I carbanioni sono ioni contenenti un atomo di carbonio con carica negativa.
Si formano a seguito della rottura eterolitica di un legame covalente tra un atomo di carbonio e un altro atomo o gruppo.[1]
Avendo una coppia di elettroni non condivisa, sono nucleofili potenti e basi forti e attaccano, al fine di formare un legame covalente, un protone o un centro elettrofilo, come un centro polarizzato o carico positivamente.[2]
I carbanioni sono estremamente reattivi. Pertanto, per consentire loro l’attacco ai centri nucleofili, devono essere stabilizzati.[3] La stabilizzazione può avvenire per effetto induttivo, per risonanza, e può anche dipendere dall’ibridazione dell’atomo di carbonio portatore di carica negativa.[1][2]
Sono intermedi in molte reazioni catalizzate da enzimi.
Indice
- Eterolisi e omolisi
- Stabilizzazione dei carbanioni
- I carbanioni nelle reazioni enzimatiche
- Bibliografia
Eterolisi e omolisi
Considerando due atomi o gruppi, indicati come A e B, uniti da legame covalente, esistono due modi per rompere suddetto legame: l’eterolisi e l’omolisi.
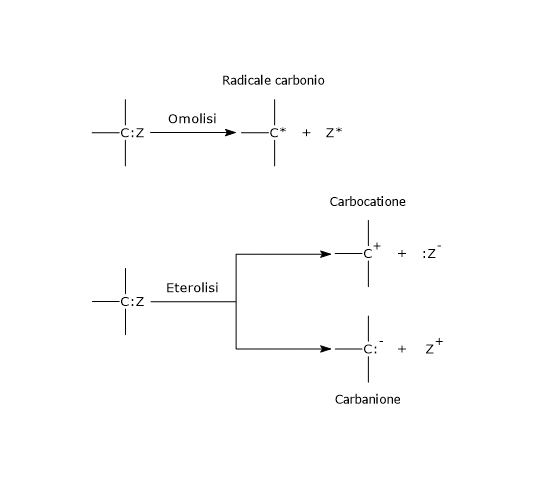 Nell’eterolisi, la rottura del legame covalente porta alla formazione di due atomi dotati di carica, ossia due ioni, un catione e un anione, poiché entrambi gli elettroni di legame vengono presi da uno dei due atomi precedentemente legati, quello più elettronegativo.[4] La reazione può procedere in due modi:
Nell’eterolisi, la rottura del legame covalente porta alla formazione di due atomi dotati di carica, ossia due ioni, un catione e un anione, poiché entrambi gli elettroni di legame vengono presi da uno dei due atomi precedentemente legati, quello più elettronegativo.[4] La reazione può procedere in due modi:
A:B → :A– + B+, se A è più elettronegativo di B;
A:B → A+ + :B–, se B è più elettronegativo di A.
Nell’eterolisi di un legame covalente che coinvolge un atomo di carbonio, se entrambi gli elettroni di legame vengono presi dall’atomo di carbonio, l’atomo avrà una carica negativa, quindi è un anione, ed è definito carbanione. Se invece il carbonio perde entrambi gli elettroni di legame avrà carica positiva, quindi è un catione, ed è definito carbocatione.[5]
Nell’omolisi, la rottura del legame covalente tra A e B porta alla formazione di due radicali liberi, poiché ciascun atomo o gruppo prende uno dei due elettroni di legame.[6] Anche i radicali liberi, che sono elettricamente neutri, sono molecole molto instabili. Si noti che la scissione omolitica è meno comune della scissione eterolitica.[7]
Stabilizzazione dei carbanioni
I carbanioni sono specie chimiche estremamente reattive e, come i carbocationi e i radicali liberi, sono quasi sempre intermedi transienti in alcune reazioni organiche. Per permettere il loro attacco ai centri elettrofili è necessario che siano stabilizzati. La loro stabilizzazione dipende dalla dispersione della carica negativa, dispersione che può avvenire per effetto induttivo, per risonanza, e può anche dipendere dal carattere s degli orbitali ibridi dell’atomo di carbonio con carica negativa.
L’effetto induttivo è dovuto alla presenza nella molecola di uno o più dipoli permanenti in uno o più legami, dipoli a loro volta derivanti dalla differenza di elettronegatività tra due gruppi che determina una distribuzione non uniforme degli elettroni di legame. L’effetto induttivo può essere positivo, detto anche effetto +I, caratteristico di atomi o gruppi che tendono a respingere gli elettroni, oppure negativo, detto anche effetto –I, caratteristico di atomi o gruppi che tendono ad attrarre gli elettroni. Gli atomi o gruppi con effetto +I tendono a diminuire la stabilità dei carbanioni, mentre quelli con effetto –I, quindi più elettronegativi, tendono a stabilizzarli.[1]
La stabilità dei carbanioni aumenta quando sono legati a una struttura elettrofila dove la coppia di elettroni non condivisi può delocalizzarsi per risonanza, quindi una struttura che agisca da trappola per gli elettroni. Le strutture aromatiche, come il gruppo fenile, sono particolarmente efficaci.[2]
Infine la stabilità è anche una funzione del carattere s degli orbitali ibridi dell’atomo di carbonio caricato negativamente, aumentando all’aumentare della percentuale del suddetto carattere s. Pertanto aumenterà passando dall’ibridazione sp3, che ha il 25% di carattere s, a sp2, con il 33% di carattere s, a sp, con il 50% di carattere s.[1]
R-CH2– < R1R2C=CH– < RC≡C–
I carbanioni nelle reazioni enzimatiche
Esempi di reazioni enzimatiche che hanno carbanioni tra gli intermedi sono quelle catalizzata da tre complessi multienzimatici appartenenti alla famiglia delle 2-ossiacido deidrogenasi o alfa-chetoacido deidrogenasi, i quali sono coinvolti nelle decarbossilazioni ossidative di chetoacidi, in particolare degli alfa-chetoacidi, di seguito brevemente descritti.
- Il complesso della piruvato deidrogenasi, che catalizza la decarbossilazione ossidativa del piruvato, la base coniugata dell’acido piruvico, in acetil-CoA, agendo così da ponte di collegamento tra glicolisi e ciclo dell’acido conversione citrico;
- il complesso della ossoglutarato deidrogenasi o alfa-chetoglutarato deidrogenasi, che catalizza la decarbossilazione ossidativa dell’alfa-chetoglutarato a succinil-CoA nel corso del ciclo dell’acido citrico;
- il complesso della alfa-chetoacido deidrogenasi a catena ramificata, che catalizza la decarbossilazione ossidativa degli aminoacidi ramificati valina, leucina e isoleucina in acetil-CoA e succinil-CoA, il cui scheletro carbonioso rimanente può quindi entrare nel ciclo dell’acido citrico.[3]
I tre complessi multienzimatici hanno strutture e meccanismi di reazione molto simili. Nello specifico, sono le loro subunità E1, che hanno come cofattore la tiamina pirofosfato, che catalizzano una reazione in cui si viene a formare un intermedio carbanionico, la cui formazione e stabilizzazione per risonanza coinvolge direttamente la tiamina.[8]
Anche la transchetolasi (EC 2.2.1.1) catalizza una reazione che comporta la formazione di un intermedio carbanionico. Questo enzima, che catalizza le tappe 6 e 8 della via del pentoso fosfato, richiede come cofattore la tiamina pirofosfato e ha un meccanismo di reazione simile a quello delle subunità E1 dei complessi multienzimatici visti in precedenza.[7]
Altro esempio di enzima che catalizza una reazione che vede la formazione di un intermedio carbanionico è l’acetil-CoA carbossilasi (EC 6.4.1.2), che catalizza la tappa di comando della sintesi degli acidi grassi, ossia la carbossilazione dell’acetil-CoA a malonil-CoA.[9]
Bibliografia
- ^ a b c d Soderberg T. Organic chemistry with a biological emphasis. Volume I. Chemistry Publications. 2019
- ^ a b c Solomons T. W.G., Fryhle C.B., Snyder S.A. Solomons’ organic chemistry. 12th Edition. John Wiley & Sons Incorporated, 2017
- ^ a b Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011
- ^ Heterolysis, in IUPAC Compendium of Chemical Terminology, 3rd ed. International Union of Pure and Applied Chemistry; 2006. Online version 3.0.1, 2019. doi:1351/goldbook.H02809
- ^ Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
- ^ Homolysis, in IUPAC Compendium of Chemical Terminology, 3rd ed. International Union of Pure and Applied Chemistry; 2006. Online version 3.0.1, 2019. doi:1351/goldbook.H02851
- ^ a b Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. H. Freeman and Company, 2012
- ^ Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002
- ^ Garrett R.H., Grisham C.M. Biochemistry. 4th Edition. Brooks/Cole, Cengage Learning, 2010
 In questi casi il
In questi casi il 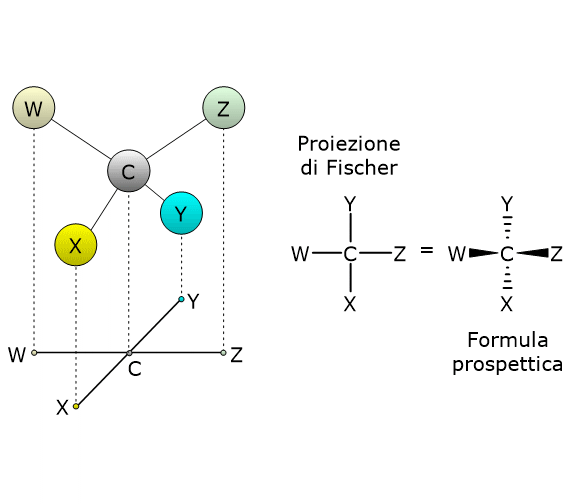
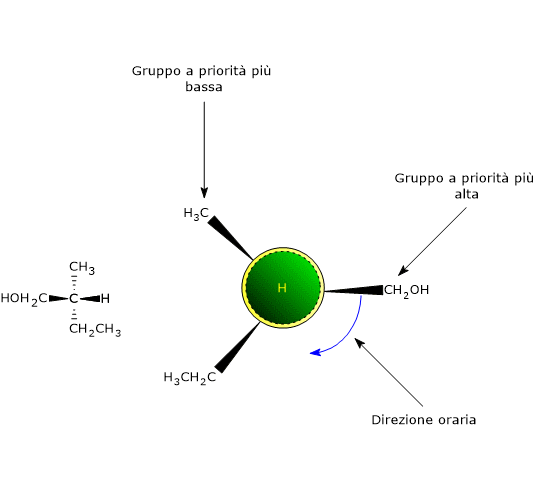
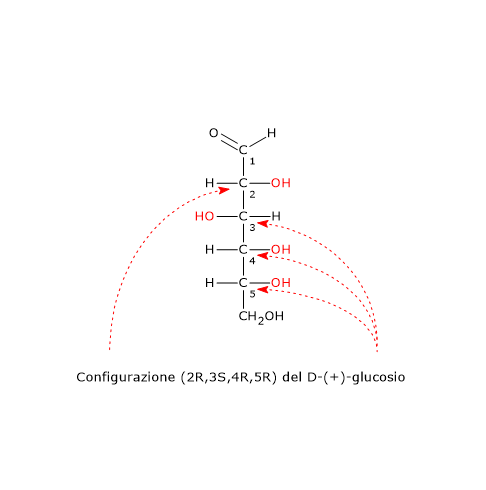
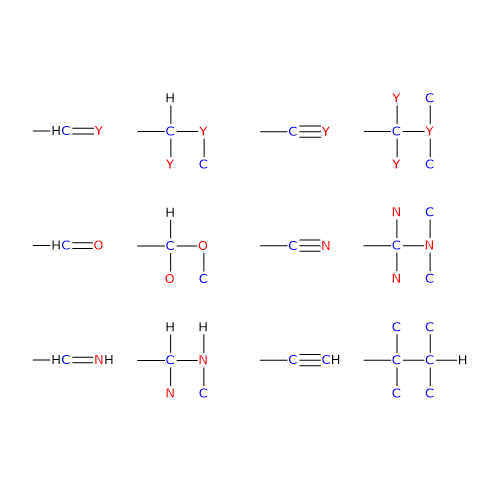 Nel caso di doppio legame C=Y, un atomo Y sarà da legare sull’atomo di carbonio, e un atomo di carbonio sull’atomo Y.
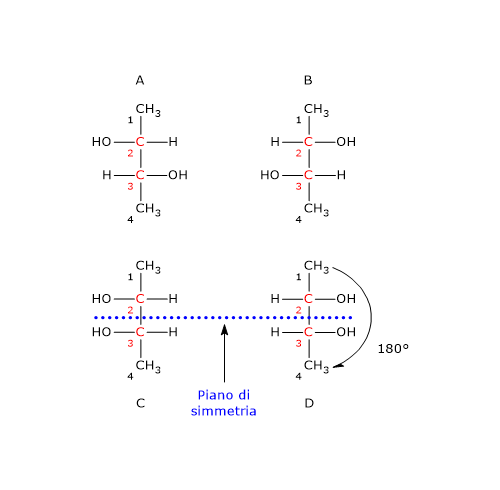
Nel caso di doppio legame C=Y, un atomo Y sarà da legare sull’atomo di carbonio, e un atomo di carbonio sull’atomo Y.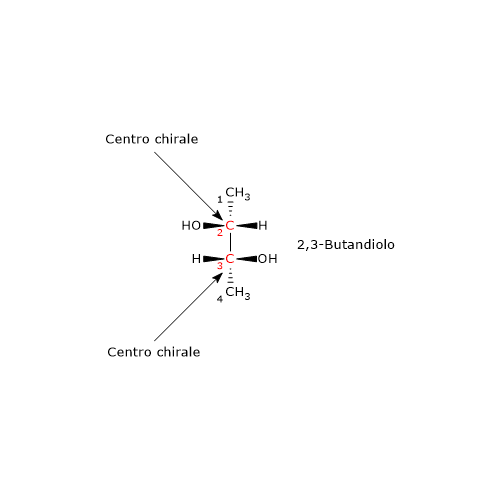 Si consideri il carbonio 2. L’ordine di priorità dei gruppi legati è: –OH > –CH2OHCH3 > –CH3 > –H. Si ruoti la molecola in modo che l’idrogeno, il gruppo con la priorità più bassa, sia diretto in direzione opposta all’osservatore. Disegnando una circonferenza partendo dal gruppo –OH, il gruppo con la priorità più alta, verso il gruppo –CH3, il gruppo a priorità più bassa, ci si muove in senso antiorario, per cui la configurazione del carbonio 2 è R. Applicando la stessa procedura al carbonio 3, si scopre che la sua configurazione è R. Quindi l’enantiomero in figura è il (2R,3R)-2,3-butandiolo.
Si consideri il carbonio 2. L’ordine di priorità dei gruppi legati è: –OH > –CH2OHCH3 > –CH3 > –H. Si ruoti la molecola in modo che l’idrogeno, il gruppo con la priorità più bassa, sia diretto in direzione opposta all’osservatore. Disegnando una circonferenza partendo dal gruppo –OH, il gruppo con la priorità più alta, verso il gruppo –CH3, il gruppo a priorità più bassa, ci si muove in senso antiorario, per cui la configurazione del carbonio 2 è R. Applicando la stessa procedura al carbonio 3, si scopre che la sua configurazione è R. Quindi l’enantiomero in figura è il (2R,3R)-2,3-butandiolo.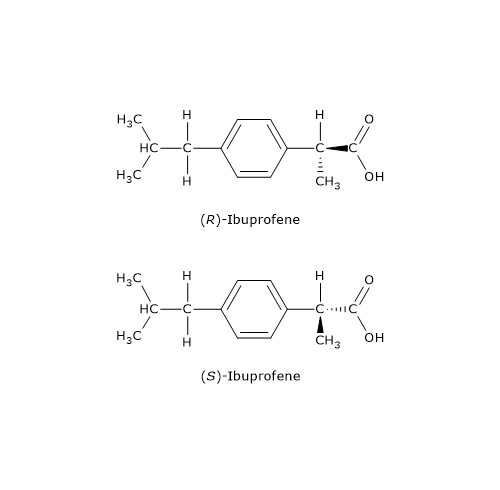
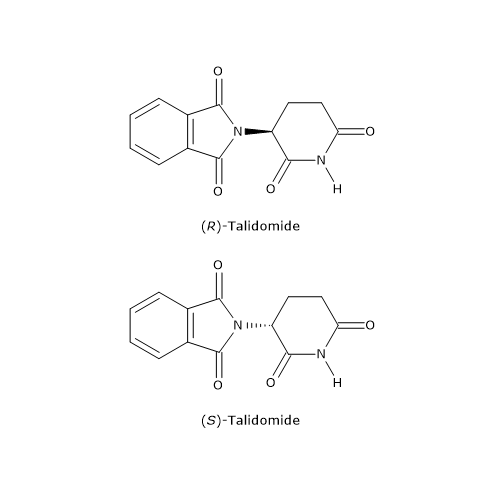 Il distomero, l’enantiomero S, poteva causare gravi difetti alla nascita, in particolare la focomelia. Questo è forse l’esempio più eclatante dell’importanza delle proprietà chirali delle molecole, che ha poi spinto gli organismi di salute pubblica a promuovere, da parte dell’industria farmaceutica, la sintesi di farmaci, talidomide compresa, contenenti un singolo enantiomero.
Il distomero, l’enantiomero S, poteva causare gravi difetti alla nascita, in particolare la focomelia. Questo è forse l’esempio più eclatante dell’importanza delle proprietà chirali delle molecole, che ha poi spinto gli organismi di salute pubblica a promuovere, da parte dell’industria farmaceutica, la sintesi di farmaci, talidomide compresa, contenenti un singolo enantiomero.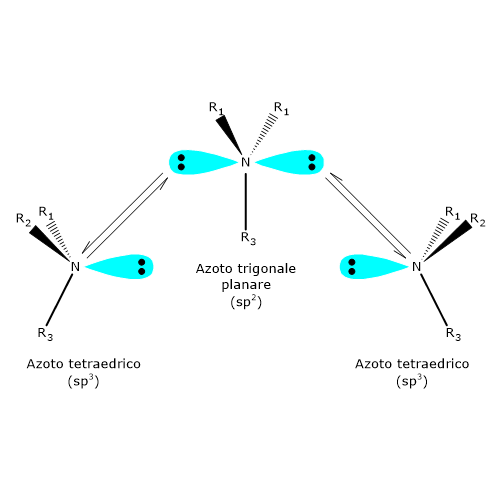 A temperatura ambiente, l’azoto inverte rapidamente la sua configurazione. Il fenomeno è noto come inversione dell’azoto, ossia, una rapida oscillazione dell’atomo e dei suoi ligandi, nel corso della quale l’azoto passa attraverso uno stato di transizione planare in cui ha ibridazione sp2. Come conseguenza, se l’azoto è il solo centro chirale della molecola, non c’è attività ottica in quanto si viene a formare una miscela racemica. L’inversione di configurazione viene evitata solamente in alcuni casi in cui l’azoto fa parte di una struttura ciclica che la impedisce. Dunque, la presenza di un centro chirale può non essere sufficiente per permettere la separazione dei rispettivi enantiomeri.
A temperatura ambiente, l’azoto inverte rapidamente la sua configurazione. Il fenomeno è noto come inversione dell’azoto, ossia, una rapida oscillazione dell’atomo e dei suoi ligandi, nel corso della quale l’azoto passa attraverso uno stato di transizione planare in cui ha ibridazione sp2. Come conseguenza, se l’azoto è il solo centro chirale della molecola, non c’è attività ottica in quanto si viene a formare una miscela racemica. L’inversione di configurazione viene evitata solamente in alcuni casi in cui l’azoto fa parte di una struttura ciclica che la impedisce. Dunque, la presenza di un centro chirale può non essere sufficiente per permettere la separazione dei rispettivi enantiomeri.
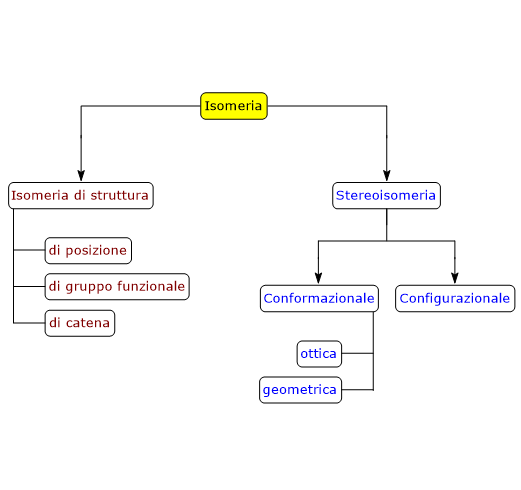
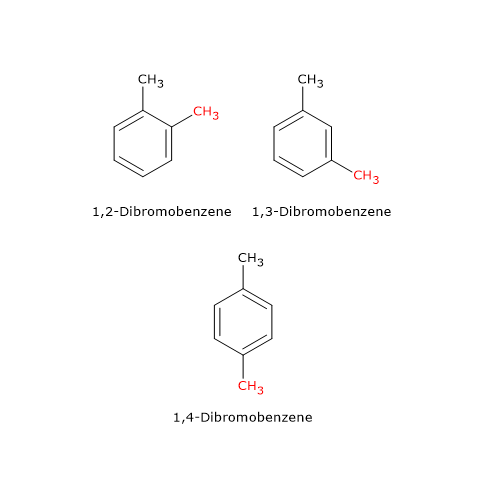
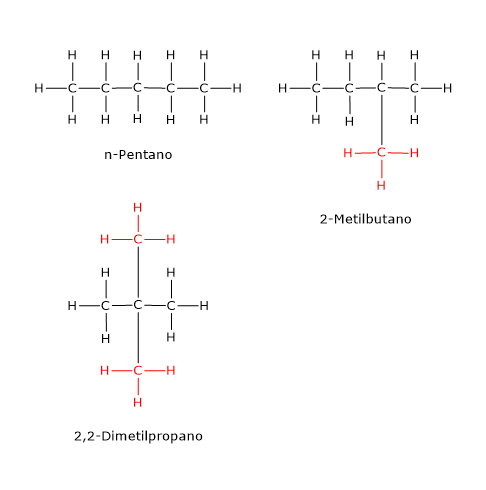 Esempi di isomeria di catena si hanno anche tra
Esempi di isomeria di catena si hanno anche tra