e la deconiugazione hanno come effetto quello di aumentare il pKa e dunque l’idrofobicità degli acidi biliari, permettendone un certo grado di recupero passivo attraverso l’epitelio colonico.
L’aumento di idrofobicità è associato anche ad un aumento della loro citotossicità. E una elevata concentrazione di acidi biliari secondari nelle feci, sangue e bile è stata associata alla patogenesi del cancro al colon.
Riassorbimento e fibre solubili
Il riassorbimento degli sali biliari può essere ridotto dall’azione chelante delle fibre solubili, come quelle presenti nella frutta fresca, legumi, avena e crusca d’avena. Tutto questo ha come effetto quello di incrementare la loro sintesi de novo, up-regolando l’espressione della colesterolo 7alfa-idrossilasi e della sterolo 12alfa-idrossilasi, e quindi ridurre la concentrazione del colesterolo negli epatociti.
La deplezione del colesterolo epatico aumenta l’espressione del recettore per le LDL, e quindi abbassa la concentrazione plasmatica del colesterolo LDL. Di contro però stimola anche la sintesi della HMG-CoA reduttasi, l’enzima chiave nella sintesi dello steroide.
Anche diversi farmaci agiscono legando gli acidi biliari a livello intestinale, impedendone così il riassorbimento.
Sintesi
Dal punto di vista quantitativo, gli acidi biliari sono il prodotto principale del metabolismo del colesterolo.
Come detto in precedenza, il circolo enteroepatico e la loro sintesi de novo assicurano la costanza del pool corporeo. In particolare, la sintesi de novo permette la sostituzione di quel 5-10%, circa 0,5 g/die, che viene perduto con le feci.
Di seguito verrà presa in esame la sintesi dell’acido colico e dell’acido chenodesossicolico, e la loro coniugazione con gli aminoacidi taurina e glicina.
Esistono due vie principali per la sintesi dei suddetti acidi: la via classica e quella alternativa. A queste si aggiungono alcune vie minori.
Sintesi de Novo degli Acidi Biliari Primari e dei loro Coniugati
La via classica o neutra
Nell’uomo fino al 90% dei sali biliari sono sintetizzati attraverso la via classica, definita anche “neutra” in quanto i suoi intermedi sono steroli neutri.
E’ una via presente solo nel fegato, cui prendono parte numerosi enzimi localizzati nel citosol, nel reticolo endoplasmatico, nei mitocondriale e perossisomi, e i cui prodotti finali sono i coniugati degli acidi colico e chenodesossicolico.
La prima reazione è l’idrossilazione in posizione 7 del colesterolo, a dare il 7alfa-idrossicolesterolo. La reazione è catalizzata dalla colesterolo 7alfa-idrossilasi o CYP7A1 (E.C. 1.14.14.23). E’ un enzima localizzato sul reticolo endoplasmatico, e catalizza il passaggio limitante dell’intera via.
Il 7alfa-idrossicolesterolo subisce l’ossidazione del gruppo 3beta-ossidrilico e lo spostamento del doppio legame dalla posizione 5,6 alla posizione 4,5 a dare il 7alfa-idrossi-4-colesten-3-one. La reazione è catalizzata dalla 3beta-idrossi-Δ5-C27-steroide ossidoreduttasi o HSD3B7 (E.C. 1.1.1.181), un enzima del reticolo endoplasmatico.
Il 7alfa-idrossi-4-colesten-3-one può seguire due vie:
entrare nella via che porta alla sintesi dell’acido colico, attraverso la reazione catalizzata dalla 7alfa-idrossi-4-colesten-3-one 12alfa-monoossigenasi o sterolo 12alfa-idrossilasi o CYP8B1 (E.C. 1.14.18.8), enzima del reticolo endoplasmatico;
entrare nella via che porta la sintesi dell’acido chenodesossicolico, attraverso la reazione catalizzata dalla 3-osso-Δ4-steroide 5β-reduttasi o AKR1D1 (E.C. 1.3.1.3), enzima citosolico.
Ed è l’attività della sterolo 12alfa-idrossilasi che determinerà il rapporto tra gli acidi colico e chenodesossicolico prodotti, e, in definitiva, quella che sarà la potenza detergente del pool degli acidi biliari. E infatti la regolazione della trascrizione del gene corrispondente è uno dei punti di regolazione principali dell’intera via di sintesi.
Dunque, se il 7alfa-idrossi-4-colesten-3-one fluisce attraverso la reazione catalizzata dalla sterolo 12alfa-idrossilasi si avranno le seguenti reazioni.
La molecola è idrossilata in posizione 12 dall’enzima suddetto, a dare il 7alfa,12alfa-diidrossi-4-colesten-3-one.
Il 7alfa,12alfa-diidrossi-4-colesten-3-one subirà la riduzione del doppio legame in posizione 4,5, nella reazione catalizzata dalla 3-osso-Δ4-steroide 5beta-reduttasi, a dare 5beta-colestan-7alfa,12alfa-diol-3-one.
Il 5beta-colestan-7alfa,12alfa-diol-3-one subirà a sua volta la riduzione a gruppo ossidrilico del gruppo in posizione 4, nella reazione catalizzata dalla 3alfa-idrossisteroide deidrogenasi o AKR1C4 (EC 1.1.1.213), enzima citosolico, a dare 5beta-colestan-3alfa,7alfa,12alfa-triolo.
Il 5beta-colestan-3alfa,7alfa,12alfa-triolo subirà l’ossidazione della catena laterale in tre reazioni catalizzate dalla sterolo 27-idrossilasi o CYP27A1 (EC 1.14.15.15). Si tratta di un enzima mitocondriale presente anche in tessuti extraepatici e nei macrofagi, che introduce un gruppo ossidrilico in posizione 27. Il gruppo ossidrilico è poi ossidato ad aldeide e quindi ad acido carbossilico, con formazione di acido 3alfa,7alfa, 12alfa-triidrossi-5beta-colestanoico.
L’acido 3alfa,7alfa, 12alfa-triidrossi-5beta-colestanoico è attivato a seguito del legame con il coenzima A, nella reazione catalizzata dagli enzimi BACS, acronimo dell’inglese bile acid CoA synthetase (EC 6.2.1.7), o VLCS, acronimo dell’inglese very long chain acyl CoA synthetase (EC 6.2.1.-), entrambe localizzati nel reticolo endoplasmatico.
Il 3alfa,7alfa,12alfa-triidrossi-5beta-colestanoilCoA è trasportato nei perossisomi dove va incontro a 5 reazioni consecutive, catalizzate da altrettanti enzimi differenti. Nelle ultime due la catena laterale è accorciata a quattro atomi di carbonio e viene prodotto il colilCoA.
L’ultimo passaggio coinvolge la coniugazione, attraverso legame ammidico, del gruppo carbossilico terminale della catena laterale con l’aminoacido glicina o taurina, nella reazione catalizzata da BAAT, acronimo dell’inglese bile acid-CoA:amino acid N-acyltransferase (EC 2.3.1.65), enzima a localizzazione prevalentemente perossisomiale.
I prodotti della reazione saranno quindi gli acidi biliari coniugati glicocolico e taurocolico.
Nel caso in cui il 7alfa-idrossi-4-colesten-3-one non entri nella reazione catalizzata dalla sterolo 12alfa-idrossilasi prenderà la via che porta alla sintesi dei coniugati dell’acido chenodesossicolico attraverso le reazioni di seguito descritte.
Il 7alfa-idrossi-4-colesten-3-one viene convertito in 7alfa-idrossi-5β-colestan-3-one nella reazione catalizzata dall’enzima 3-osso-Δ4-steroide 5beta-reduttasi.
Il 7alfa-idrossi-5β-colestan-3-one nella reazioni dalla 3alfa-idrossisteroide deidrogenasi, viene convertito in 5beta-colestan-3alfa,7alfa-diolo.
Quindi, a seguito di modificazione analoghe a quelle viste per la sintesi dei coniugati dell’acido colico, e catalizzate per la maggior parte dagli stessi enzimi, si formeranno gli acidi biliari coniugati glicochenodesossicolico e taurochenodesossicolico.
Nota: gli acidi biliari deconiugati a livello intestinale dovranno raggiungere il fegato per essere riconiugati.
La via alternativa o acida
E’ prevalente nel feto e nel neonato, mentre nell’adulto porta alla sintesi di meno del 10% dei sali biliari.
Questa via si differenzia da quella classica, in quanto:
i prodotti intermedi sono acidi, per cui viene definita anche come “via acida”;
l’ossidazione della catena laterale precede le modificazioni del nucleo steroideo;
i prodotti finali sono i coniugati dell’acido chenodesossicolico.
Il primo passaggio comporta la conversione del colesterolo in 27-idrossicolesterolo nella reazione catalizzata dalla sterolo 27-idrossilasi.
A questo punto sono possibili due vie.
Via A
In una reazione catalizzata dalla sterolo 27-idrossilasi, il 27-idrossicolesterolo è convertito in acido 3beta-idrossi-5-colestenoico.
L’acido 3beta-idrossi-5-colestenoico nella reazione catalizzata dalla ossisterolo 7α-idrossilasi o CYP7B1 (EC 1.14.13.100), un enzima del reticolo endoplasmatico, viene idrossilato in posizione 7 a dare l’acido 3beta-7alfa-diidrossi-5-colestenoico.
L’acido 3beta-7alfa-diidrossi-5-colestenoico viene convertito in acido 3-osso-7alfa-idrossi-4-colestenoico, nella reazione catalizzata dalla 3beta-idrossi-Δ5-C27-steroide ossidoreduttasi.
L’acido 3-osso-7alfa-idrossi-4-colestenoico, a seguito di modificazioni della catena laterale, darà origine all’acido chenodesossicolico e quindi ai suoi coniugati.
Via B
Il 27-idrossicolesterolo è convertito in 7alfa,27-diidrossicolesterolo nella reazione catalizzata dalla ossisterolo 7alfa-idrossilasi e colesterolo 7alfa-idrossilasi.
Il 7alfa,27-diidrossicolesterolo è convertito in 7alfa,26-diidrossi-4-colesten-3-one nella reazione catalizzata dalla 3beta-idrossi-Δ5-C27-steroide ossidoreduttasi.
Il 7alfa,26-diidrossi-4-colesten-3-one può ora proseguire come tale o essere convertito in acido 3-osso-7alfa-idrossi-4-colestenoico e subire le modifiche alla catena laterale e le altre reazioni che portano alla sintesi dei coniugati dell’acido chenodesossicolico.
Le vie minori
Esistono anche vie minori che possono contribuire, sebbene in misura ridotta, alla sintesi degli acidi biliari.
Ad esempio:
Nel fegato viene espressa una colesterolo 25-idrossilasi (EC 1.14.99.38).
Nel cervello è espressa una colesterolo 24- idrossilasi o CYP46A1 (EC 1.14.14.25), e quindi, sebbene l’organo non sia in grado di esportare colesterolo, può esportare ossisteroli.
E’ stata scoperta anche una 7alfa-idrossilasi non specifica espressa in tutti i tessuti, che sembra essere coinvolta nella generazione di ossisteroli che possono poi essere trasportati al fegato per essere convertiti in acido chenodesossicolico.
Inoltre, la sterolo 27-idrossilasi è espressa in vari tessuti, per cui i suoi prodotti di reazione possono essere trasportati al fegato ed essere convertiti in sali biliari.
Regolazione della sintesi degli acidi biliari
La regolazione avviene attraverso un feedback negativo, in particolare sull’espressione degli enzimi colesterolo 7alfa-idrossilasi e sterolo 12alfa-idrossilasi.
In caso di eccesso di acidi biliari, sia coniugati che liberi, gli stessi si vanno a legare al recettore nucleare FRX, acronimo dell’inglese farnesoid X receptor, attivandolo. L’acido biliare più efficace nell’attivare FRX è l’acido chenodesossicolico, mentre altri, come l’acido ursodesossicolico non lo attivano.
FRX induce l’espressione del repressore trascrizionale SHP, acronimo dell’inglese small heterodimer partner, che a sua volta interagisce con altri fattori trascrizionali, come LRH-1, acronimo dell’inglese liver receptor homolog-1, e HNF-4alfa, acronimo dell’inglese hepatocyte nuclear factor-4alfa, che si legano ad una sequenza presente nella regione del promotore dei geni per la 7alfa-idrossilasi e 12alfa-idrossilasi, regione definita BAREs, acronimo dell’inglese bile acid response elements, inibendone la trascrizione.
Uno dei motivi per cui la sintesi dei sali biliari è strettamente regolata è perché molti dei loro metaboliti sono tossici.
Bibliografia
Chiang J.Y.L. Bile acids: regulation of synthesis. J Lipid Res 2009;50(10):1955-1966. doi:10.1194/jlr.R900010-JLR200
Gropper S.S., Smith J.L. Advanced nutrition and human metabolism. 6th Edition. Cengage Learning, 2012
Moghimipour E., Ameri A., and Handali S. Absorption-enhancing effects of bile salts. Molecules 2015;20(8); 14451-14473. doi:10.3390/molecules200814451
Monte M.J., Marin J.J.G., Antelo A., Vazquez-Tato J. Bile acids: Chemistry, physiology, and pathophysiology. World J Gastroenterol 2009;15(7):804-816. doi:10.3748/wjg.15.804
Rosenthal M.D., Glew R.H. Medical biochemistry – Human metabolism in health and disease. John Wiley J. & Sons, Inc., Publication, 2009
Sundaram S.S., Bove K.E., Lovell M.A. and Sokol R.J. Mechanisms of Disease: inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol 2008;5(8):456-468. doi:10.1038/ncpgasthep1179
L’alfa-amilasi (EC 3.2.1.1) è un enzima che catalizza l’idrolisi di legami glicosidici α-(1→4) presenti all’interno di catene lineari di amilosio e amilopectina, i due principali costituenti dei granuli di amido.[2]
L’enzima è ampiamente diffuso negli animali, piante, funghi, nei phyla Ascomiceti e Basidiomiceti, batteri, nel genere Bacillus, e negli Archea. La sua ampia distribuzione suggerisce un suo ruolo centrale nel metabolismo dei carboidrati.[15]
Nell’uomo l’alfa-amilasi è codificata da due geni differenti, ed è prodotta principalmente delle ghiandole parotidi e dal pancreas esocrino.[9] Nelle piante sei differenti geni codificano per altrettante isoforme dell’enzima.[11]
Negli animali è coinvolta nel primo passaggio della digestione dell’amido, con liberazione del disaccaride maltosio, degli oligosaccaridi maltotriosio e destrine alfa-limite, e di piccole quantità di glucosio.[5]
Poiché l’amido è la principale fonte di alimentare di carboidrati, e quindi di glucosio ed energia, l’attività catalitica dell’alfa-amilasi può influenzare la regolazione della glicemia postprandiale.[8]
L’alfa-amilasi è utilizzata in diversi settori industriali, nonché nella diagnostica clinica.[7]
Nelle piante, e in particolare nelle graminacee quali grano, orzo e riso, è cruciale per la germinazione dei semi e la maturazione dei chicchi.[11][17]
La prima rilevazione di un’attività amilasica risale al 1811, a opera del chimico russo Konstantin Sigizmundovič Kirchhoff, che, lavorando con il grano, scoprì la prima attività enzimatica capace di degradare l’amido del cereale.[16]
A questo lavoro seguirono, nel 1831, gli studi Erhard Friedrich Leuchs che osservò la degradazione dell’amido quando veniva miscelato con la saliva umana. Leuchs chiamò ptialina l’agente responsabile di questa reazione chimica.
Nel 1833, Anselme Payen e Jean-François Persoz, chimici francesi che lavoravano in uno zuccherificio, isolarono dall’orzo un enzima in grado di degradare l’amido, enzima che fu chiamato distasi.[5]
Il nome di alfa-amilasi si deve a Richard Kuhn, che nel 1925 scoprì che i prodotti dell’idrolisi catalizzata dall’enzima erano in configurazione alfa.[13]
L’alfa-amilasi fu anche il primo enzima a essere prodotto industrialmente, nel 1894, estratto da un fungo e utilizzato per il trattamento di disordini digestivi. In seguito, a partire dal 1917, venne estratto anche da Bacillus subtilis e Bacillus mesentericus. Attualmente la maggior parte viene prodotta da organismi geneticamente modificati.[15]
L’alfa-amilasi, e più in generale le amilasi, di cui ne esistono almeno altri due tipi, la beta-amilasi (EC 3.2.1.2) e la gamma-amilasi (EC 3.2.1.3), trovano largo impiego in settori industriali differenti, come quello alimentare, dei mangimi, il settore tessile, la produzione delle birra e del whisky, l’industria farmaceutica, cosmetica e la diagnostica clinica.[7]
Le alfa-amilasi possono essere classificate, sulla base del grado d’idrolisi del substrato, come liquefanti, se idrolizzano il 30-40% dei legami glicosidici dell’amido, e saccarificanti, se l’idrolisi arriva al 50-60%.[15]
Geni dell’alfa-amilasi umana
In molti mammiferi, compreso l’uomo, l’alfa-amilasi è codificata da due geni distinti ma strettamente correlati, presenti sul braccio corto del cromosoma 1, e indicati come:
AMY1, che codifica per l’isoforma presente nella saliva, nella ghiandola mammaria, e in alcuni tipi di tumore, come quello del polmone;[5]
AMY2, che codifica per l’enzima prodotto dal pancreas. AMY2 è presente in due forme, AMY2A e AMY2B, il cui numero di copie per gene differisce nelle diverse popolazioni, sebbene due copie per gene sembra essere un pattern frequente. Sembra anche che il numero di copie di AMY2 sia associato a quello di AMY1.[9]
Azione catalitica
L’alfa-amilasi catalizza il primo passaggio nella digestione dell’amido, vale a dire l’idrolisi dei legami glicosidici α-(1→4) presenti all’interno delle catene lineari di amilosio e amilopectina, agendo su posizioni casuali lungo gli α-glucani. L’enzima è quindi una endoglicosidasi, ha un pH ottimale di circa 7, richiede la presenza di ioni calcio, e porta al rilascio di disaccaridi e oligosaccaridi in configurazione alfa, nello specifico, maltosio e maltotriosio dall’amilosio, e maltosio, destrine alfa-limite e glucosio, quest’ultimo in minima quantità, dall’amilopectina.[2]
Alfa-amilasi
L’alfa-amilasi, al pari di altri enzimi coinvolti nel metabolismo dell’amido, come la pullulanasi (EC 3.2.1.41), la isoamilasi (EC 3.2.1.68), gli enzimi ramificanti l’amido (EC 2.4.1.18) e gli enzimi deramificanti l’amido, appartiene alla famiglia delle glicosil idrolasi 13 o GH-13, enzimi in grado di agire su substrati contenenti legami α-glicosidici.[10]
A questa famiglia appartengono anche due enzimi coinvolti nel metabolismo del trealosio, la trealosio sintasi (EC 5.4.99.16), che catalizza l’interconversione tra maltosio e trealosio, e la trealosio-6-fosfato idrolasi (EC 3.2.1.93), che idrolizza il trealosio a dare una molecola di glucosio e una di glucosio-6-fosfato.
Alfa-amilasi salivare
L’alfa-amilasi salivare o ptialina è sintetizzata principalmente dalle ghiandole parotidi, e in misura minore dalle ghiandole sottomandibolari.
Presente in due forme, una glicosilata e una non glicosilata, è coinvolta nella prima, limitata, digestione dell’amido che avviene nella cavità orale.
L’enzima è una delle proteine salivari più abbondanti, rappresentando il 40-50% di tutte quelle presenti, e, con la lipasi linguale (EC 3.1.1.3), coinvolta nella digestione dei lipidi, quali i trigliceridi e gli esteri del colesterolo, è uno dei più importanti enzimi salivari.[3]
Alfa-amilasi pancreatica
L’alfa-amilasi pancreatica è prodotta dalle cellule acinose del pancreas.
E’ una delle principali proteine del succo pancreatico, e con esso viene secreta nel duodeno dove attacca amilosio e amilopectina, portando a termine la prima parte dell’amilolisi. I prodotti dell’idrolisi sono quindi attaccati dalle alfa-glucosidasi dell’orletto a spazzola degli enterociti, ossia la saccarasi-isomaltasi (EC 3.2.1.48) e la maltasi-glucoamilasi (EC 3.2.1.20), enzimi che idrolizzano anche il legame glicosidico α-(1→2) del saccarosio.[14]
L’alfa-amilasi pancreatica dei mammiferi è in grado di legarsi a N-glicani di glicoproteine dell’orletto a spazzola.[1] Questo legame consente la cooperazione con la saccarasi-isomaltasi, il che determina un notevole aumento del rilascio di glucosio. Tuttavia è stato osservato che l’uptake del glucosio da parte di SGLT1, uno dei trasportatori intestinali coinvolti nell’assorbimento dei monosaccaridi, è inibito da elevate concentrazioni intestinali di alfa-amilasi. E’ stato ipotizzato che questo processo possa rappresentare un meccanismo di regolazione innata per evitare improvvisi aumenti dell’assorbimento del glucosio, e quindi facilitare la regolazione della glicemia postprandiale.[6]
Controllo della glicemia
Il controllo della glicemia postprandiale è ritenuto una valida strategia nella prevenzione dello sviluppo del diabete di tipo II. Tra le metodiche non farmacologiche che è possibile adottare c’è la riduzione della capacità dell’alfa-amilasi di attaccare l’amido attraverso una lavorazione degli alimenti che porti alla retrogradazione dell’amido stesso, o a mezzo di molecole presenti nei cibi che, con diverse modalità d’azione, ne riducano l’attività.[8]
Lavorazione dei cibi
I processi idrotermici cui i cibi ricchi di amido sono sottoposti, ad esempio la cottura in acqua bollente, fanno si che i granuli di amido vadano incontro al fenomeno della gelatinizzazione, ossia assorbano acqua, si rigonfino e perdano buona parte delle loro strutture cristalline, sostituite da strutture amorfe, più facilmente attaccabili dalle alfa-amilasi.[4] Se l’amido gelatinizzato viene fatto raffreddare si può verificare una ricristallizzazione che porta alla formazione di quello che è conosciuto come amido retrogradato, un tipo di amido resistente all’azione dell’alfa-amilasi e che è in grado di raggiungere praticamente intatto il colon, dove viene fermentato dai batteri del microbiota intestinale, al pari della fibra solubile.
Ruolo dei polifenoli
La digestione dell’amido da parte dell’alfa-amilasi è influenzata da molecole presenti negli alimenti, quali i polisaccaridi non amidacei della parete cellulare, proteine, lipidi, e, tra i fitochimici, diversi tipi di polifenoli.[4] Questi composti possono limitare l’azione dell’enzima in almeno quattro modi differenti.[8]
Le proteine e i polisaccaridi non amidacei sono in grado di limitare la perdita della struttura ordinata dell’amido nativo nel corso dei processi idrotermici cui è sottoposto il cibo durante la lavorazione.
I lipidi, i polifenoli, le proteine e i polisaccaridi non amidacei possono formare strutture ordinate con l’amido.
Le proteine, i polisaccaridi non amidacei e i polifenoli possono formare barriere fisiche che riducono l’accesso degli enzimi idrolitici alle catene degli alfa-glucani.
I polifenoli e i polisaccaridi non amidacei sono in grado di ridurre l’attività dell’alfa-amilasi e delle alfa-glucosidasi.
Tra i polifenoli sembrano essere importanti alcuni tipi di flavonoidi come le proantocianidine o tannini condensati.[8] Per essere efficace, l’assunzione di alimenti ricchi di polifenoli, come il tè nero o il te verde, deve essere associata al pasto ricco in carboidrati. Il glutine sembra in grado di ostacolare l’interazione tra polifenoli e granuli di amido, e dunque ridurne l’effetto inibitorio.
Dosaggio dell’amilasi sierica
Il dosaggio dell’attività sierica dell’alfa-amilasi viene utilizzato come test diagnostico di laboratorio.[7]
L’aumento dei livelli sierici dell’attività dell’enzima può derivare da:
una pancreatite acuta o una riacutizzazione di una pancreatite cronica;
un’infiammazione a carico delle ghiandole parotidi;
un’ostruzione dei dotti secretori delle ghiandole parotidi o del dotto pancreatico.
I test attualmente in uso non distinguono tra le isoforme salivare e pancreatica. Tuttavia, se l’aumento dell’attività amilasica è associato a quello dell’attività lipasica, la patologia sottostante è probabilmente di origine pancreatica.
Alfa-amilasi del grano
Nelle piante, e in particolare nel grano, l’alfa-amilasi ha un ruolo nella germinazione e nel processo di maltazione, ossia una germinazione controllata che porta alla conversione del grano in malto che viene utilizzato principalmente per la produzione di whisky e birra.[11]
Nei chicchi di grano l’enzima viene sintetizzato principalmente nelle prime fasi della germinazione, che sono eterotrofiche in quanto dipendono fortemente dalle risorse immagazzinate, e quindi anche sull’amido presente nell’endosperma.[17] La sintesi dell’enzima avviene nello strato aleuronico, e da inizio alla mobilizzazione delle riserve di amido.
L’alfa-amilasi è coinvolta anche in uno dei principali difetti del grano, la germinazione pre-raccolta.
Bibliografia
^ Asanuma-Date K., Hirano Y., Le N., Sano K., Kawasaki N., Hashii N., Hiruta Y., Nakayama K., Umemura M., Ishikawa K., Sakagami H., and Ogawa H. Functional regulation of sugar assimilation by N-glycan-specific interaction of pancreatic α-amylase with glycoproteins of duodenal brush border membrane. J Biol Chem 2012;287(27):23104-18. doi:10.1074/jbc.m111.314658
^ ab Benjamin S., Smitha R.B., Jisha V.N., Pradeep S., Sajith S., Sreedevi S., Priji P., Unni K.N., Sarath Josh M.K. A monograph on amylases from Bacillus spp. Adv biosci biotechnol 2013;4:No.2. doi:10.4236/abb.2013.42032
^ Boehlke C., Zierau O., Hannig C. Salivary amylase – The enzyme of unspecialized euryphagous animals. Arch Oral Biol 2015;60(8):1162-76. doi:10.1016/j.archoralbio.2015.05.008
^ ab Butterworth P.J., Bajka B.H., Edwards C.H., Warren F.J., Ellis P.R. Enzyme kinetic approach for mechanistic insight and predictions of in vivo starch digestibility and the glycaemic index of foods. Trends Food Sci Technol 2022;120:254-264. doi:10.1016/j.tifs.2021.11.015
^ abc Butterworth P.J., Warren F.J. and Ellis P.R. Human α-amylase and starch digestion: an interesting marriage. Starch/Stärke 2011;63:395-405. doi:10.1002/star.201000150
^ Date K., Satoh A., Lida K., Ogawa H. Pancreatic α-amylase controls glucose assimilation by duodenal retrieval through N-glycan-specific binding, endocytosis, and degradation. J Biol Chem 2015;290(28):17439-50. doi:10.1074/jbc.M114.594937
abc de Souza P.M., de Oliveira Magalhães P. Application of microbial α-amylase in industry – A review. Braz J Microbiol 2010;41(4):850-61. doi:10.1590/S1517-83822010000400004
^ abcd Gong L., Feng D., Wang T., Ren Y., Liu Y., Wang J. Inhibitors of α-amylase and α-glucosidase: potential linkage for whole cereal foods on prevention of hyperglycemia. Food Sci Nutr 2020;8(12):6320-6337. doi:10.1002/fsn3.1987
^ ab Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W, Lee C. Detection of large-scale variation in the human genome. Nature Genetics. 2004;36:949–951. doi:10.1038/ng1416
^ Janeček Š., Svensson B., MacGregor E.A. α-Amylase: an enzyme specificity found in various families of glycoside hydrolases. Cell Mol Life Sci 2014;71(7):1149-70. doi:10.1007/s00018-013-1388-z
^ abc Ju L., Pan Z., Zhang H., Li Q., Liang J., Deng G., Yu M., Long H. New insights into the origin and evolution of α-amylase genes in green plants. Sci Rep 2019;9(1):4929. doi:10.1038/s41598-019-41420-w
^ Keller P.J., Allan B.J. The protein composition of human pancreatic juice. J Biol Chem 196725;242(2):281-7. doi:10.1016/S0021-9258(19)81461-8
^ Kuhn R. Mechanism of the action of amylases. Constitution of starch. Justus Liebigs Annalen der Chemie 1925;443:1-71
^ Nichols B.L., Avery S., Sen P., Swallow D.M., Hahn D., Sterchi E. The maltase-glucoamylase gene: common ancestry to sucrase-isomaltase with complementary starch digestion activities. Proc Natl Acad Sci U S A. 2003 Feb 4;100(3):1432-7. doi:10.1073/pnas.0237170100
^ abc Tiwari S.P., Srivastava R., Singh C.S., Shukla K., Singh R.K., Singh P., Singh R., Singh N.L., and Sharma R. Amylases: an overview with special reference to alpha amylase. Journal of Global Biosciences 2015;4(SI1):1886-1901.
^ Zakowski J.J., Bruns D.E. Biochemistry of human alpha amylase isoenzymes. Crit Rev Clin Lab Sci 1985;21(4):283-322. doi:10.3109/10408368509165786
^ ab Zhang Q., Pritchard J., Mieog J., Byrne K., Colgrave M.L., Wang J.R., Ral J.F. Over-expression of a wheat late maturity alpha-amylase type 1 impact on starch properties during grain development and germination. Front Plant Sci 2022;13:811728. doi:10.3389/fpls.2022.811728
Una delle funzioni più importanti del fegato è quella di partecipare alla regolazione della glicemia, ossia al mantenimento dei livelli di glucosio nel sangue entro un intervallo ben definito, che in condizioni normali è pari a 60-100 mg/dL o 3,33-5,56 mmol/L. Per fare ciò rilascia glucosio nel sangue:
nel digiuno;
nell’intervallo tra i pasti;
durante l’attività muscolare.
Regolazione della glicemia: ruolo della glucosio-6-fosfatasi epatica
Nel fegato il glicogeno è la forma di deposito del glucosio il quale non viene rilasciato come tale ma in forma fosforilata, ossia dotata di carica, il glucosio-1-fosfato, nel processo detto glicogenolisi.
La molecola fosforilata non può diffondere liberamente dalla cellula: a livello epatico è presente l’enzima glucosio-6-fosfatasi che idrolizza il glucosio-6-fosfato, prodotto dal glucosio-1-fosfato nella reazione catalizzata dalla fosfoglucomutasi, a glucosio, una defosforilazione irreversibile.
glicogeno(n residui di glucosio) + Pi → glucosio-1-fosfato + glicogeno(n-1 residui di glucosio)
glucosio-1-fosfato ↔ glucosio-6-fosfato
glucosio-6-fosfato + H2O → glucosio + Pi
Il glucosio potrà così lasciare l’epatocita, per mezzo di un trasportatore di membrana detto GLUT2, e riversarsi nel circolo sanguigno per essere distribuito alle cellule extraepatiche, in primis neuroni e globuli rossi per i quali rappresenta la principale, e per i globuli rossi l’unica fonte di energia. I neuroni, con l’esclusione di quelli presenti in alcune zone del cervello che non possono fare a meno del glucosio, sono in grado di utilizzare un’altra fonte di energia che diviene preponderante nei periodi di digiuno prolungato, i corpi chetonici.
Il fegato ricava la maggior parte dell’energia di cui necessita dall’ossidazione degli acidi grassi, una classe di lipidi, e non dal glucosio.
La glucosio-6-fosfatasi oltre che nel fegato è presente anche nel rene e nell’intestino mentre è assente nel muscolo e nel cervello. Pertanto in queste sedi il glucosio-6-fosfato non potrà essere rilasciato dalla cellula.
La glucosio-6-fosfatasi ha un ruolo importante anche nella gluconeogenesi.
La glucosio-6-fosfatatasi è presente nella membrana del reticolo endoplasmatico. L’idrolisi del glucosio-6-fosfato avviene nel lume del reticolo, quindi questa reazione è separata dalla via glicolitica, ed è necessaria la presenza di uno specifico trasportatore, la glucosio-6-fosfato traslocasi, per portare lo zucchero fosforilato dal citosol nel lume del reticolo stesso. Sebbene nella membrana del reticolo endoplasmatico sia presente un trasportatore per il glucosio, la maggior parte del glucosio liberato non è ritrasportato nel citosol della cellula ma secreto nel circolo ematico. Infine un trasportatore anionico trasloca il fosfato inorganico liberato dal lume del reticolo endoplasmatico nel citosol.
Bibliografia
Nelson D.L., M. M. Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
Roach P.J., Depaoli-Roach A.A., Hurley T.D and Tagliabracci V.C. Glycogen and its metabolism: some new developments and old themes. Biochem J 2012;441:763-787. doi:10.1042/BJ20111416
Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
Nel caso in cui si consumi un eccesso di calorie in forma di carboidrati o proteine, tale surplus è utilizzato per la sintesi degli acidi grassi e quindi trigliceridi, cosa che non accade se l’eccesso di calorie proviene dai grassi stessi.
Biosintesi degli Acidi Grassi Saturi e Insaturi a Catena Lunga
Sintesi de novo degli acidi grassi negli animali e piante
La sintesi de novo degli acidi grassi, molecole appartenenti al gruppo die lipidi, è in gran parte simile tra le piante e gli animali.
Avviene nei cloroplasti delle cellule fotosintetiche delle piante superiori, e nel citosol delle cellule animali, attraverso l’azione concertata di due enzimi: l’acetil-CoA carbossilasi (EC 6.4.1.2) e l’acido grasso sintetasi (EC 2.3.1.85).
L’acido grasso sintetasi catalizza una sequenza ripetuta di 4 passaggi tramite la quale la catena acilica è allungata di due atomi di carbonio, all’estremità carbossilica, a ogni passaggio attraverso il ciclo; questo processo in quattro tappe è il medesimo in tutti gli organismi.
Negli animali il sito primario per il metabolismo lipidico non è il tessuto adiposo ma il fegato. Il tessuto adiposo è comunque un organo importante in cui si verifica la sintesi degli acidi grassi, anche se negli esseri umani è meno attivo rispetto a molte altre specie animali.
Da notare che in alcune piante, quali la palma e la noce di cocco, si verifica la terminazione della catena acilica prima che sia rilasciato l’acido palmitico: fino al 90% degli acidi grassi prodotti, e quindi presenti negli oli ricavati da questi vegetali, ha tra gli 8, atomi di carbonio, l’acido caprilico, un degli acidi grassi a catena media, e i 14 atomi di carbonio, ossia l’acido miristico.
Sintesi degli acidi grassi a catena lunga saturi e insaturi
L’acido palmitico è, tra gli acidi grassi saturi, il più comune nei lipidi di piante e animali, sebbene generalmente non sia presente in grandi quantità poiché può entrare in diverse vie metaboliche.
Infatti:
è il precursore dell’acido stearico;
può essere desaturato a dare acido palmitoleico, il precursore di tutti gli acidi grassi della famiglia omega-7, in una reazione catalizzata dall’enzima Δ9-desaturasi (EC 1.14.19.1). Questo enzima è ubiquitario sia nel regno animale che vegetale, è il più attivo del metabolismo lipidico nei tessuti di mammifero, ed è lo stesso che catalizza la desaturazione dell’acido stearico ad acido oleico.
La Δ9-desaturasi inserisce un doppio legame nella posizione 9-10 della catena carboniosa dell’acido grasso, posizione numerata a partire dall’estremità carbossilica della molecola. Se il substrato è l’acido palmitico, il doppio legame apparirà tra la posizione posizione n-7 e n-8 della catena, stavolta numerate a partire dall’estremità metilica, producendo acido palmitoleico, dunque il capostipite della famiglia omega-7. Se il substrato è l’acido stearico il doppio legame apparirà tra la posizione posizione n-9 e n-10 della catena, e verrà prodotto l’acido oleico.
Può essere esterificato in lipidi complessi.
Naturalmente nelle piante e negli animali ci sono acidi grassi sia con catene più lunghe sia con gradi di insaturazione maggiori rispetto a quelli visti, il tutto grazie a sistemi enzimatici (di nuovo di desaturazione e allungamento), che catalizzano reazioni di sintesi degli acidi grassi che sono organismo-, tessuto- e cellula specifiche.
Ad esempio l’acido stearico può essere:
allungato ad acido arachidico, acido beenico e acido lignocerico, tutti acidi grassi saturi, in reazioni catalizzate da elongasi.
Anche in questo caso, l’allungamento della catena si verifica, sia nei mitocondri che nel reticolo endoplasmatico liscio, attraverso l’aggiunta di unità a due atomi di carbonio alla volta all’estremità carbossilica dell’acido grasso, tramite l’azione dei sistemi di allungamento degli acidi grassi (in particolare gli acidi grassi a catena lunga e molto lunga, da 18 a 24 atomi di carbonio, sono sintetizzati sulla superficie citosolica del reticolo endoplasmatico liscio);
desaturato, come visto, nella reazione catalizzata dalla Δ9-desaturasi, a dare acido oleico, un acido grasso omega-9.
Diversi ricercatori hanno supposto che la ragione per cui l’acido stearico non concorra allo sviluppo della ipercolesterolemia sia la sua rapida conversione in acido oleico.
L’acido oleico è il punto di partenza per la sintesi di molti acidi grassi insaturi attraverso reazioni di allungamento e/o desaturazione.
Infatti:
l’allungamento della sua catena acilica per mezzo di un altro sistema enzimatico, l’acido grasso elongasi, produce acido gadoleico, acido erucico e acido nervonico, tutti acidi grassi monoinsaturi.
Gli acidi grassi saturi e i monoinsaturi della famiglia omega-9, in genere l’acido oleico, ma anche l’acido palmitoleico e gli altri omega-7, sono gli unici acidi grassi prodotti attraverso la sintesi de novo nei mammiferi.
Grazie all’azione consecutiva degli enzimi Δ12-desaturasi (1.14.19.6) e Δ15-desaturasi (EC 1.14.19.25), che inseriscono un doppio legame rispettivamente nella posizione posizione 12-13 e nella posizione 15-16 della catena carboniosa dell’acido grasso, l’acido oleico è convertito prima in acido linoleico, precursore di tutti gli acidi grassi polinsaturi omega-6, e quindi in acido alfa-linolenico, precursore di tutti gli acidi grassi polinsaturi omega-3 (acidi grassi polinsaturi che saranno prodotti a partire da questi precursori attraverso reazione ripetute di allungamento e desaturazione).
In caso di deficienza di acidi grassi essenziali, l’acido oleico può essere desaturato e allungato a dare la serie di acidi grassi polinsaturi omega-9, con accumulo soprattutto di acido Mead.
Sintesi degli omega-3 e omega-6
I tessuti animali possono desaturare gli acidi grassi in posizione 9-10 della catena, grazie alla presenza della Δ9 desaturasi; come visto in precedenza se il substrato della reazione è l’acido palmitico il doppio legame apparirà tra le posizioni n-7 e n-8 della catena, con l’acido stearico tra le posizioni n-9 e n-10 portando così alla formazione rispettivamente di acido palmitoleico e oleico.
Gli animali non possiedono la Δ12- e la Δ15-desaturasi, enzimi in grado desaturare i legami carbonio carbonio oltre la posizione 9-10 della catena, e non sono quindi in grado di sintetizzare de novo gli acidi grassi polinsaturi omega-3 e omega-6, che posseggono doppi legami anche oltre la posizione 9-10, che sono pertanto acidi grassi essenziali.
La Δ12- e la Δ15-desaturasi sono presenti nelle piante; sebbene molte piante terrestri manchino della Δ15-desaturasi, detta anche omega-3 desaturasi, il plancton e le piante acquatiche delle acque fredde la posseggono e producono quantità abbondanti di acidi grassi omega-3.
Bibliografia
Akoh C.C. and Min D.B. “Food lipids: chemistry, nutrition, and biotechnology”. CRC Press Taylor & Francis Group, 2008 3th ed. 2008
Bender D.A. “Benders’ dictionary of nutrition and food technology”. 2006, 8th Edition. Woodhead Publishing. Oxford
Burr G.O. and Burr M.M. A new deficiency disease produced by the rigid exclusion of fat from the diet. Nutr Rev 1973;31(8):148-149. doi:10.1111/j.1753-4887.1973.tb06008.x
Chow Ching K. “Fatty acids in foods and their health implication”. 3rd Edition. CRC Press Taylor & Francis Group, 2008
Rosenthal M.D., Glew R.H. Mediacal biochemistry. Human metabolism in health and disease. John Wiley & Sons, Inc. 2009
Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
Biochemistry and metabolism
Utilizziamo i cookie per essere sicuri che tu possa avere la migliore esperienza sul nostro sito. Se continui ad utilizzare questo sito noi assumiamo che tu ne sia felice.OkPrivacy policy
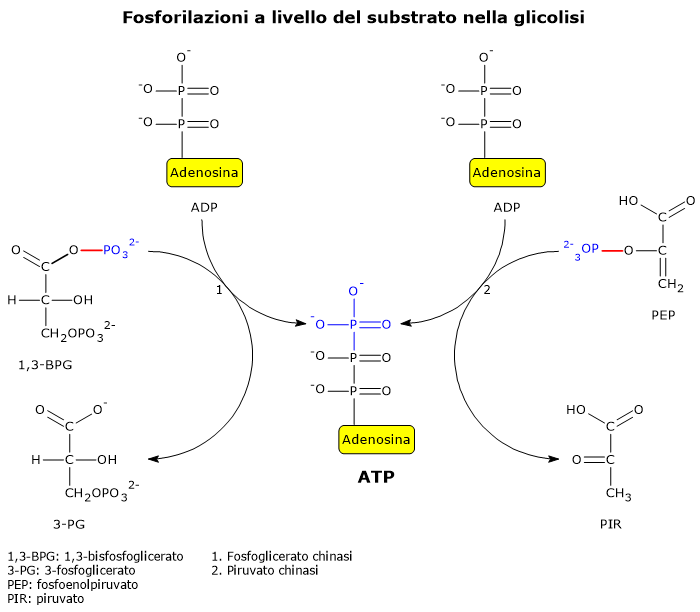 Le reazioni coinvolte sono quelle catalizzate dalla fosfoglicerato chinasi (EC 2.7.2.3), e dalla piruvato chinasi (EC 2.7.1.40).
Le reazioni coinvolte sono quelle catalizzate dalla fosfoglicerato chinasi (EC 2.7.2.3), e dalla piruvato chinasi (EC 2.7.1.40).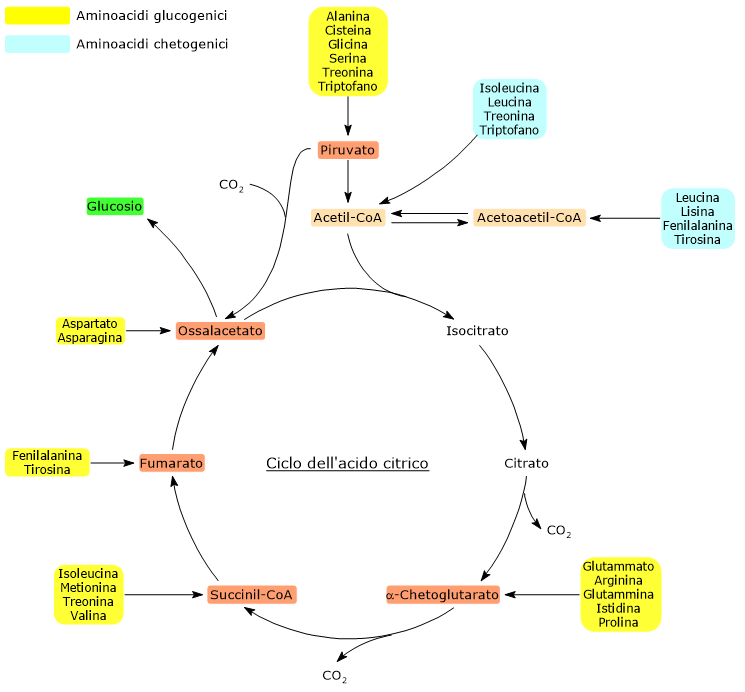
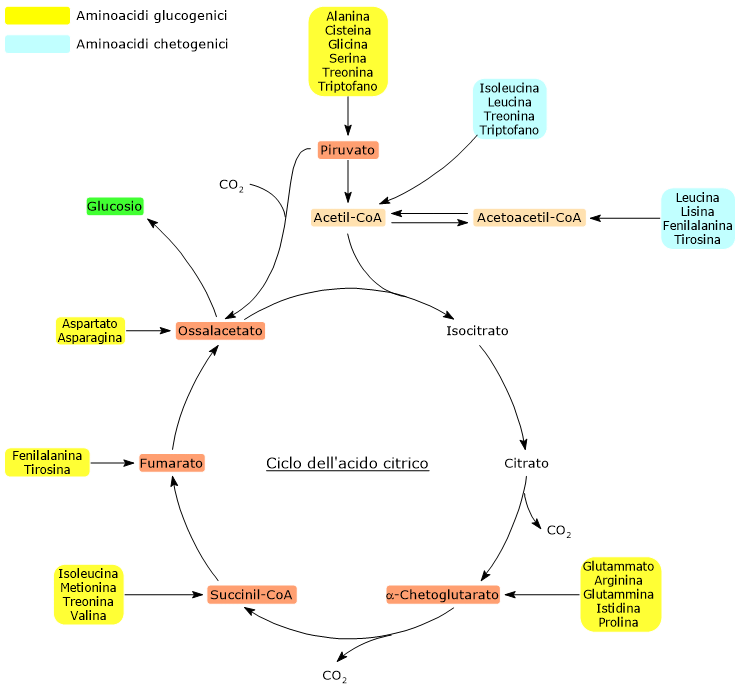
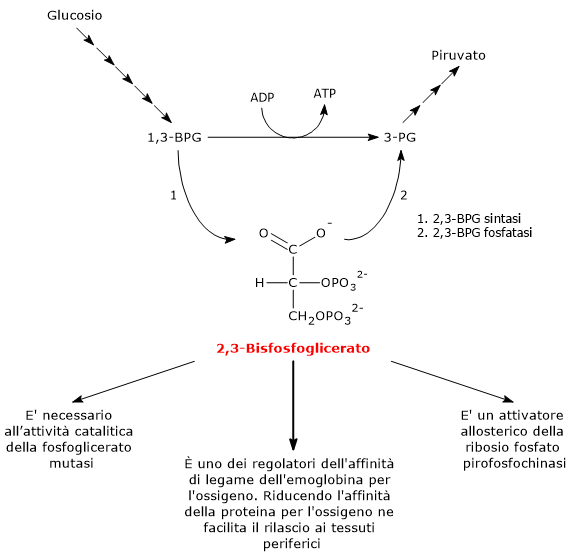 Nella seconda reazione dello shunt, il 2,3-bisfosfoglicerato viene defosforilato a 3-fosfoglicerato. La reazione è catalizzata dall’attività fosfatasica della bisfosfoglicerato mutasi. Il 3-fosfoglicerato rientra quindi nella via glicolitica a livello dell’ottava tappa, catalizzata dalla fosfoglicerato mutasi (EC 5.4.2.1).
Nella seconda reazione dello shunt, il 2,3-bisfosfoglicerato viene defosforilato a 3-fosfoglicerato. La reazione è catalizzata dall’attività fosfatasica della bisfosfoglicerato mutasi. Il 3-fosfoglicerato rientra quindi nella via glicolitica a livello dell’ottava tappa, catalizzata dalla fosfoglicerato mutasi (EC 5.4.2.1).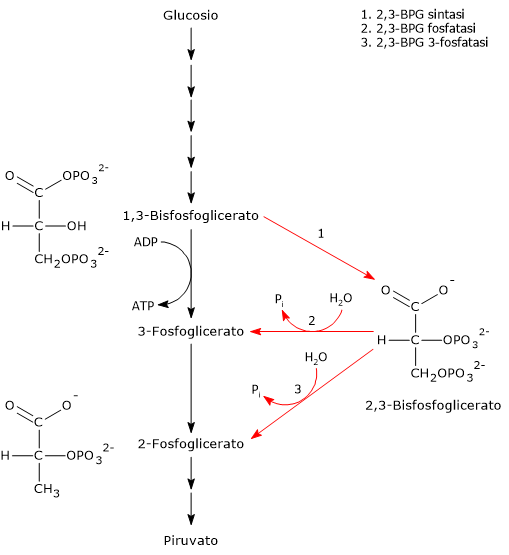 L’attività di fosfatasi è bassa, circa 1000 volte inferiore all’attività di sintasi, ed è stimolata da diversi anioni, come cloruro, fosfato, solfito e, in modo più marcato, dal 2-fosfoglicolato.
L’attività di fosfatasi è bassa, circa 1000 volte inferiore all’attività di sintasi, ed è stimolata da diversi anioni, come cloruro, fosfato, solfito e, in modo più marcato, dal 2-fosfoglicolato.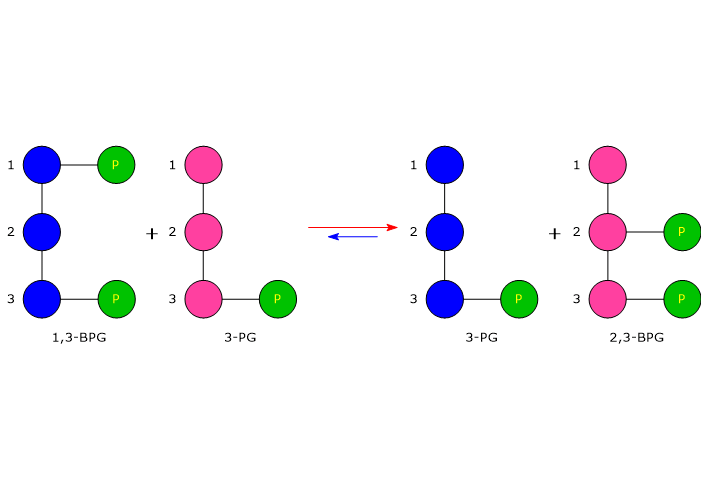 Nel secondo passaggio l’attività fosfatasica della bisfosfoglicerato mutasi catalizza la sintesi del 3-fosfoglicerato, un intermedio glicolitico, a seguito dell’idrolisi del gruppo fosforico in posizione C-2 del 2,3-BPG.Il 3-fosfoglicerato prodotto può quindi rientrare nella via glicolitica a livello della reazione catalizzata dalla fosfoglicerato mutasi, ossia l’ottava tappa della via.
Nel secondo passaggio l’attività fosfatasica della bisfosfoglicerato mutasi catalizza la sintesi del 3-fosfoglicerato, un intermedio glicolitico, a seguito dell’idrolisi del gruppo fosforico in posizione C-2 del 2,3-BPG.Il 3-fosfoglicerato prodotto può quindi rientrare nella via glicolitica a livello della reazione catalizzata dalla fosfoglicerato mutasi, ossia l’ottava tappa della via.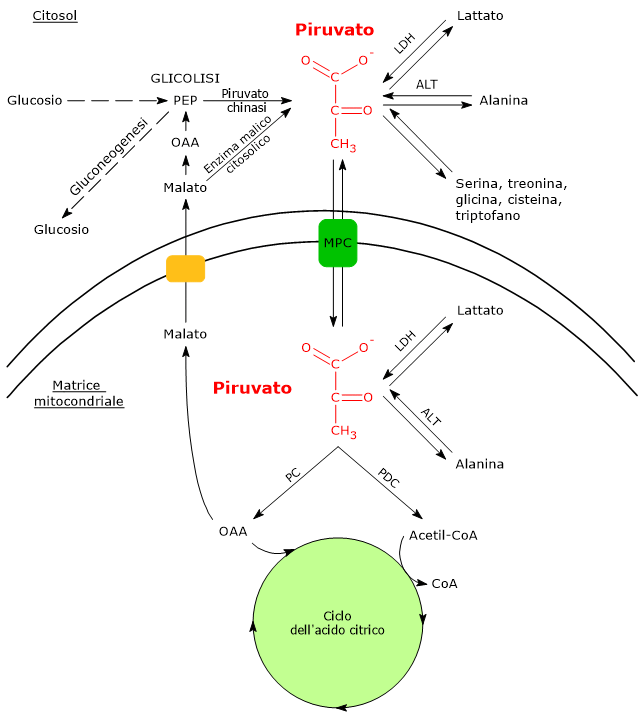 L’acido piruvico prodotto nel citosol, a mezzo di trasportatori specifici, entra nel mitocondrio dove potrà essere utilizzato a fini energetici, entrando nel ciclo dell’acido citrico, e/o biosintetici/anaplerotici, sulla base delle necessità della cellula.
L’acido piruvico prodotto nel citosol, a mezzo di trasportatori specifici, entra nel mitocondrio dove potrà essere utilizzato a fini energetici, entrando nel ciclo dell’acido citrico, e/o biosintetici/anaplerotici, sulla base delle necessità della cellula.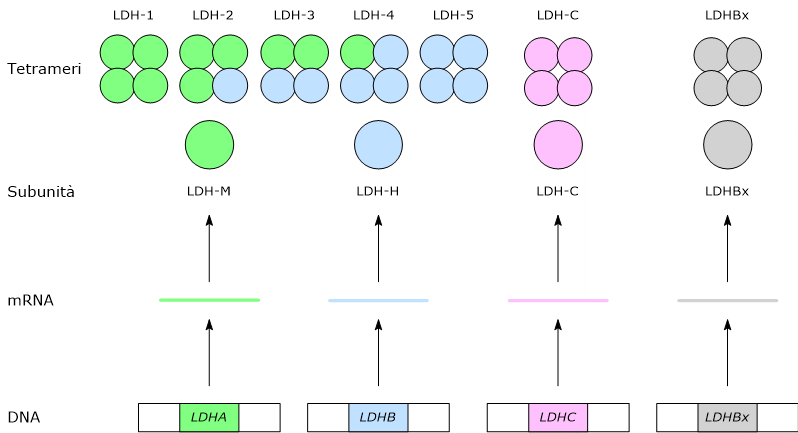 La subunità C va a formare una sesta isoforma, omotetramerica, indicata come LDH-6 o C4.
La subunità C va a formare una sesta isoforma, omotetramerica, indicata come LDH-6 o C4.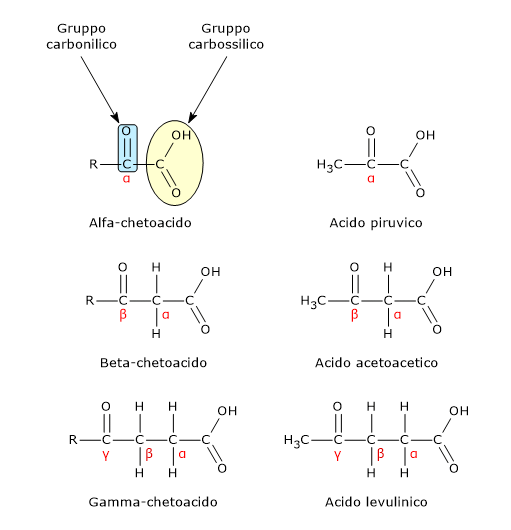 I chetoacidi, e in particolare gli alfa-chetoacidi, sono molto importanti in biochimica, essendo coinvolti in molte vie metaboliche.
I chetoacidi, e in particolare gli alfa-chetoacidi, sono molto importanti in biochimica, essendo coinvolti in molte vie metaboliche.
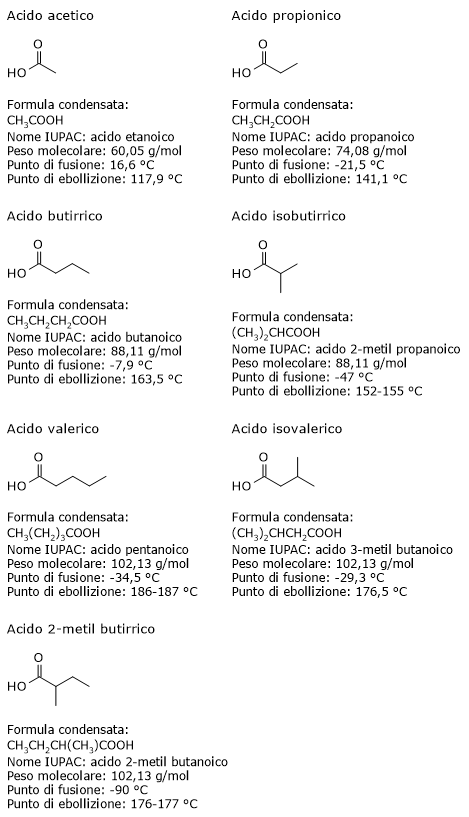
 L’enzima, al pari delle
L’enzima, al pari delle 
 Le poche ramificazioni sono connesse alla catena lineare da legami glicosidici α-(1→6), al pari di quanto accade con l’amilopectina e il
Le poche ramificazioni sono connesse alla catena lineare da legami glicosidici α-(1→6), al pari di quanto accade con l’amilopectina e il 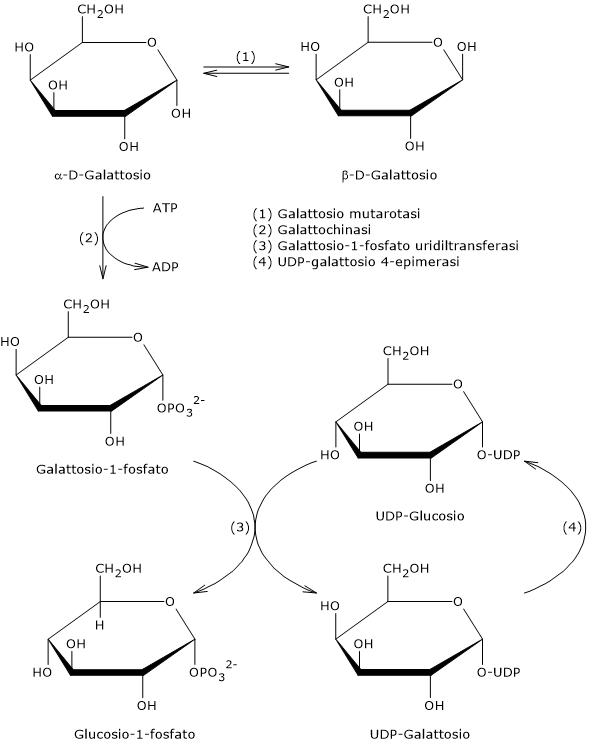
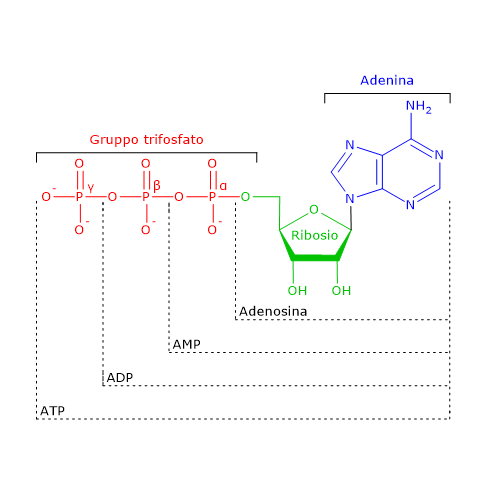
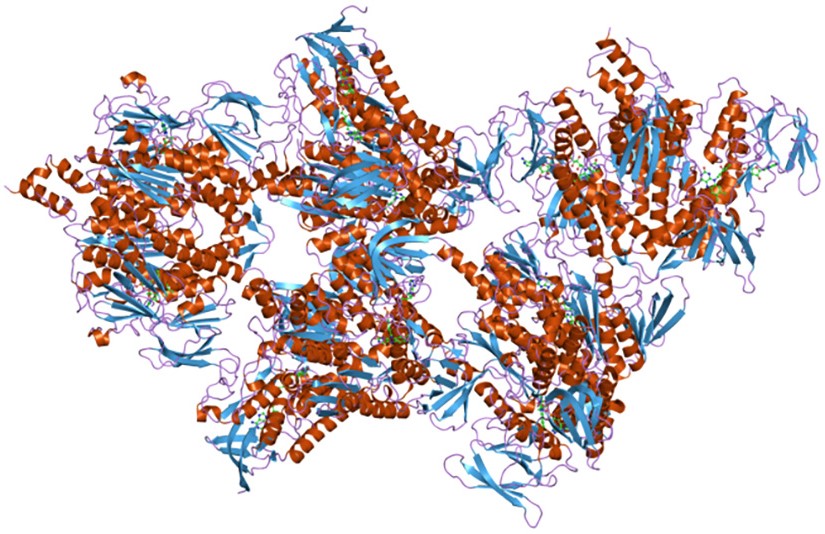
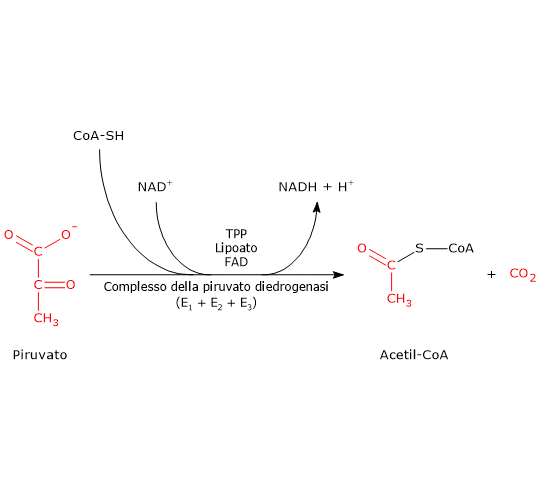
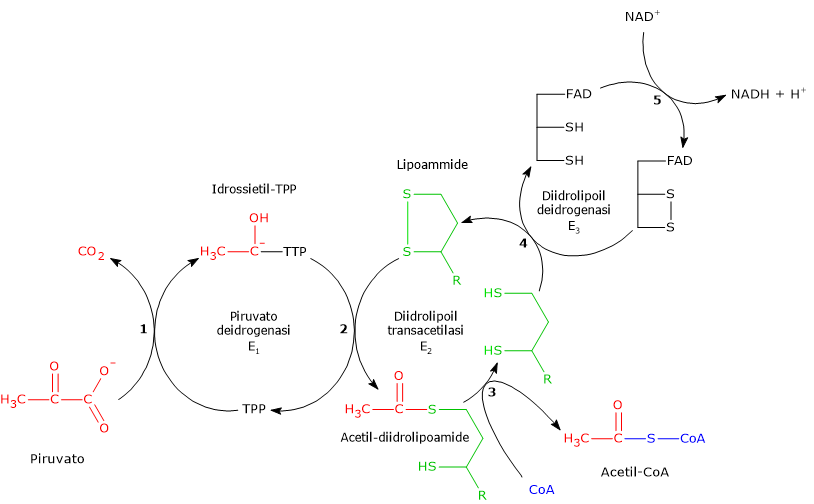
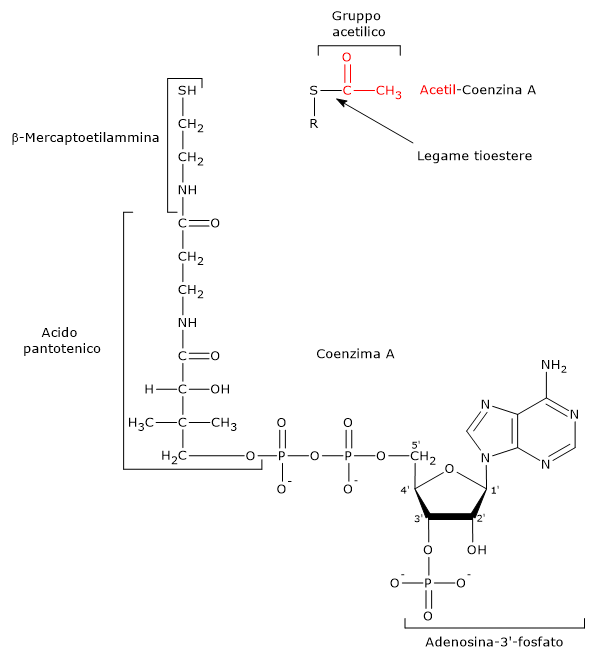

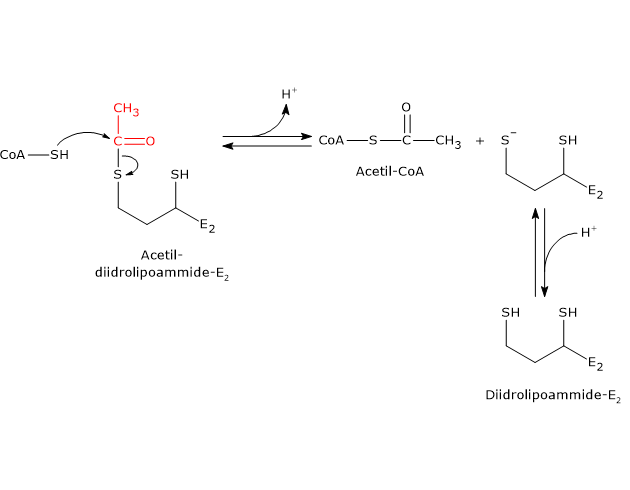 Meccanismo Catalitico della Diidrolipoil Transacetilasi
Meccanismo Catalitico della Diidrolipoil Transacetilasi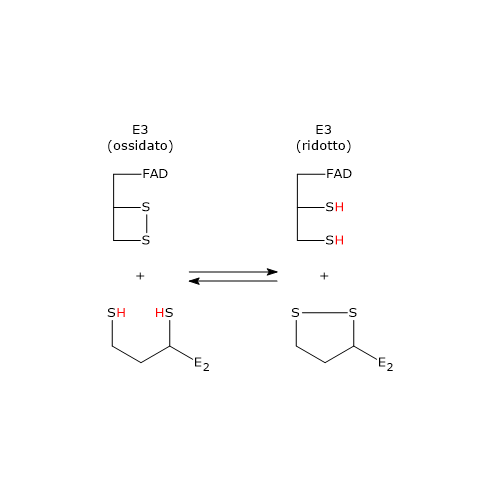
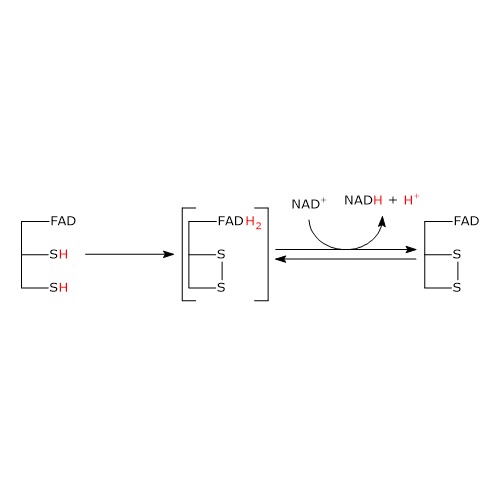
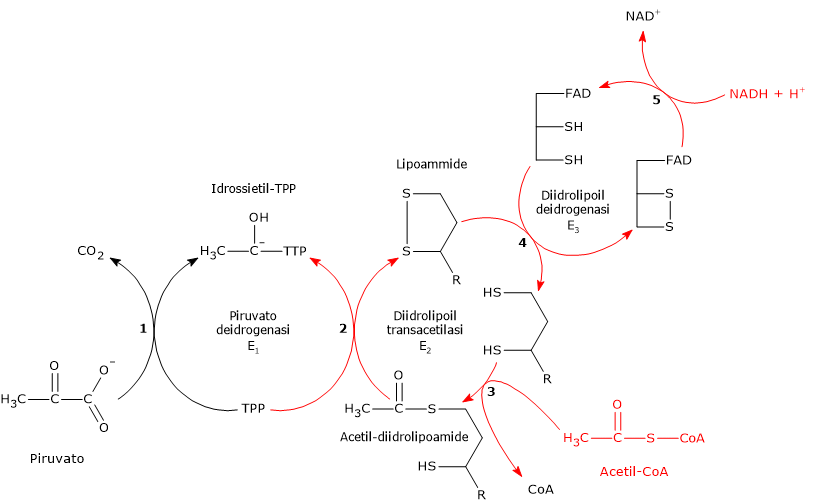
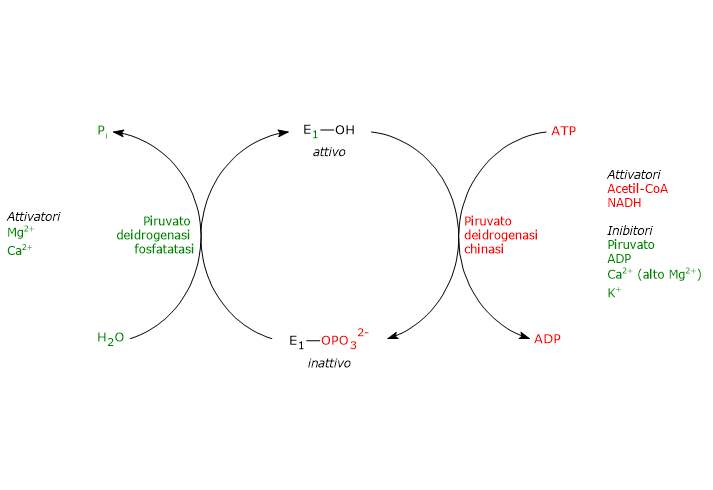
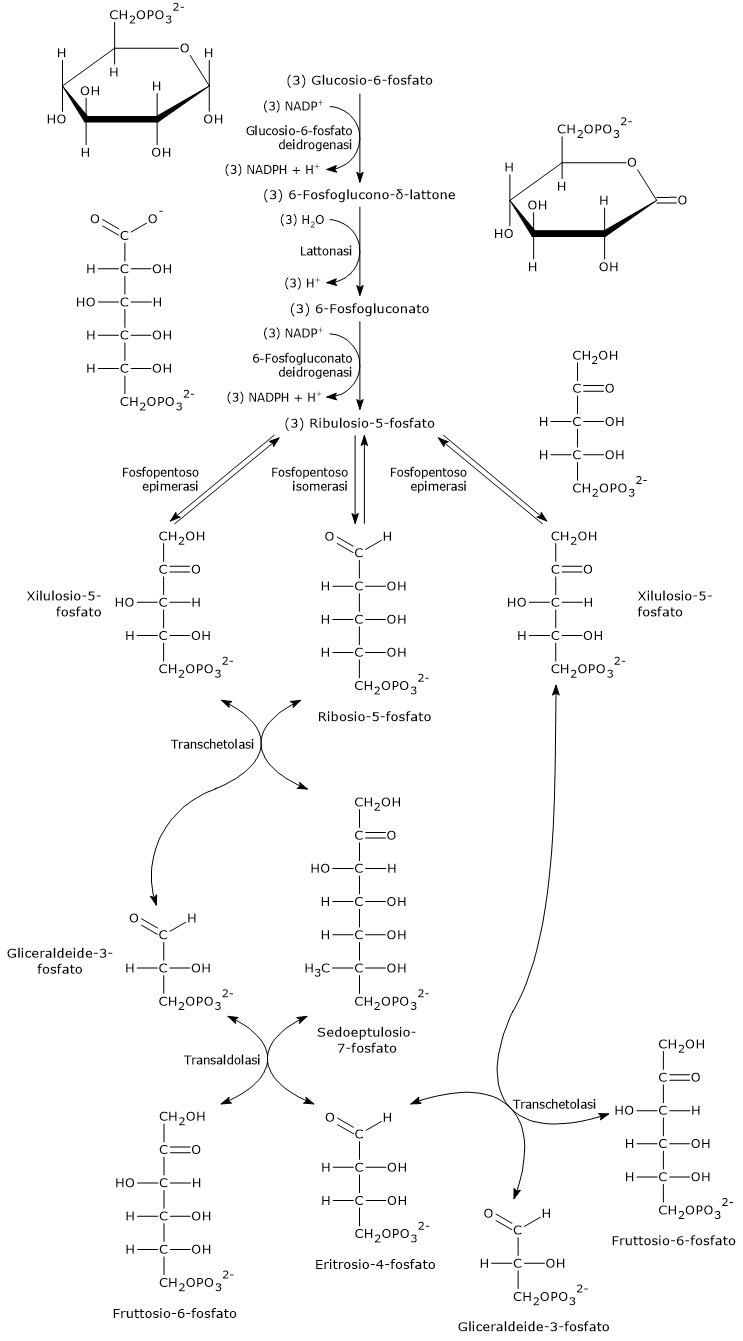
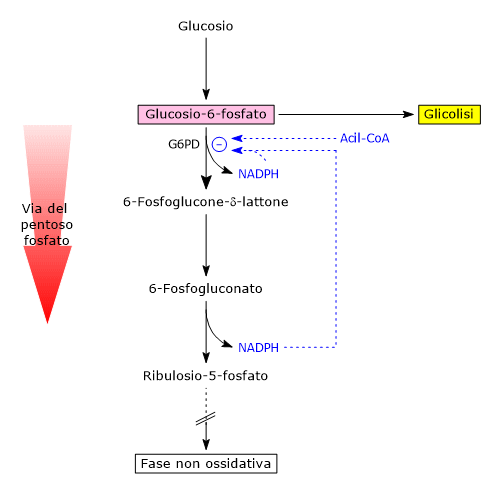
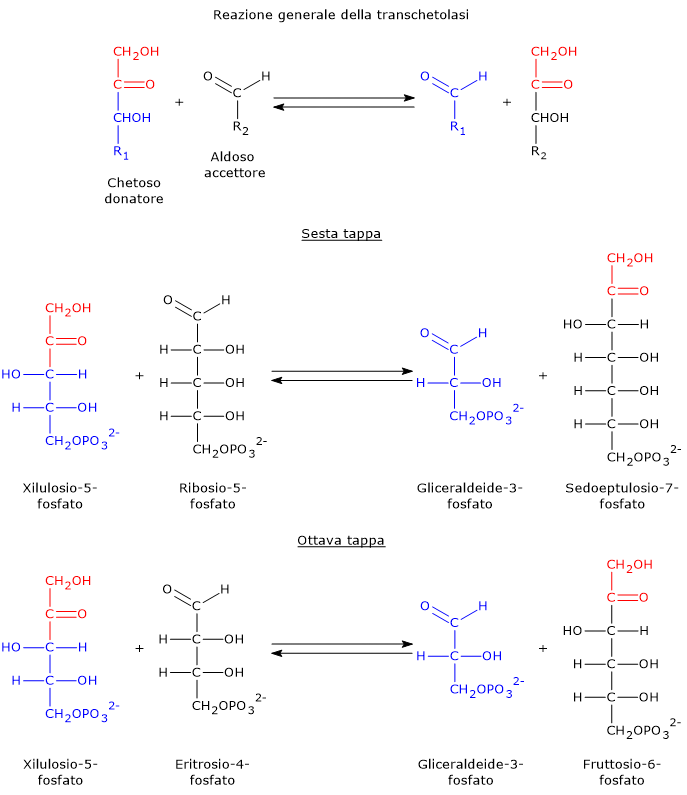
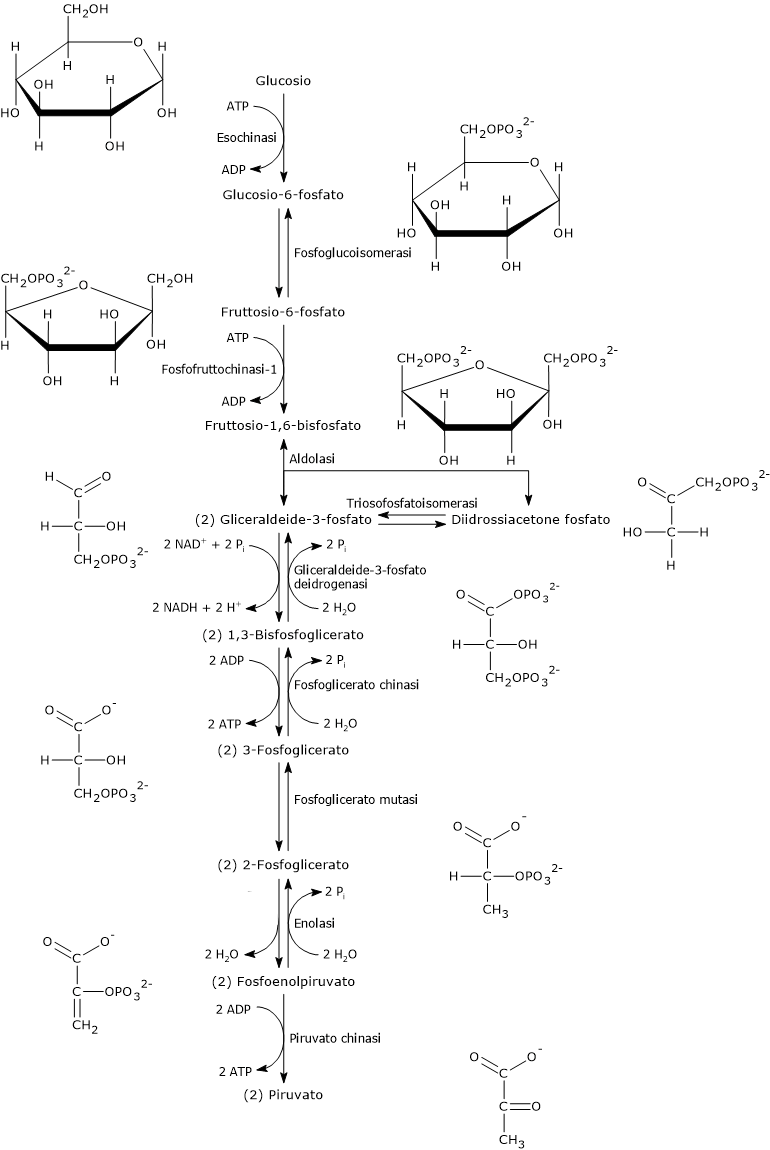
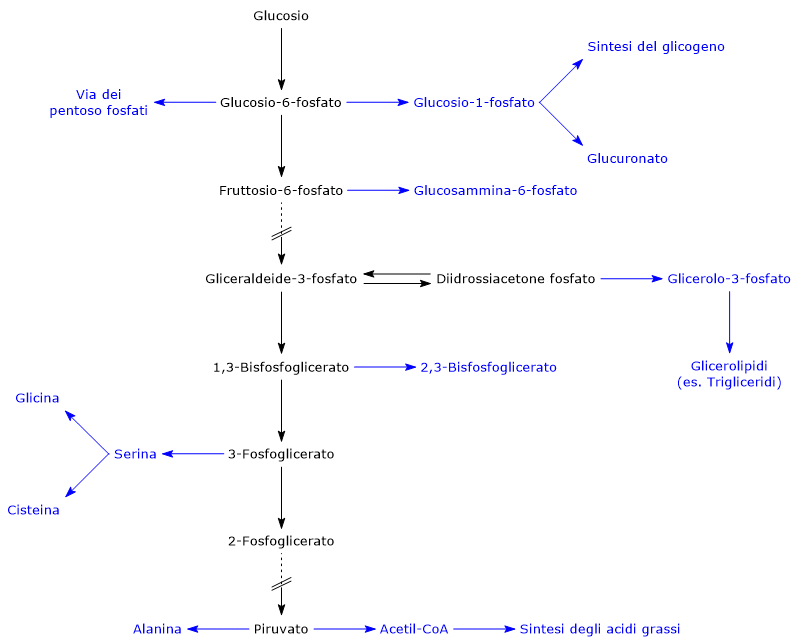
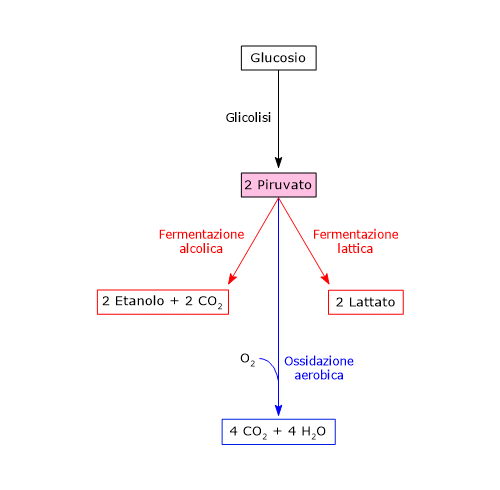
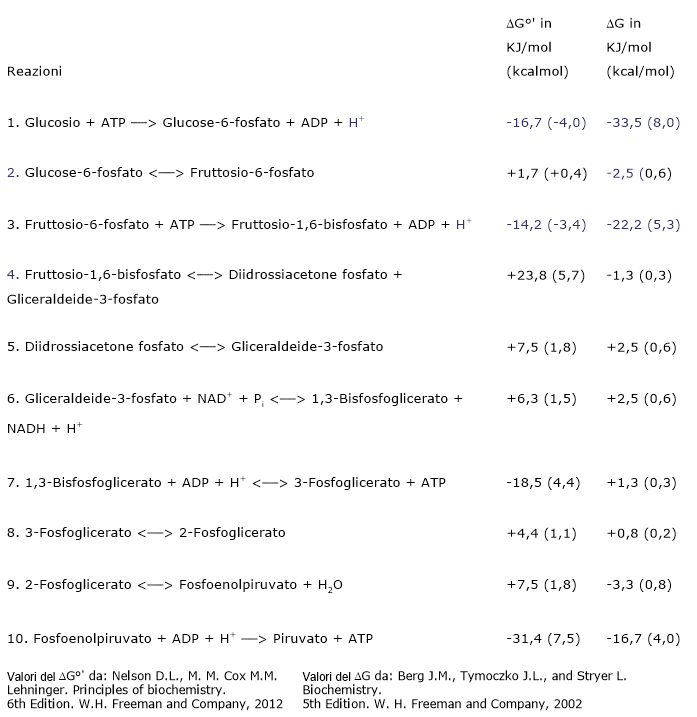
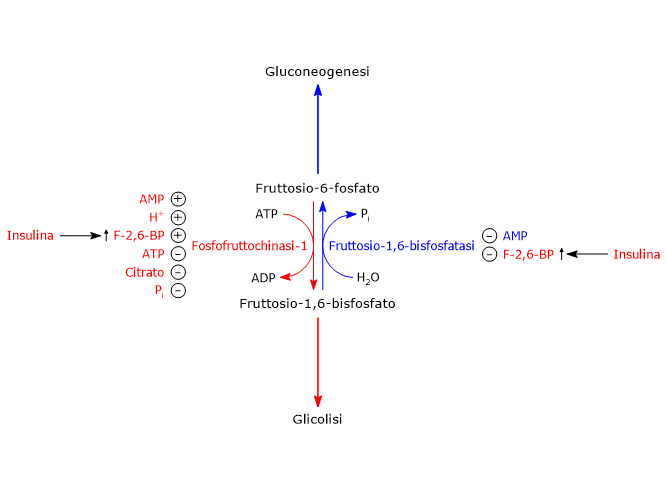
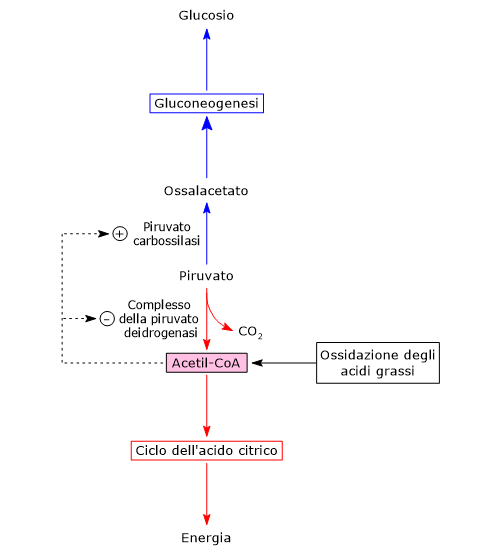
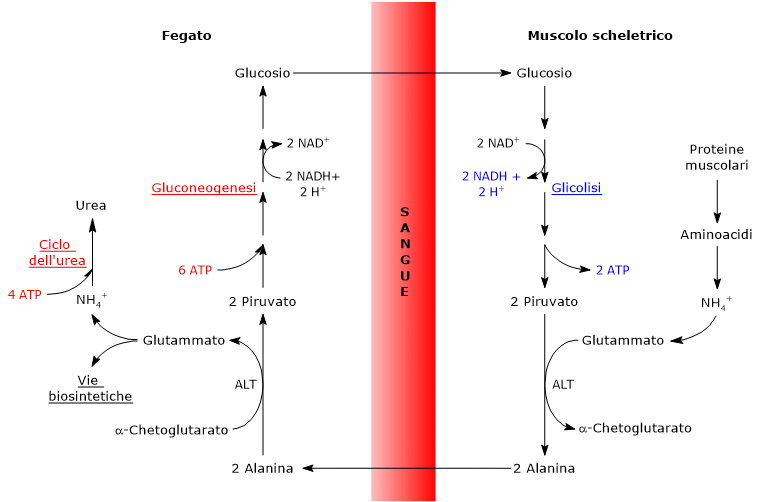
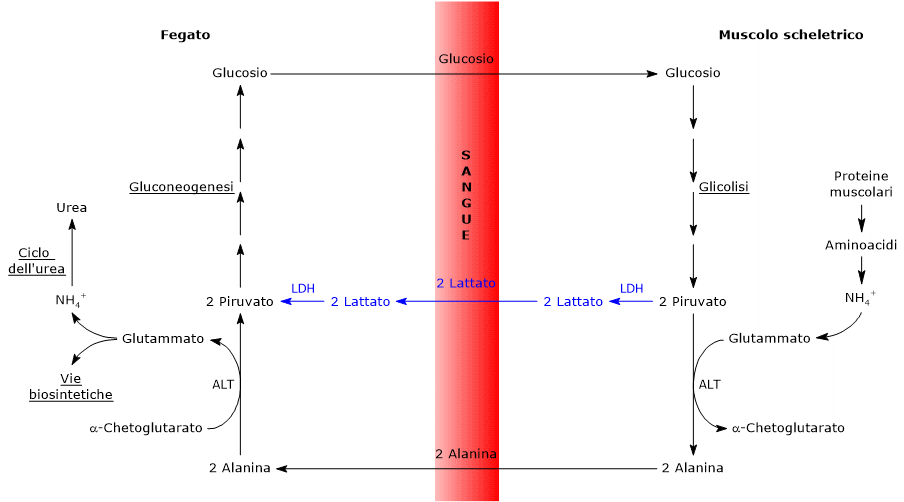
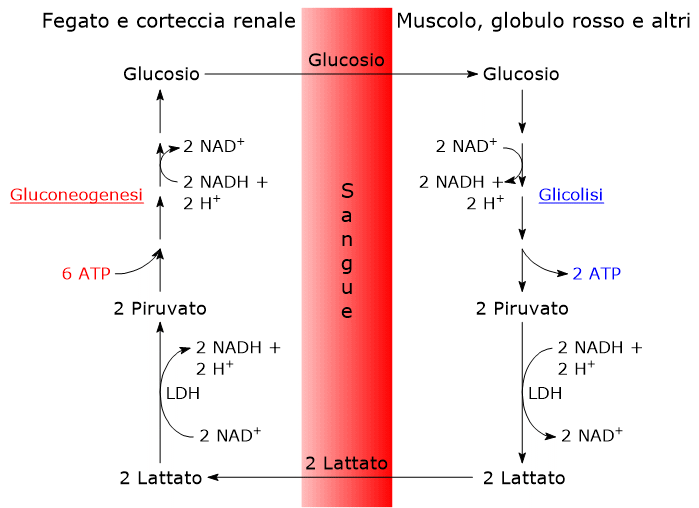
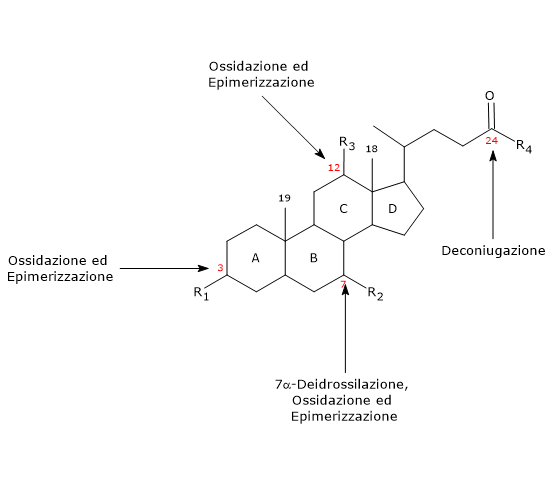
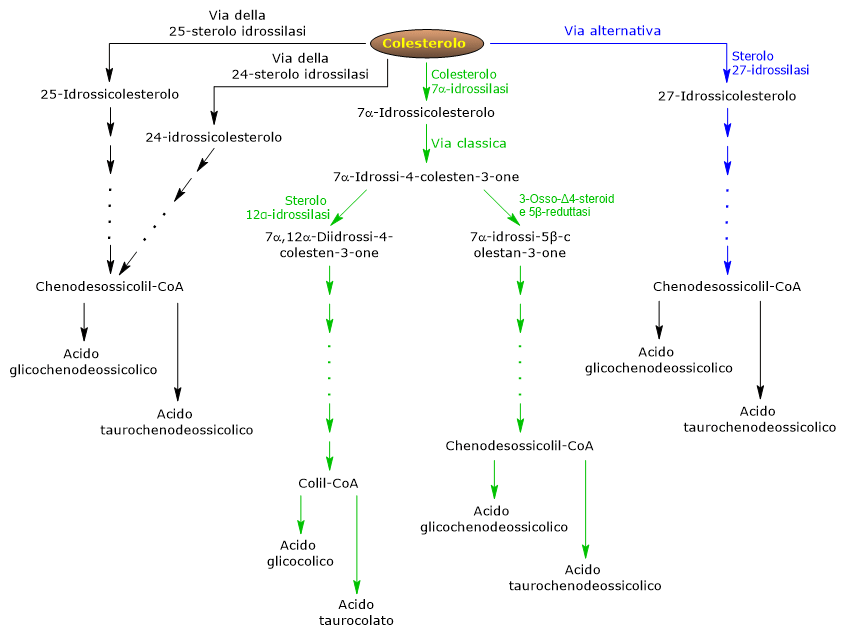
 Il glucosio potrà così lasciare l’epatocita, per mezzo di un trasportatore di membrana detto GLUT2, e riversarsi nel circolo sanguigno per essere distribuito alle cellule extraepatiche, in primis neuroni e globuli rossi per i quali rappresenta la principale, e per i globuli rossi l’unica fonte di energia. I neuroni, con l’esclusione di quelli presenti in alcune zone del cervello che non possono fare a meno del glucosio, sono in grado di utilizzare un’altra fonte di energia che diviene preponderante nei periodi di digiuno prolungato, i corpi chetonici.
Il glucosio potrà così lasciare l’epatocita, per mezzo di un trasportatore di membrana detto GLUT2, e riversarsi nel circolo sanguigno per essere distribuito alle cellule extraepatiche, in primis neuroni e globuli rossi per i quali rappresenta la principale, e per i globuli rossi l’unica fonte di energia. I neuroni, con l’esclusione di quelli presenti in alcune zone del cervello che non possono fare a meno del glucosio, sono in grado di utilizzare un’altra fonte di energia che diviene preponderante nei periodi di digiuno prolungato, i corpi chetonici.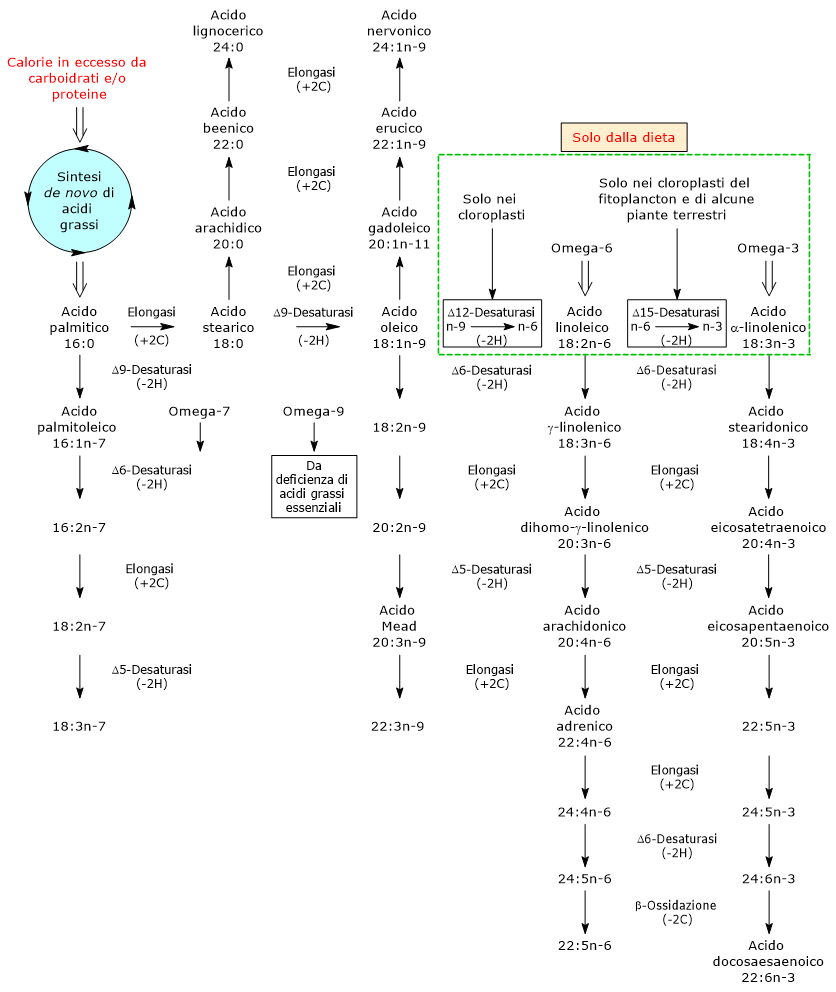
 Se il substrato è l’acido stearico il doppio legame apparirà tra la posizione posizione n-9 e n-10 della catena, e verrà prodotto l’acido oleico.
Se il substrato è l’acido stearico il doppio legame apparirà tra la posizione posizione n-9 e n-10 della catena, e verrà prodotto l’acido oleico.