Nicola Tazzini, Biologo
Il Dott. Tazzini si è laureato con lode all’Università degli Studi di Pisa l’undici novembre del 1996 dopo aver svolto l’internato di tesi nel laboratorio di Biochimica del Dipartimento di Fisiologia e Biochimica della Facoltà di Scienze Naturali, Fisiche e Matematiche della medesima Università.
L’argomento della tesi è stato: “Studi sul meccanismo di citotossicità della combinazione di deossiadenosina e deossicoformicina su una linea cellulare derivante da carcinoma del colon umano”.
Ha superato l’esame di stato per l’abilitazione alla professione di Biologo presso l’Università degli Studi di Pisa il 10 maggio del 1998.
Si è specializzato con lode in Biochimica e Chimica Clinica il 3 ottobre del 2001 presso il Dipartimento di Chimica Biologica della Facoltà di Medicina e Chirurgia dell’Università degli Studi di Parma. L’argomento della tesi era: “Analisi dei parametrici ematochimici, enzimatici e non enzimatici, ad attività antiossidante in giovani atleti professionisti”.
Ha iniziato l’attività di Nutrizionista, come libera professione, il 2 febbraio del 2002.
CORSI DI AGGIORNAMENTO
2000
1. La pasta nell'alimentazione umana. Ancona 28 ottobre 2000. Associazione Biologi Nutrizionisti Italiani.
2. Corso di formazione ed aggiornamento in nutrizione e salute: il ruolo del Biologo. Associazione Scientifica Biologi Pisa.
2001
Elementi di nutrizione. Associazione Scientifica Biologi Pisa.
2002
L’alimentazione come fattore di salute: aspetti metodologici e aggiornamento professionale. Associazione Biologi Nutrizionisti Italiani.
2003
Alimentazione come fattore di salute - parte I. Associazione Biologi Nutrizionisti Italiani, evento formativo n. 4387-86945.
2004
1. Alimentazione come fattore di salute - parte II. Associazione Biologi Nutrizionisti Italiani, evento formativo n. 4387-103887.
2. Alimentazione ed età evolutiva. Associazione Biologi Nutrizionisti Italiani, evento formativo n. 4387-103200.
3. Attività sportiva, accrescimento e corretta alimentazione. Associazione Biologi Nutrizionisti Italiani, evento formativo n. 4387-130785.
4. Nutrizione e tumori. Prevenzione e stili di vita. PLANNING congressi Srl., evento formativo n. 1884-163563.
2005
1. Il doping. Linee guida e percorsi diagnostici: aspetti giuridici, biochimici, medici e tossicologici. Restless Architech of Human Possibilities S.a.s., evento formativo n. 6815-184678.
2. L’alimentazione nella terza età: problematiche nutrizionali e corretto stile alimentare. Associazione Biologi Nutrizionisti Italiani, evento formativo n. 4387-161719.
3. Evoluzione tecnico-normativa ed etica nello sviluppo della professione. Ordine Nazionale dei Biologi, evento formativo n. 1315-117400.
2006
1. Sport e nutrizione. Syntonie S.r.l., evento formativo n. 11670-227471.
2. Alimentazione e prevenzione: scegliere per stare bene. Ordine Nazionale dei Biologi, evento formativo n. 1315-230429.
3. Patologia, nutrizione e aspetti legislativi. Syntonie S.r.l., evento formativo n. 11670-227802.
4. Nutrizione: linee guida. Ordine Nazionale dei Biologi, evento formativo codice 8984.
2007
Argomenti di nutrizione. Alimenti come strumenti di salute e benessere. Ordine Nazionale dei Biologi, evento formativo n. 1315-294699.
2008
1. Ambiente esterno ed indoor. Risorse ed equilibri. Ordine Nazionale dei Biologi, evento formativo n. 1315-8019210.
2. La professione del Biologo nell'attuale evoluzione tecnico normativa. Ordine Nazionale dei Biologi, evento formativo n. 1315-8007383.
3. Prevenzione dell’obesità infantile: strategie nutrizionali dalla gravidanza all’età scolare. Ordine Nazionale dei Biologi, evento formativo n. 1315-8034743.
2009
1. Nutrizione, la pietra d’angolo. Fabbisogni nutrizionali e salute nell’epoca del genoma. Società Italiana di Nutrizione Umana, evento formativo n. 853-9038605.
2. Inglese scientifico. Med3, evento formativo FAD.
2010
1. L’evoluzione della sicurezza alimentare. Ordine Nazionale dei Biologi, evento formativo n. 1315-9019674.
2. Sicurezza alimentare e corretta nutrizione. Associazione Scientifica Biologi Pisa ed Ordine Nazionale dei Biologi, evento formativo n. 1315-10003500.
2011
Ruolo del caffè negli stati fisiologici e patologici. CMGRP Italia S.p.A.
2012
1. Nutrigenetica ed obesità. A.I.Nu.C. S.r.l.
2. Il senso della danza ormonale nella complessità del femminile: il ruolo dell’alimentazione. A.I.Nu.C. S.r.l.
3. Nutrizione nello sport: dall’allenamento al recupero post-gara. DocLeader S.r.l., evento formativo FAD n. 44-44292.
2013
1. Interpretazione delle analisi cliniche e consigli nutrizionali. A.I.Nu.C. S.r.l.
2. L’alimentazione nelle patologie cardiovascolari: prevenzione e strategie nutrizionali. Akesios group S.r.l., evento formativo n. 64355.
3. Il corretto uso dei probiotici. ALFA FCM S.r.l., evento formativo n. 3282-53306.
2014
1. L’alimentazione nelle patologie metaboliche. Prevenzione e strategie nutrizionali. A.C.S.I.A.N., evento formativo FAD n. 87271.
2. I disturbi glutine correlati: inquadramento, diagnosi, terapia. DNA Medical Communication, evento formativo FAD.
3. Nutrizione e laboratorio. Allmeetings S.r.l., evento formativo FAD n. 2828-85181.
4. La salute passa per l’intestino: il ruolo della permeabilità intestinale. A.I.Nu.C. S.r.l., evento formativo FAD.
2015
1. The best way per il biologo professionista. Provider Dynamicom Education S.r.l., evento formativo FAD n. 141172.
2. Gonfiore e discomfort addominale: intolleranza al lattosio, SIBO e sindrome dell’intestino irritabile. Allmeetings S.r.l., evento formativo FAD n. 2828-115855.
2016
Alimentazione consapevole e sana nutrizione avente come obiettivo didattico/formativo generale: sicurezza alimentare e/o patologie correlate. B.B.C. By Business Center S.r.l., evento formativo FAD n. 158730.
2017
La Medicina di Genere: oltre la pillola rosa e la pillola blu. FIB - Fondazione Italiana Biologi, evento formativo FAD n. 183576.
2018
La comprensione del paziente: dalla composizione corporea agli aspetti nutrizionali in condizioni fisio-patologiche. AKESIOS GROUP S.r.l., evento formativo FAD.
2020
1. Vitamina D e malattie cardiovascolari: mito o realtà? Ordine Nazionale dei Biologi, evento formativo FAD n. 284317.
2. La nutrizione oncologica. Ordine Nazionale dei Biologi, evento formativo FAD n. 286818.
3. Diabete e patologia cardiovascolare. Ordine Nazionale dei Biologi, evento formativo FAD n. 292248.
4. Colesterolo, malattie cardiovascolari e nuove linee guida: c’è ancora spazio per una sana alimentazione? Ordine Nazionale dei Biologi, evento formativo FAD n. 295801.
5. Antiossidanti e sport. Ordine Nazionale dei Biologi, evento formativo FAD n. 296268.
6. Inquinanti ambientali e malattie cardiovascolari. Ordine Nazionale dei Biologi, evento formativo FAD n. 306564.
2021
1. La promozione della salute e la valutazione di impatto sanitario (VIS): nuovi orizzonti sanitari per il Biologo. Ordine Nazionale dei Biologi, evento formativo FAD n. 316057.
2. Attività antivirale delle proteine del siero di latte. Ordine nazionale dei Biologi, evento formativo FAD n.324065.
3. Acido urico: dai meccanismi molecolari alle prospettive cliniche. Ordine Nazionale dei Biologi, evento formativo FAD n. 326154.
4. Efficienza metabolica. Ordine Nazionale dei Biologi, evento formativo FAD n. 334256.
5. Dieta chetogenica in pazienti con obesità e comorbilità obesità-associate: management nel setting ambulatoriale. Ordine Nazionale dei Biologi, evento formativo FAD n. 336621.
6. La meningite meningococcica: caratteristiche, diagnosi e vaccini. Ordine Nazionale dei Biologi, evento formativo FAD n. 329225.
BIBLIOGRAFIA E CITAZIONI
1. Bemi V., Tazzini N., Banditelli S., Giorgelli F., Pesi R., Turchi G., Mattana A., Sgarrella F., Tozzi M.G., Camici M. Deoxyadenosine metabolism in a human colon-carcinoma cell line (LoVo) in relation to its cytotoxic effect in combination with deoxycoformycin. Int J Cancer 1998;75(5):713-20 doi:https://doi.org/10.1002/(SICI)1097-0215(19980302)75:53.0.CO;2-1
2. Cassandra Studio. Nutraceutici e cibi funzionali. Youcanprint, 2015
3. Singh A.N., Baruah M.M. & Sharma N. Structure based docking studies towards exploring potential anti-androgen activity of selected phytochemicals against prostate cancer. Sci Rep 2017;7(1):1955. doi:https://doi.org/10.1038/s41598-017-02023-5
4. Wee T.T., Lun K.R. Teaching science in culturally relevant ways: ideas from Singapore teachers. World Scientific, 2014. doi:https://doi.org/10.1142/9252
Viene definita fosforilazione a livello del substrato la produzione di ATP o GTP a seguito del trasferimento di un gruppo fosfato da un substrato all’ADP o al GDP.[1]
L’energia rilasciata dalla rottura del legame ad alta energia del substrato rende possibile la formazione del legame fosfoanidridico.[2] Parte di questa energia agisce come una sorta di forza trainante che spinge la reazione verso la formazione dei nucleotidi trifosfato, mentre parte è immagazzinata nel legame neoformato.[3]
E’ un processo che coinvolge enzimi solubili e intermedi chimici, differenziandosi in questo dalla fosforilazione ossidativa e dalla fotofosforilazione, processi in cui la sintesi dei nucleotidi trifosfati coinvolge enzimi di membrana e gradienti protonici transmembrana.[2]
Esempi di fosforilazione a livello del substrato si hanno nella via glicolitica e nel ciclo dell’acido citrico.[3]
Sebbene la maggior parte dell’ATP prodotto in condizioni aerobiche derivi dalla fosforilazione ossidativa e, negli organismi fotosintetizzanti, anche dalla fotofosforilazione, la fosforilazione a livello del substrato è comunque essenziale nel metabolismo energetico, specialmente in condizioni anaerobiche.[4][5]
Due reazioni della glicolisi sono esempi ben noti di fosforilazione a livello del substrato.
Nella seconda fase della via glicolitica, la fase di recupero energetico, due delle cinque reazioni che la compongono permettono l’estrazione diretta e l’immagazzinamento nella molecola di ATP di parte dell’energia chimica della molecola di glucosio.[5]
Le reazioni coinvolte sono quelle catalizzate dalla fosfoglicerato chinasi (EC 2.7.2.3), e dalla piruvato chinasi (EC 2.7.1.40).
Nelle reazione un gruppo fosfato ad alta energia è trasferito all’ADP:
dall’1,3-bisfosfoglicerato, con produzione di una molecola di ATP e una molecola di 3-fosfoglicerato;
dal fosfoenolpiruvato, con produzione di una seconda molecola di ATP e una molecola di piruvato.[6]
Queste due reazioni di fosforilazione a livello del substrato consentono la sintesi di ATP anche in condizioni anaerobiche.[3]
Ciclo dell’acido citrico e fosforilazione del GDP
Nel ciclo dell’acido citrico si trova un altro esempio di fosforilazione a livello del substrato nella reazione catalizzata dalla succinil-CoA sintetasi, anche detta succinato-CoA ligasi (EC 6.2.1.4). L’enzima catalizza la scissione del legame tioestere ad alta energia del succinil-CoA, analogo a quello presente nell’acetil-CoA. L’energia rilasciata rende possibile la formazione di un legame fosfoanidridico, quindi la sintesi del GTP dal GDP.[5]
Bibliografia
^ Ha C.E., Bhagavan N.V.. Chapter 11 – Carbohydrate metabolism I: glycolysis and the tricarboxylic acid cycle. Editor(s): Chung Eun Ha, N.V. Bhagavan. Essentials of Medical Biochemistry. 3rd Edition. Academic Press, 2023, pages 203-227. doi:10.1016/B978-0-323-88541-6.00030-2
Gli amminoacidi chetogenici sono amminoacidi il cui scheletro carbonioso può essere catabolizzato per intero o almeno in parte ad acetoacetil-CoA o acetil-CoA, molecole che possono essere utilizzate come precursori per la sintesi dei corpi chetonici, da cui il nome, o degli acidi grassi.[1]
Gli amminoacidi sono i costituenti fondamentali delle proteine. Una volta che il loro pool disponibile è sufficiente a soddisfare le necessità della sintesi proteica, non disponendo l’organismo di riserve specifiche di amminoacidi paragonabili a quelle del glicogeno per il glucosio o dei trigliceridi per gli acidi grassi, ne converte l’eccesso in intermedi del ciclo dell’acido citrico, impiegandoli nella produzione di energia.[2]
Quando il fabbisogno energetico cellulare è stato soddisfatto, i residui carboniosi derivanti dalla catabolismo amminoacidico vengono reindirizzati verso la sintesi del glucosio, degli acidi grassi, o dei corpi chetonici.[3]
Gli amminoacidi proteinogenici possono essere classificati come amminoacidi chetogenici o amminoacidi glucogenici sulla base del destino metabolico del loro scheletro carbonioso.[4] Lo scheletro carbonioso degli amminoacidi chetogenici viene catabolizzato ad acetoacetil-CoA e/o acetil-CoA, mentre quello degli amminoacidi glucogenici in uno o più dei seguenti cinque metaboliti precursori glucogenici: piruvato, ossalacetato, α-chetoglutarato, succinil-CoA e fumarato.[5]
Questa modalità di classificazione non è però univoca in quanto cinque amminoacidi sono sia chetogenici che glucogenici, poiché dal loro catabolismo si origina almeno un precursore glucogenico e uno chetogenico.[6]
Solamente due dei venti amminoacidi che compongono le proteine sono esclusivamente chetogenici: la leucina e la lisina. Dal catabolismo del loro scheletro carbonioso si origina acetoacetil-CoA e acetil-CoA.[6]
Amminoacido
Catabolita
chetogenico
glucogenico
Fenilalanina
Acetoacetil-CoA
Fumarato
Isoleucina
Acetil-CoA
Succinil-CoA
Leucina *
Acetil-CoA
Acetoacetil-CoA
Lisina *
Acetoacetil-CoA
Tirosina
Acetoacetil-CoA
Fumarato
Treonina
Acetil-CoA
Succinil-CoA
Triptofano
Acetil-CoA
Acetoacetil-CoA
Piruvato
* Amminoacidi esclusivamente chetogenici
Altri cinque amminoacidi, ossia isoleucina, fenilalanina, treonina, triptofano, e tirosina, sono invece sia chetogenici che glucogenici in quanto il catabolismo dello scheletro carbonioso porta alla formazione di acetil-CoA e/o acetoacetil-CoA e un precursore glucogenico.[5]
L’utilizzo degli scheletri carboniosi degli amminoacidi è preceduto dalla rimozione del gruppo amminico. Alanina e glutammato, amminoacidi glucogenici, svolgono un ruolo chiave nel trasporto dei gruppi amminici dai tessuti extraepatici al fegato. In particolare, l’alanina viene trasportata dal muscolo e da altri tessuti periferici al fegato attraverso il ciclo glucosio-alanina.[7]
Basi biochimiche
Acetil-CoA e acetoacetil-CoA non sono precursori glucogenici. La spiegazione risiede nella stechiometria del ciclo dell’acido citrico e nell’incapacità degli animali di convertire l’acetil-CoA in piruvato.[8]
L’acetil-CoA entra nel ciclo dell’acido citrico a livello della reazione catalizzata dalla citrato sintasi (EC 2.3.3.1). L’enzima catalizza la condensazione dell’acetil-CoA con l’ossalacetato a formare il citrato. Dunque si passa da un composto a quattro atomi di carbonio a uno a sei atomi di carbonio, con un guadagno netto di due atomi carboni.
Tuttavia, nelle due successive decarbossilazioni ossidative del ciclo, catalizzate dalla isocitrico deidrogenasi (EC 1.1.1.42) e dal complesso multienzimatico dell’α-chetoglutarato deidrogenasi, sono perduti due atomi di carbonio.[4]
Quindi l’ingresso dell’acetil-CoA nel ciclo non comporta alcun guadagno netto di carbonio.[6]
La discriminante è quindi rappresentata dal punto d’ingresso delle unità carboniose a monte o a valle delle decarbossilazioni ossidative del ciclo dell’acido citrico.[8]
A tutto ciò va aggiunto che negli animali non esiste una via metabolica che permetta di produrre piruvato dall’acetil-CoA, data anche l’irreversibilità della reazione catalizzata dal complesso della piruvato deidrogenasi, la decarbossilazione ossidativa del piruvato ad acetil-CoA.[8]
Amminoacidi chetogenici nel digiuno prolungato
A differenza degli amminoacidi glucogenici, gli amminoacidi chetogenici non hanno un ruolo cruciale durante il digiuno prolungato o nelle diete con forte restrizione dei carboidrati.
Durante il digiuno prolungato, la principale fonte di acetil-CoA per la chetogenesi è la beta-ossidazione degli acidi grassi, mentre il contributo degli amminoacidi chetogenici è marginale. In queste condizioni, il ruolo centrale è svolto dagli amminoacidi glucogenici, che, attraverso la gluconeogenesi, garantiscono il mantenimento dell’omeostasi glicemica quando le riserve di glicogeno epatico sono esaurite.[5]
Ciclo del gliossilato
Piante, lieviti, e molti batteri possono utilizzare l’acetil-CoA per la sintesi del glucosio grazie alla via metabolica chiamata ciclo del gliossilato. Tale ciclo ha alcune reazioni in comune con il ciclo dell’acido citrico, due esclusive catalizzate dalla isocitrato liasi (EC 4.1.3.1) e malato sintasi (EC 2.3.3.9), ma non ha reazioni di decarbossilazione. Quindi gli organismi che possiedono il ciclo del gliossilato sono in grado di utilizzare gli acidi grassi e gli amminoacidi chetogenici per la sintesi del glucosio.[9]
Bibliografia
^ D’Andrea G. Classifying amino acids as gluco(glyco)genic, ketogenic, or both. Biochem Educ 2000;28(1):27-28. doi:10.1016/s0307-4412(98)00271-4
^ Brosnan J.T. Interorgan amino acid transport and its regulation. J Nutr 2003;133(6 Suppl 1):2068S-2072S. doi:10.1093/jn/133.6.2068S
^ Litwack G. Human biochemistry. 2nd Edition. Academic Pr, 2021.
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ abc Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009.
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012.
^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
^ abc Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
^ Kondrashov F.A., Koonin E.V., Morgunov I.G., Finogenova T.V., Kondrashova M.N. Evolution of glyoxylate cycle enzymes in Metazoa: evidence of multiple horizontal transfer events and pseudogene formation. Biol Direct 2006;1:31. doi:10.1186/1745-6150-1-31
Sono definiti amminoacidi glucogenici quegli amminoacidi il cui scheletro carbonioso può essere catabolizzato in toto o in parte in precursori della sintesi del glucosio o gluconeogenesi, da cui il nome.[1]
Gli amminoacidi sono i mattoni costituenti le proteine. Quando il loro pool soddisfa le esigenze cellulari per la sintesi proteica, non esistendo nell’organismo depositi di amminoacidi analoghi al glicogeno per il glucosio o ai trigliceridi per gli acidi grassi, l’eccesso amminoacidico viene catabolizzato a precursori del ciclo dell’acido citrico e utilizzato per la produzione di energia.[2]
Tuttavia, nel momento in cui le esigenze energetiche della cellula sono soddisfatte, gli scheletri carboniosi derivanti dal catabolismo amminoacidico sono utilizzati per la sintesi di acidi grassi, corpi chetonici, o glucosio.[3]
Sulla base del destino metabolico dello scheletro carbonioso si individuano amminoacidi glucogenici e amminoacidi chetogenici. Tale classificazione non è però netta in quanto, dei venti amminoacidi standard che si ritrovano nelle proteine, cinque sono sia glucogenici che chetogenici.[4]
La sintesi del glucosio dagli amminoacidi glucogenici è resa possibile dal fatto che il loro scheletro carbonioso viene catabolizzato, per intero o in parte, in piruvato, ossalacetato, α-chetoglutarato, succinil-CoA, o fumarato.[5]
Piruvato e ossalacetato sono intermedi della gluconeogenesi, mentre l’ingresso degli altri tre metaboliti nel ciclo dell’acido citrico permette un guadagno netto di unità carboniose per il ciclo medesimo, unità che quindi potranno entrare nella gluconeogenesi come ossalacetato.[6]
Gli amminoacidi glucogenici sono particolarmente importanti nel digiuno prolungato, rappresentando una fonte rilevante di precursori per la gluconeogenesi, secondi solo al glicerolo derivante dalla lipolisi nel tessuto adiposo.[7]
Dei venti amminoacidi costituenti le proteine tredici sono esclusivamente glucogenici, ossia dal catabolismo del loro scheletro carbonioso si originano solo precursori del glucosio.[4]
Amminoacido
Catabolita
chetogenico
glucogenico
Alanina
Piruvato
Arginina
α-Chetoglutarato
Asparagina
Fumarato
Aspartato
Ossalacetato
Cisteina
Piruvato
Fenilalanina *
Acetoacetil-CoA
Fumarato
Glicina
Piruvato
Glutammato
α-Chetoglutarato
Glutammina
α-Chetoglutarato
Isoleucina *
Acetil-CoA
Succinil-CoA
Istidina
α-Chetoglutarato
Metionina
Succinil-CoA
Prolina
α-Chetoglutarato
Serina
Piruvato
Tirosina *
Acetoacetil-CoA
Fumarato
Treonina *
Acetil-CoA
Succinil-CoA
Triptofano *
Acetil-CoA
Acetoacetil-CoA
Piruvato
Valina
Succinil-CoA
* Amminoacidi sia glucogenici che chetogenici
Cinque amminoacidi, ossia isoleucina, fenilalanina, treonina, triptofano, e tirosina, sono invece sia glucogenici che chetogenici in quanto parte del loro scheletro carbonioso viene catabolizzato a precursori glucogenici e parte ad acetil-CoA e/o acetoacetil-CoA.[8]
L’utilizzo degli scheletri carboniosi degli amminoacidi deve essere preceduto dalla rimozione del loro gruppo amminico. Alanina e glutammato, le principali molecole responsabili del trasporto dei gruppi amminici dai tessuti extraepatici al fegato, sono amminoacidi glucogenici particolarmente importanti nei mammiferi. L’alanina è il principale substrato gluconeogenetico per il fegato, e arriva all’organo dal muscolo e da altri tessuti periferici seguendo la via del ciclo glucosio-alanina.[9]
Basi biochimiche
Come con gli amminoacidi chetogenici, anche con gli amminoacidi glucogenici l’analisi della stechiometria del ciclo dell’acido citrico chiarisce perché il loro scheletro carbonioso sia un precursore per la sintesi del glucosio. La discriminante è rappresentata dal punto d’ingresso nel ciclo dell’acido citrico.[6]
L’ingresso nel ciclo dei carboni di derivazione amminoacidica in forma di α-chetoglutarato, succinil-CoA, e fumarato comporta un guadagno netto di unità carboniose. Infatti, con l’esclusione dell’α-chetoglutarato, gli altri metaboliti vi entrano a valle delle due reazioni di decarbossilazione ossidativa catalizzate dalla isocitrico deidrogenasi (EC 1.1.1.42) e dal complesso multienzimatico dell’α-chetoglutarato deidrogenasi.[5]
Questo permette al ciclo un guadagno netto di un’unità carboniosa in caso di ingresso a livello dell’α-chetoglutarato, o due se l’ingresso avviene come succinil-CoA, e fumarato. Le unità carboniose potranno quindi entrare nella gluconeogenesi in forma di ossalacetato.[4]
Inoltre, anche l’irreversibilità della reazione catalizzata dal complesso della piruvato deidrogenasi, ossia la decarbossilazione ossidativa del piruvato ad acetil-CoA, e l’assenza negli animali di altre vie metaboliche che permettano la sintesi di piruvato dall’acetil-CoA, non permettono all’acetil-CoA di essere un substrato glucogenico.[6]
Amminoacidi glucogenici nel digiuno prolungato
Gli amminoacidi glucogenici sono particolarmente importanti nel digiuno prolungato e nelle diete con forte restrizione dell’apporto di carboidrati. In queste condizioni sono tra i più importanti precursori per la sintesi del glucosio.[6] L’utilizzo del loro scheletro carbonioso, assieme al glicerolo e al propionato, un acido grasso a catena corta, concorre infatti al mantenimento dell’omeostasi glicemica, via gluconeogenesi, quando le riserve epatiche di glicogeno sono esaurite.[7]
Tuttavia, anche in condizioni fisiologiche, ad esempio nel turn-over delle proteine cellulari, o a seguito di un pasto ricco in proteine, gli amminoacidi in eccesso rispetto alle necessità della sintesi proteica saranno catabolizzati e utilizzati, sulla base dello stato metabolico, a fini energetici o anabolici, per la sintesi del glucosio e di seguito per la glicogenosintesi, la sintesi dei corpi chetonici o degli acidi grassi, a seconda dell’amminoacido in questione.[2]
Bibliografia
^ D’Andrea G. Classifying amino acids as gluco(glyco)genic, ketogenic, or both. Biochem Educ 2000;28(1):27-28. doi:10.1016/s0307-4412(98)00271-4
^ ab Brosnan J.T. Interorgan amino acid transport and its regulation. J Nutr 2003;133(6 Suppl 1):2068S-2072S. doi:10.1093/jn/133.6.2068S
^ Litwack G. Human biochemistry. 2nd Edition. Academic Pr, 2021.
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012.
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ abcd Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
^ ab Kuriyama H., Shimomura I., Kishida K., Kondo H., Furuyama N., Nishizawa H., Maeda N., Matsuda M., Nagaretani H., Kihara S., Nakamura T., Tochino Y., Funahashi T., Matsuzawa Y. Coordinated regulation of fat-specific and liver-specific glycerol channels, aquaporin adipose and aquaporin 9. Diabetes 2002;51(10):2915-2921. 10.2337/diabetes.51.10.2915
^ Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009.
^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
Il 2,3-bisfosfoglicerato (2,3-BPG) è la base coniugata dell’acido 2,3-bisfosfoglicerico.[1]
Nel globulo rosso, a livello del mare, è presente in concentrazioni vicine a quella dell’emoglobina (Hb), circa 5 mM. Negli altri tipi cellulari è invece presente in tracce.[2]
Nell’eritrocita viene sintetizzato a partire dall’1,3-bisfosfoglicerato (1,3-BPG) nella prima reazione dello shunt di Rapoport-Luebering.[3][4] Essendo lo shunt di Rapoport-Luebering una deviazione dalla via glicolitica a monte delle reazioni che portano alla sintesi dell’ATP, la produzione di 2,3-bisfosfoglicerato comporta un costo energetico di due molecole di ATP per molecola prodotta.[5]
Il 2,3-BPG ed è uno dei regolatori dell’affinità dell’emoglobina per l’ossigeno.[6] E’ inoltre necessario all’attività catalitica della fosfoglicerato mutasi (EC 5.4.2.1), ed è un attivatore allosterico della ribosio fosfato pirofosfochinasi (EC 2.7.6.1).[2][7]
Legandosi all’emoglobina ne riduce l’affinità per l’ossigeno facilitandone il rilascio a livello dei tessuti periferici.[6]
Nel globulo rosso la concentrazione del 2,3-bisfosfoglicerato può variare in risposta a modifiche nel flusso di carbonio attraverso la via glicolitica. Tali modificazioni possono essere conseguenti a variazioni di altitudine, a malattie che compromettano l’ossigenazione del sangue, o difetti a carico di enzimi glicolitici, e si riflettono sull’affinità di legame tra emoglobina e ossigeno.[8][9][10]
Il 2,3-bisfosfoglicerato è la base coniugata dell’acido 2,3-bisfosfoglicerico. Ha un peso molecolare di 261,00 g/mol e formula molecolare C3H3O10P2-5.[1]
Secondo la nomenclatura IUPAC il suo nome è (2R)-2,3-bis(fosfonoossi)propanoato.[11]
Il pKa dell’acido 2,3-bisfosfoglicerico è pari a 0,48. Ne consegue che a pH fisiologico è presente quasi esclusivamente in forma ionizzata, il 2,3-BPG.[11]
E’ solubile in acqua, con un valore pari a 9,69 g/L.[1]
Il 2,3-bisfosfoglicerato è un isomero dell’1,3-bisfosfoglicerato.
Metabolismo
Il 2,3-bisfosfoglicerato viene prodotto a partire dall’intermedio glicolitico 1,3-bisfofoglicerato nella prima delle due tappe che compongono lo shunt di Rapoport-Luering.[3][4]
Entrambe le reazioni dello shunt sono catalizzate dalla bisfosfoglicerato mutasi (EC 5.4.2.4), enzima multifunzionale in grado di svolgere tre attività principali, potendo agire come 2,3-BPG sintasi, l’attività principale, 2,3-BPG fosfatasi, e 2,3-BPG mutasi.[12]
Nella prima reazione dello shunt di Rapoport-Luebering l’enzima agisce come sintasi e catalizza l’isomerizzazione dell’1,3-bisfosfoglicerato a 2,3-bisfosfoglicerato. L’enzima catalizza il trasferimento intermolecolare di un gruppo fosforico dal C-1 dell’1,3-bisfosfoglicerato al C-2 del 3-fosfoglicerato, che quindi deve essere presente nel sito attivo. La reazione comporta la conversione del 3-fosfoglicerato a 2,3-bisfosfoglicerato mentre l’1,3-BPG diviene il nuovo 3-fosfoglicerato.[6]
Nella seconda reazione dello shunt, il 2,3-bisfosfoglicerato viene defosforilato a 3-fosfoglicerato. La reazione è catalizzata dall’attività fosfatasica della bisfosfoglicerato mutasi. Il 3-fosfoglicerato rientra quindi nella via glicolitica a livello dell’ottava tappa, catalizzata dalla fosfoglicerato mutasi (EC 5.4.2.1).[5]
Costo energetico della sintesi del 2,3-bisfosfoglicerato
La sintesi del 2,3-bisfosfoglicerato comporta un costo energetico per il globulo rosso pari a due molecole di ATP per molecola prodotta.[5]
L’1,3-BPG è il substrato della settima reazione della glicolisi, il trasferimento catalizzato dalla fosfoglicerato chinasi (EC 2.7.2.3) del gruppo fosforico ad alta energia legato al C-1 all’ADP, con formazione di 3-fosfoglicerato e ATP. La fosfoglicerato chinasi catalizza infatti la prima delle due fosforilazioni a livello del substrato che, lungo la via glicolitica, portano alla conservazione di parte dell’energia chimica presente nel glucosio in forma di ATP, la moneta di scambio energetico della cellula.[6]
Dato che, in condizioni fisiologiche, lo shunt di Rapoport-Luebering intercetta circa il 20% del flusso glicolitico, il costo energetico pagato dal globulo rosso per la sintesi del 2,3-bisfosfoglicerato implica l’esistenza di un equilibrio finemente regolato tra il fabbisogno energetico della cellula e la necessità di mantenere l’emoglobina in un adeguato stato di deossigenazione e ossigenazione.[13][14]
Ruolo del 2,3-bisfosfoglicerato
Il 2,3-bisfosfoglicerato svolge almeno tre funzioni.
E’ necessario per l’attività catalitica della fosfoglicerato mutasi. Ne consegue che la molecola ha anche un ruolo nel controllo del livello degli intermedi glicolitici.[7]
E’ uno degli attivatori allosterici della ribosio fosfato pirofosfochinasi o PRPP sintetasi. L’enzima catalizza l’attivazione del ribosio-5-fosfato, uno dei due prodotti della via del pentoso fosfato, a 5-fosforibosil-1-pirofosfato (PRPP), intermedio nella sintesi de novo delle purine.[2]
Il 2,3-bisfosfoglicerato è coinvolto, assieme ad altri fattori, nella regolazione allosterica dell’affinità dell’emoglobina per l’ossigeno, concorrendo a regolarne il rilascio a livello dei tessuti periferici.[15] Nell’uomo, nella maggior parte dei primati e in molti altri mammiferi il 2,3-bisfosfoglicerato riduce l’affinità dell’emoglobina per l’ossigeno. In questo modo la molecola favorisce la dissociazione dell’ossigeno dall’emoglobina, facilitandone la cessione ai tessuti.[6] L’azione del 2,3-BPG è un esempio di regolazione allosterica eterotropica, ossia un regolazione allosterica in cui l’effettore è diverso dal normale ligando della proteina.[5]
Emoglobina
L’emoglobina è presente nei globuli rossi, e, in quasi tutti i vertebrati, ha il compito di trasportare l’ossigeno molecolare ai tessuti.
La proteina ha una struttura quaternaria formata da quattro subunità e contiene quattro gruppi prostetici eme, uno per subunità, ognuno dei quali in grado di legare, reversibilmente, una molecola di ossigeno.[13]
In condizioni fisiologiche nell’uomo si distinguono due tipi di emoglobina: quella presente nell’adulto, indicata come emoglobina A, e quella fetale, l’emoglobina F.
L’emoglobina A è formata da due catene α, di 141 residui amminoacidici, e due catene β, di 146 residui amminoacidici. Dunque è un tetramero α2β2.
Nell’emoglobina F le catene β sono sostituite da catene γ, molto simili, ma non identiche alle β. Ne risulta un tetramero α2γ2.[13] Questa differenza ha una grande influenza sull’affinità della proteina per l’ossigeno.[5]
L’emoglobina può esistere in due conformazioni indicate come stato T, teso, e stato R, rilassato.
Sebbene l’ossigeno si leghi all’emoglobina sia nello stato T che in quello R, ha un’affinità maggiore per lo stato R, che viene stabilizzato dal legame. Il legame dell’ossigeno da parte dell’emoglobina è modulato da diversi fattori, alcuni con azione a breve termine e altri, come il 2,3-bisfosfoglicerato, che agiscono più a lungo termine.[6]
Cooperatività del legame emoglobina-ossigeno
L’emoglobina nello stato deossigenato si trova in conformazione T.
Il legame dell’ossigeno all’emoglobina nello stato T può avvenire solo sul gruppo eme di una delle subunità α, essendo i gruppi eme delle subunità β nello stato T virtualmente inaccessibili.[6]
Il legame della prima molecola di ossigeno innesca una variazione conformazionale nella subunità α che la converte nello stato R. Tale modifica è trasmessa alle subunità adiacenti mediante interazioni subunità-subunità, modifica che determina il passaggio della seconda subunità α allo stato R. Ciò innesca cambiamenti conformazionali sequenziali che portano al passaggio della struttura quaternaria dell’emoglobina che lega due molecole di ossigeno, Hb:(O2)2, dallo stato T a quello R.
A questo punto anche le subunità β possono legare l’ossigeno.[6]
Questo tipo di legame è detto cooperativo ed è alla base dell’andamento sigmoidale della curva di saturazione dell’emoglobina.[16]
Affinità dell’emoglobina per l’ossigeno
Nei capillari polmonari l’ossigeno si lega all’emoglobina, e, a mezzo della circolazione sanguigna, raggiunge i tessuti periferici cui viene rilasciato.
L’affinità di legame tra emoglobina e ossigeno è determinata principalmente dalla struttura dell’emoglobina. Tuttavia risulta modulata da vari fattori quali:
Le modifiche nell’affinità del legame Hb-O2 che si verificano nel sistema circolatorio ad opera dei suddetti fattori sono tali da ottimizzare il carico di ossigeno a livello polmonare e la sua cessione nei tessuti periferici.[10]
Graficamente, le modifiche dell’affinità di legame indotte dagli effettori allosterici e dalla temperatura, si traducono in spostamenti della curva di saturazione dell’emoglobina verso destra, nel caso di una diminuzione dell’affinità di legame, o verso sinistra, quando l’affinità di legame aumenta.[5]
Temperatura, pH, e CO2 operano un regolazione a breve termine.[10] Se ad esempio si considera il muscolo scheletrico durante l’esercizio fisico, vi si osserva un aumento della temperatura, della concentrazione degli ioni idrogeno e della pressione parziale della CO2. Alte concentrazioni di H⁺ e CO2 riducono l’affinità dell’emoglobina per l’ossigeno, un effetto noto come effetto Bohr. Anche l’aumento della temperatura diminuisce l’affinità. Tutto ciò favorisce il rilascio di ossigeno al tessuto muscolare.[17]
Anche gli ioni cloro e il 2,3-bisfosfoglicerato riducono l’affinità dell’emoglobina per l’ossigeno, ma si associano a una regolazione più a lungo termine.[10]
2,3-Bisfosfoglicerato ed emoglobina A
Il 2,3-bisfosfoglicerato si va a legare all’emoglobina A a mezzo di legami ionici che si stabiliscono con residui delle catene β carichi positivamente.
I legami tra il 2,3-bisfosfoglicerato e l’emoglobina si stabiliscono con le catene laterali della lisina 82, istidina143, e il gruppo ammino-terminale di ciascuna della due catene β. Questi residui vanno a formare una tasca elettrostatica complementare sia alla conformazione che alla distribuzione di carica del 2,3-BPG.[5]
Il 2,3-BPG si lega all’emoglobina deossigenata e stabilizza lo stato T. Anche gli ioni idrogeno, cloro e l’anidride carbonica stabilizzano lo stato T.[10]
Nel passaggio dallo stato T a quello R, tra le varie modifiche conformazionali che occorrono nella struttura quaternaria dell’emoglobina, si verifica anche il restringimento della tasca di legame per 2,3-BPG tra le subunità β, impedendone quindi il legame. Degno di nota è il fatto che mentre l’emoglobina ha quattro siti di legame per l’ossigeno molecolare, ne ha solo uno per il 2,3-BPG.[6] Inoltre, il sito di legame per il 2,3-BPG è lontano da quello per l’ossigeno.[5]
2,3-Bisfosfoglicerato ed emoglobina F
Le catene γ dell’emoglobina F, analogamente alle catene β dell’emoglobina A, vanno a formare la tasca di legame per il 2,3-BPG.[5]
Tuttavia, nella tasca di legame sono presenti due cariche positive in meno rispetto all’emoglobina A. Infatti, in posizione 143 delle catene γ l’istidina è sostituita da una serina.[2]
Questa piccola differenza nella struttura primaria fa si che il 2,3-bisfosfoglicerato si leghi con minor affinità, e di conseguenza che l’emoglobina F abbia un’affinità per l’ossigeno maggiore rispetto all’emoglobina materna. Ciò concorre ad assicurare il trasferimento dell’ossigeno dall’emoglobina A a quella F, quindi dalla circolazione materna a quella fetale.[6]
Variazioni della concentrazione del 2,3-bisfosfoglicerato
La concentrazione del 2,3-bisfosfoglicerato nel globulo rosso può essere influenzata da diversi fattori, sia fisiologici che patologici.[2]
Tra le condizioni fisiologiche c’è l’adattamento all’alta montagna, mentre tra le condizioni patologiche si hanno difetti genetici a carico di enzimi glicolitici sia a valle che a monte della reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12), o malattie che causano ipossia.[18][19]
In tutti i casi le variazioni sono conseguenti a modifiche nella sua velocità di sintesi, e si riflettono sull’affinità dell’emoglobina per l’ossigeno.
Adattamento all’alta montagna
L’adattamento all’alta montagna è un processo piuttosto complesso che comporta anche un aumento del numero dei globuli rossi e del loro contenuto in emoglobina. Queste modifiche richiedono, per completarsi, settimane. Tuttavia, già dopo un giorno di permanenza in alta quota viene percepito un certo grado di adattamento. Questo effetto è conseguenza dell’aumento della concentrazione eritrocitaria del 2,3-bisfosfoglicerato.[2]
Ciò che accade è che la bassa pressione parziale dell’ossigeno attiva la glicolisi. A sua volta questo comporta un aumento del flusso di carbonio attraverso lo shunt di Rapoport-Luebering con conseguente aumento della sintesi del 2,3-bisfosfoglicerato, la cui concentrazione che può arrivare sino a 8 mM.
Tale aumento determina una riduzione dell’affinità dell’emoglobina per l’ossigeno, che ne facilita il rilascio ai tessuti periferici, concorrendo così all’adattamento all’alta montagna.[5]
Anemia
Variazioni della concentrazione eritrocitaria del 2,3-bisfosfoglicerato si hanno anche in due malattie a trasmissione autosomica recessiva: l’anemia emolitica non sferocitica da deficit di esochinasi e l’anemia emolitica da deficit di piruvato chinasi negli eritrociti.[18][19]
Nell’anemia emolitica dovuta a un deficit di piruvato chinasi negli eritrociti (EC 2.7.1.40), la carenza dell’enzima causa un incremento della sintesi di 2,3-BPG e, di conseguenza, una riduzione dell’affinità dell’emoglobina per l’ossigeno.[8] Un incremento della concentrazione del 2,3-BPG si verifica anche nei soggetti affetti da condizioni che limitano l’ossigenazione del sangue, che causano cioè ipossia. Un esempio è l’insufficienza cardiopolmonare.[2]
Nell’anemia emolitica non sferocitica causata da un deficit di esochinasi (EC 2.7.1.1), il deficit enzimatico eritrocitario causa una riduzione della concentrazione del 2,3-bisfosfoglicerato, portando così a un aumento dell’affinità dell’emoglobina per l’ossigeno.[9]
^ abcdefg Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
^ ab Rapoport S., Luebering J. Glycerate-2,3-diphosphatase. J Biol Chem 1951;189(2):683-694.
^ ab Rapoport S., Luebering J. The formation of 2,3-diphosphoglycerate in rabbit erythrocytes: the existence of a diphosphoglycerate mutase. J Biol Chem 1950;183(2):507-516.
^ abcdefghi Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012.
^ ab Oslund R.C., Su X., Haugbro M., Kee J-M., Esposito M., David Y., Wang B., Ge E., Perlman D.H., Kang Y., Muir T.W., Rabinowitz J.D. Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat Chem Biol 2017;13:1081-1087. doi:10.1038/nchembio.2453
^ ab Enegela O.A., Anjum F. Pyruvate Kinase Deficiency. [Updated 2023 Apr 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560581/
^ ab Paglia D.E., Shende A., Lanzkowsky P., Valentine W.N.. Hexokinase “New Hyde Park”: a low activity erythrocyte isozyme in a Chinese kindred. Am J Hematol 1981;10(2):107-17. doi:10.1002/ajh.2830100202
^ abcde Webb K.L., Dominelli P.B., Baker S.E., Klassen S.A., Joyner M.J., Senefeld J.W., Wiggins C.C. Influence of high hemoglobin-oxygen affinity on humans during hypoxia. Front Physiol 2022;12:763933. doi:10.3389/fphys.2021.763933
^ Aljahdali A.S., Musayev F.N., Burgner J.W. 2nd, Ghatge M.S., Shekar V., Zhang Y., Omar A.M., Safo M.K. Molecular insight into 2-phosphoglycolate activation of the phosphatase activity of bisphosphoglycerate mutase. Acta Crystallogr D Struct Biol 2022;78(Pt 4):472-482. doi:10.1107/S2059798322001802
^ abc Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ Hodgkins S.R. 6 – Erythrocyte metabolism and membrane structure and function. Editor(s): Keohane E.M., Otto C.N., Walenga J.M. Rodak’s Hematology (Sixth Edition), Elsevier. 2020:78-90. doi:10.1016/B978-0-323-53045-3.00015-5
^ ab Mairbäurl H., Oelz O., Bärtsch P. Interactions between Hb, Mg, DPG, ATP, and Cl determine the change in Hb-O2 affinity at high altitude. J Appl Physiol 1993;74(1):40-8. doi:10.1152/jappl.1993.74.1.40
^ Henry E.R., Bettati S., Hofrichter J., Eaton W.A. A tertiary two-state allosteric model for hemoglobin. Biophys Chem 2002;98(1-2):149-64. doi:10.1016/s0301-4622(02)00091-1
^ Böning D., Schwiegart U., Tibes U., Hemmer B. Influences of exercise and endurance training on the oxygen dissociation curve of blood under in vivo and in vitro conditions. Eur J Appl Physiol Occup Physiol 1975;34(1):1-10. doi:10.1007/BF00999910
Lo shunt di Rapoport-Luebering è una via metabolica presente prevalentemente nei globuli rossi ed è una deviazione dalla via glicolitica, da cui si diparte a livello dell’1,3-bisfosfoglicerato.[1][2]
Le due reazioni dello shunt sono catalizzate da un enzima multifunzionale, la bisfosfoglicerato mutasi (EC 5.4.2.4). Tuttavia di recente è stato identificato un secondo enzima che partecipa allo shunt, la 2,3-bisfosfoglicerato 3-fosfatasi (EC 3.1.3.80).[3]
Il prodotto più importante è il 2,3-bisfosfoglicerato (2,3-BPG), un effettore allosterico dell’emoglobina di cui riduce l’affinità per l’ossigeno molecolare, concorrendo così a regolarne il rilascio a livello tissutale.[4][5]
Essendo una deviazione dalla via glicolitica a monte delle reazioni che portano alla sintesi di ATP, lo shunt di Rapoport-Luebering comporta un costo energetico per la cellula.[3]
Difetti a carico degli enzimi glicolitici dei globuli rossi determinano anomalie nella sintesi del 2,3-BPG, e di conseguenza nell’affinità dell’emoglobina per l’ossigeno molecolare.[6]
La bisfosfoglicerato mutasi è un enzima centrale nello shunt di Rapoport-Luebering, e si ritrova nei globuli rossi.[4]
E’ un enzima multifunzionale essendo una 2,3-bisfosfoglicerato sintasi e una 2,3-bisfosfoglicerato fosfatasi. Inoltre è anche una 2,3-bisfosfoglicerato mutasi, attività analoga a quella della fosfoglicerato mutasi glicolitica (EC 5.4.2.1), ma che non partecipa allo shunt. Le tre attività catalitiche avvengono nello stesso sito attivo, con l’attività di sintasi come attività principale, che è inibita dal 2,3-bisfosfoglicerato libero.[7][8]
L’attività di fosfatasi è bassa, circa 1000 volte inferiore all’attività di sintasi, ed è stimolata da diversi anioni, come cloruro, fosfato, solfito e, in modo più marcato, dal 2-fosfoglicolato.[9]
Sembra infine che l’attività di mutasi sia fisiologicamente non significativa, catalizzando circa il 5% della conversione reversibile del 3-fosfoglicerato a 2-fosfoglicerato.[10][11]
La bisfosfoglicerato mutasi è una delle tre isoforme con cui nei mammiferi è presente la fosfoglicerato mutasi. Le altre due isoforme sono la fosfoglicerato mutasi 1 o tipo M, presente nel muscolo, e la fosfoglicerato mutasi 2 o tipo B presente in tutti gli altri tessuti. Anche queste due isoforme sono enzimi trifunzionali.[4]
Reazioni
Lo shunt di Rapoport-Luebering si compone di due reazioni.
Nella prima la bisfosfoglicerato mutasi, agendo come sintasi, catalizza l’isomerizzazione dell’1,3-bisfosfoglicerato (1,3-BPG) a 2,3- bisfosfoglicerato (2,3-BPG). L’enzima necessita della presenza del 3-fosfoglicerato in quanto catalizza il trasferimento intermolecolare di un gruppo fosforico dal C-1 dell’1,3-bisfosfoglicerato al C-2 del 3-fosfoglicerato. Nella reazione quindi il 3-fosfoglicerato di partenza diviene il 2,3-BPG, mentre l’1,3-BPG diviene il nuovo 3-fosfoglicerato.[12]
Nel secondo passaggio l’attività fosfatasica della bisfosfoglicerato mutasi catalizza la sintesi del 3-fosfoglicerato, un intermedio glicolitico, a seguito dell’idrolisi del gruppo fosforico in posizione C-2 del 2,3-BPG.Il 3-fosfoglicerato prodotto può quindi rientrare nella via glicolitica a livello della reazione catalizzata dalla fosfoglicerato mutasi, ossia l’ottava tappa della via.[5]
2,3-Bisfosfoglicerato 3-fosfatasi
E’ stato identificato un secondo enzima coinvolto nello shunt Rapoport-Luebering.
Oltre alla bisfosfoglicerato mutasi, è stata scoperta un’attività di 2,3-bisfosfoglicerato 3-fosfatasi catalizzata da una inositolo polifosfato fosfatasi multipla evolutivamente conservata, nota come MIPP1, che è stata trovata nelle amebe del genere Dictyostelium, negli uccelli e nei mammiferi.
Questa scoperta rivela quindi che la formazione di 3-fosfoglicerato, un precursore per la biosintesi della serina e un attivatore della proteina chinasi attivata da AMP, può essere bypassata. Manipolando geneticamente l’espressione di MIPP1 in Dictyostelium, è stato dimostrato il ruolo fisiologicamente rilevante dell’enzima nella regolazione del contenuto cellulare di 2,3-BPG.[3]
Ruolo dello shunt di Rapoport-Luebering
Lo shunt di Rapoport-Luebering porta alla formazione del 2,3-bisfosfoglicerato, che con l’anidride carbonica e gli ioni idrogeno, è uno degli effettori allosterici dell’emoglobina.[5]
Il 2,3-bisfosfoglicerato è presente in elevate concentrazioni nel globulo rosso dell’uomo, mentre nelle altre cellule è presente in tracce.[6] Nella maggior parte dei primati e molti altri mammiferi riduce l’affinità dell’emoglobina per l’ossigeno molecolare, facilitandone così il rilascio ai tessuti.[3]
Il 2,3-BPG si lega alla deossiemoglobina a mezzo di legami ionici con le catene β nello stato T, stato nel quale queste subunità formano una tasca elettrostatica complementare sia alla conformazione che alla distribuzione di carica della molecola. Il legame stabilizza lo stato T, quindi la deossiemoglobina, ed è principalmente responsabile della natura cooperativa del legame dell’ossigeno molecolare da parte dell’emoglobina.[12]
In tracce, il 2,3-bisfosfoglicerato è anche necessario all’attività catalitica della fosfoglicerato mutasi. Ne consegue che un’altra funzione della bisfosfoglicerato mutasi, e quindi dello shunt di Rapoport-Luebering, è quella di controllare il livello degli intermedi glicolitici.[13]
Costo energetico dello shunt di Rapoport-Luebering
L’1,3-bisfoglicerato è un intermedio della via glicolitica. Infatti, nella settima tappa della glicolisi, la fosfoglicerato chinasi (EC 2.7.2.3) catalizzata il trasferimento del gruppo fosforico ad alta energia presente sul C-1 dell’1,3-bisfosfoglicerato all’ADP, con formazione di ATP e 3-fosfoglicerato. La reazione, una fosforilazione a livello del substrato, è la prima delle due reazioni della via glicolitica in cui parte dell’energia chimica contenuta nel glucosio viene conservata in forma di ATP.[5]
Pertanto, l’ingresso dell’1,3-bisfosfoglicerato nel ciclo di Rapoport-Leubering comporta un costo energetico pari due molecole di ATP per molecola di 1,3-bisfosfoglicerato che devia dalla glicolisi ed entra nello shunt.[3] Ne consegue che esiste un delicato equilibrio tra il fabbisogno energetico del globulo rosso e la necessità di mantenere l’emoglobina in un adeguato stato di ossigenazione e deossigenazione.[14]
Rapporto tra glicolisi e shunt di Rapoport-Luebering
In condizioni fisiologiche, nei globuli rossi lo shunt di Rapoport-Luebering intercetta circa il 20% del flusso glicolitico.[8] Pertanto difetti a carico di enzimi glicolitici eritrocitari influenzano lo shunt, e quindi l’affinità dell’emoglobina per l’ossigeno molecolare.[6]
Due condizioni che evidenziano tale connessione sono l’anemia emolitica non sferocitica da deficit di esochinasi e l’anemia emolitica da deficit di piruvato chinasi negli eritrociti, malattie a trasmissione autosomica recessiva.[15][16]
Nell’anemia emolitica non sferocitica da deficit di esochinasi c’è una carenza di esochinasi eritrocitaria (EC 2.7.1.1).[17] L’enzima catalizza la fosforilazione del glucosio a glucosio-6-fosfato, e un suo deficit comporta una diminuzione della concentrazione eritrocitaria di 2,3-BPG, e quindi un aumento dell’affinità dell’emoglobina per l’ossigeno molecolare.[6]
Di contro, nell’anemia emolitica da deficit di piruvato chinasi negli eritrociti c’è una carenza della piruvato chinasi (EC 2.7.1.40), che ha come conseguenze un aumento della sintesi del 2,3-BPG e una diminuzione dell’affinità dell’emoglobina per l’ossigeno molecolare.[18]
Bibliografia
^ Rapoport S., Luebering J. Glycerate-2,3-diphosphatase. J Biol Chem 1951;189(2):683-694.
^ Rapoport S., Luebering J. The formation of 2,3-diphosphoglycerate in rabbit erythrocytes: the existence of a diphosphoglycerate mutase. J Biol Chem 1950;183(2):507-516.
^ abcde Cho J., King J.S., Qian X., Harwood A.J., Shears S.B. Dephosphorylation of 2,3-bisphosphoglycerate by MIPP expands the regulatory capacity of the Rapoport-Luebering glycolytic shunt. Proc Natl Acad Sci U S A 2008;105(16):5998-6003. doi:10.1073/pnas.0710980105
^ abc DiMauro S., Miranda A.F., Khan S., Gitlin K., Friedman R. Human muscle phosphoglycerate mutase deficiency: newly discovered metabolic myopathy. Science 1981;212(4500):1277-9. doi:10.1126/science.6262916
^ abcd Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012.
^ abcd Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
^ Aljahdali A.S., Musayev F.N., Burgner J.W. 2nd, Ghatge M.S., Shekar V., Zhang Y., Omar A.M., Safo M.K. Molecular insight into 2-phosphoglycolate activation of the phosphatase activity of bisphosphoglycerate mutase. Acta Crystallogr D Struct Biol 2022;78(Pt 4):472-482. doi:10.1107/S2059798322001802
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
^ Reynolds C.H. Activation of human erythrocyte 2,3-bisphosphoglycerate phosphatase at physiological concentrations of substrate. Arch Biochem Biophys 1986;250(1):106-11. doi:10.1016/0003-9861(86)90706-x
^ Lemarchandel V., Joulin V., Valentin C., Rosa R., Galactéros F., Rosa J., Cohen-Solal M. Compound heterozygosity in a complete erythrocyte bisphosphoglycerate mutase deficiency. Blood 1992;80(10):2643-9. doi:10.1182/blood.V80.10.2643.2643
^ Sasaki R., Chiba H. Role and induction of 2,3-bisphosphoglycerate synthase. Mol Cell Biochem 1983;53-54(1-2):247-56. doi:10.1007/BF00225257
^ Oslund R.C., Su X., Haugbro M., Kee J-M., Esposito M., David Y., Wang B., Ge E., Perlman D.H., Kang Y., Muir T.W., Rabinowitz J.D. Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat Chem Biol 2017;13:1081-1087. doi:10.1038/nchembio.2453
^ Hodgkins S.R. 6 – Erythrocyte metabolism and membrane structure and function. Editor(s): Keohane E.M., Otto C.N., Walenga J.M. Rodak’s Hematology (Sixth Edition), Elsevier. 2020:78-90. doi:10.1016/B978-0-323-53045-3.00015-5
^ Paglia D.E., Shende A., Lanzkowsky P., Valentine W.N. Hexokinase “New Hyde Park”: a low activity erythrocyte isozyme in a Chinese kindred. Am J Hematol 1981;10(2):107-17. doi:10.1002/ajh.2830100202
^ Enegela O.A., Anjum F. Pyruvate Kinase Deficiency. [Updated 2023 Apr 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560581/
L’acido piruvico, un alfa-chetoacido, è una molecola con un ruolo centrale nel metabolismo cellulare.[1][2]
Può essere prodotto attraverso diverse vie metaboliche, per la maggior parte citosoliche, tra le quali glicolisi è di solito la più importante, mentre il suo destino dipende dal tipo di cellula e dalla disponibilità di ossigeno, potendo essere utilizzato sia a fini energetici, biosintetici e anaplerotici.[3]
Dato il suo ruolo centrale nel metabolismo cellulare, mutazioni a carico dei geni che codifica per le proteine coinvolte nel suo metabolismo sono causa, nell’uomo, di malattie da lievi a gravi.[4]
L’acido piruvico o, secondo la nomenclatura IUPAC, l’acido 2-ossopropanoico, ha formula molecolare C3H4O3, formula condensata CH3COCOOH, e peso molecolare molecolare di 88,06 g/mol.
E’ classificato tra gli alfa-chetoacidi, ossia chetoacidi dove il gruppo carbonilico è adiacente al carbonio carbossilico. E, tra gli alfa-chetoacidi è quello con la struttura chimica più semplice.[1]
In forma purificata si presenta come un liquido incolore, con un odore che ricorda quello dell’acido acetico.
La sua costante di dissociazione o pKa, a 25 °C, è pari a 2,45. Si tratta dunque di un acido molto forte. Pertanto a pH fisiologico, sia nelle cellule che nei fluidi extracellulari, è presente quasi totalmente nella sua forma anionica, il piruvato.
Il suo punto di fusione è pari a 13,8 °C (56,84 °F; 286,95 K), mentre il punto di ebollizione è pari a 163,5 °C (329 °F; 438 K).[1]
Metabolismo del piruvato
Le vie metaboliche che portano alla sintesi dell’acido piruvico, nonché il suo successivo utilizzo, dipendono dal tipo di cellula e/o dalla disponibilità di ossigeno.[3]
Nelle cellule dotate di mitocondri, il metabolismo del piruvato si compone di una fase citosolica e una mitocondriale.
Nel citosol diverse vie metaboliche portano alla sua formazione. Tra queste, la glicolisi, l’ossidazione del lattato, la reazione catalizzata dall’enzima malico citosolico, e il catabolismo di almeno sei aminoacidi, tra cui l’alanina è il più importante. L’acido piruvico può essere prodotto anche nella matrice mitocondriale dall’alanina e dal lattato.[5] L’acido piruvico prodotto nel citosol, a mezzo di trasportatori specifici, entra nel mitocondrio dove potrà essere utilizzato a fini energetici, entrando nel ciclo dell’acido citrico, e/o biosintetici/anaplerotici, sulla base delle necessità della cellula.[6]
Se invece si considerano le cellule prive di mitocondri, come i globuli rossi, e, in condizioni di ipossia, anche le cellule dotate di mitocondri, il piruvato è ridotto a lattato e/o lascia la cellula come tale per essere quindi metabolizzato in altri tessuti, ad esempio il muscolo cardiaco.[7]
Glicolisi
In condizioni fisiologiche nella maggior parte delle cellule l’acido piruvico deriva principalmente dalla glicolisi, di cui ne rappresenta uno dei tre prodotti, con l’ATP e il NADH.
Nell’ultima reazione della via glicolitica la piruvato chinasi (EC 2.7.1.40) catalizza una fosforilazione a livello del substrato che porta al trasferimento del gruppo fosforico dal fosfoenolpiruvato all’ADP. La reazione, essenzialmente irreversibile, porta dalla formazione di una molecola di ATP e una di piruvato.
Fosfoenolpiruvato + ADP + H+ → Piruvato + ATP
Dunque, a mezzo della via glicolitica, da una molecola di glucosio sono prodotte due molecole di piruvato, oltre che di ATP e NADH.[2][8]
Lattato deidrogenasi
Nel citosol l’acido piruvico può essere prodotto dall’acido lattico.
L’enzima lattato deidrogenasi o LDH (EC 1.1.1.27) catalizza la conversione, reversibile, del piruvato a lattato e del NADH a NAD+.
Piruvato + NADH + H+ ⇄ Lattato + NAD+
La direzione della reazione dipende dagli isozimi della lattato deidrogenasi presenti e dal rapporto NADH/NAD+ nel citosol.[3][5]
Gli isoenzimi in cui predomina la subunità H, come LDH-1, predominano nel muscolo cardiaco, un tessuto completamente aerobico, dove catalizzano l’ossidazione del lattato, proveniente da altri tessuti, a piruvato, che sarà quindi utilizzato a fini energetici.
L’ossidazione del lattato prodotto ad esempio dai globuli rossi o dal muscolo scheletrico che lavora in condizioni ipossiche, può avvenire anche negli epatociti, favorita dal basso rapporto NADH/NAD+ nel citosol, sebbene l’isozima prevalente sia LDH-5. Nell’epatocita l’acido piruvico prodotto potrà essere utilizzato nella gluconeogenesi, che in questo caso è parte del più ampio ciclo di Cori, o essere ossidato a fini energetici.[9]
Di contro, nella fibra muscolare scheletrica che lavora in condizioni ipossiche, essendo inibito il complesso della piruvato deidrogenasi o PDC e bloccata fosforilazione ossidativa, perché la glicolisi possa procedere, il piruvato sarà ridotta a lattato con la concomitante ossidazione del NADH a NAD+.[10] Si noti che la conversione del glucosio in acido lattico è definita fermentazione lattica. Tale riduzione è favorita dal fatto che nella fibra muscolare scheletrica predominano isoenzimi con prevalenza della subunità M, quali LDH-4 e LDH-5.[11]
Alanina aminotransferasi
Un’altra fonte di acido piruvico è rappresentata dalla alanina, un aminoacido particolarmente abbondante nelle proteine muscolari.
L’utilizzazione degli scheletri carboniosi degli aminoacidi a fini energetici e anabolici implica la rimozione del gruppo amminico, che si verifica in reazioni catalizzate da enzimi chiamati transaminasi (EC 2.6.1-), e il successivo smaltimento dell’azoto in forma non tossica, attraverso il ciclo dell’urea.[2][8]
La rimozione del gruppo amminico dell’alanina è catalizzata dalla alanina aminotransferasi o ALT (EC 2.6.1.2). La reazione, reversibile, porta alla formazione di piruvato e glutammato.
Sono state identificate due forme della ALT, una a localizzazione citosolica, ALT1, e l’altra presente nella matrice mitocondriale, indicata come ALT2.[4] Ne risulta che l’acido piruvico può essere prodotto per transaminazione dell’alanina anche nella matrice mitocondriale.
L’alanina è uno dei principali precursori gluconeogenetici, e, a mezzo del ciclo glucosio-alanina, rappresenta un collegamento tra il metabolismo degli aminoacidi e quello dei carboidrati.[12]
Nel citosol gli scheletri carboniosi di altri cinque aminoacidi, ossia cisteina, glicina, serina, treonina e triptofano, possono essere convertiti in toto o in parte in piruvato.[2]
Enzima malico
Altra fonte piruvato è il malato.
Nella reazione, catalizzata dall’enzima malico citosolico, una decarbossilazione ossidativa (EC 1.1.1.40), il malato subisce una decarbossilazione ossidativa a dare piruvato.[3]
Malato + NADP+ → Piruvato + CO2 + NADPH + H+
L’enzima svolge un ruolo importante nel trasporto di intermedi del ciclo dell’acido citrico, quali, oltre al malato, l’ossalacetato e il citrato, tra il citosol e la matrice mitocondriale.[7]
Trasportatori mitocondriali del piruvato
Nelle cellule dotate di mitocondri, la maggior parte del piruvato prodotto nel citoplasma passa nella matrice mitocondriale, superando prima la membrana mitocondriale esterna e quindi quella interna.
Una volta nella matrice mitocondriale il piruvato può essere utilizzato sia per fini anabolici che catabolici.
Il passaggio attraverso la membrana mitocondriale esterna avviene per libera diffusione mediata dalle porine o VDAC, dall’inglese voltage dependent anion-selective canne, canali anionici voltaggio-dipendenti relativamente non specifici, nonché le più proteine più abbondanti della membrana mitocondriale esterna, la cui funzione è quella di mediare lo scambio di ioni e piccole molecole, tra cui oltre al piruvato anche l’ATP e il NADH, tra il citosol e lo spazio intermembrana dei mitocondri.[13][14]
La membrana mitocondriale interna è invece impermeabile alle molecole dotate di carica, il che permette il mantenimento del gradiente protonico necessario alla fosforilazione ossidativa. Ne consegue che il passaggio del piruvato necessita di specifici trasportatori, indicati come MPC, dall’inglese mitochondrial pyruvate carriers, un complesso etero-oligomerico di due subunità indicate come MPC1 e MPC2, che operano un simporto di protoni. Questi trasportatori legano quindi il metabolismo citosolico e mitocondriale del piruvato.[15][16]
Complesso della piruvato deidrogenasi
Una volta nella matrice mitocondriale, l’acido piruvico è per la maggior parte ossidato ad anidride carbonica al fine di supportare la produzione di ATP.
Il primo passaggio di questo processo di ossidazione vede l’intervento del complesso della piruvato deidrogenasi, uno tra i più importanti complessi multienzimatici presenti nelle cellule. Il complesso catalizza la decarbossilazione ossidativa del piruvato, con formazione di acetil-CoA e di una molecola di anidride carbonica, con il rilascio di due elettroni, che sono trasportati dal NADH.
Piruvato + CoA + NAD+ → Acetil-CoA + NADH + H+ + CO2
Il gruppo acetilico può ora entrare nel ciclo dell’acido citrico per essere completamente ossidato ad anidride carbonica, con produzione di una molecola di GTP, 3 molecole di NADH, una di FADH2. I due coenzimi ridotti, a mezzo della fosforilazione ossidativa, permetteranno la formazione di ulteriori molecole di ATP.[2]
In alternativa, il gruppo acetilico derivante dal piruvato può essere utilizzato a fini anabolici, tra cui la sintesi di alcuni lipidi quali gli acidi grassi, i fosfolipidi e il colesterolo, o essere utilizzato a fini regolatori a mezzo dell’acetilazione degli istoni.
Poiché la biosintesi dei lipidi si verifica nel citosol e l’acetilazione degli istoni nel nucleo, l’acetil-CoA dovrà lasciare la matrice mitocondriale, processo che necessita della formazione di citrato, a mezzo della condensazione tra il gruppo acetilico e il carbonile dell’ossalacetato, reazione catalizzata dalla citrato sintasi (EC 2.3.3.1). Una volta raggiunto il citosol, a mezzo di un trasportatore del citrato, una proteina integrale della membrana mitocondriale interna, il citrato viene scisso ad acetil-CoA e ossalacetato nella reazione catalizzata dalla citrato liasi (EC 4.1.3.6).[17]
Piruvato carbossilasi
Nella matrice mitocondriale l’acido piruvico può essere carbossilato a ossalacetato in una reazione, irreversibile catalizzata dalla piruvato carbossilasi o PC (EC 6.4.1.1).[8]
Piruvato + HCO3– + ATP ⇄ Ossalacetato + ADP + Pi
Molti intermedi del ciclo dell’acido citrico sono anche precursori per la sintesi di diverse molecole. Ne consegue che ognuno di questi intermedi rimossi dal ciclo dell’acido citrico dovrà essere reintegrato perché il ciclo possa proseguire. Le reazioni che reintegrano il ciclo sono definite anaplerotiche. In quest’ottica, la reazione catalizzata dalla piruvato carbossilasi svolge una funzione anaplerotica catalizzando la formazione di ossalacetato dal piruvato.[6]
^ abcde Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ abcd Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002
^ ab Gray L.R., Tompkins S.C., Taylor E.B. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-604. doi:10.1007/s00018-013-1539-2
^ ab Markert C.L., Shaklee J.B., Whitt G.S. Evolution of a gene. Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science 1975;189(4197):102-14. doi:10.1126/science.1138367
^ ab Owen O.E., Kalhan S.C., Hanson R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 2002;277(34):30409-12. doi:10.1074/jbc.R200006200
^ ab McCommis K.S. and Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J 2015;466(3):443-454. doi:10.1042/BJ20141171
^ abc Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
^ Gleeson T.T. Post-exercise lactate metabolism: a comparative review of sites, pathways, and regulation. Annu Rev Physiol 1996;58:565-81. doi:10.1146/annurev.ph.58.030196.003025
^ Li X., Yang Y., Zhang B., Lin X., Fu X., An Y., Zou Y., Wang J.X., Wang Z., Yu T. Lactate metabolism in human health and disease. Signal Transduct Target Ther 2022;7(1):305. doi:10.1038/s41392-022-01151-3
^ Farhana A., Lappin S.L. Biochemistry, lactate dehydrogenase. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557536/
^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
^ Colombini M. The VDAC channel: molecular basis for selectivity. Biochim Biophys Acta 2016;1863(10):2498-502. doi:10.1016/j.bbamcr.2016.01.019
^ Young M.J., Bay D.C., Hausner G., Court D.A. The evolutionary history of mitochondrial porins. BMC Evol Biol 2007;7:31. doi:10.1186/1471-2148-7-31
^ Bricker D.K., Taylor E.B., Schell J.C., Orsak T., Boutron A., Chen Y.C., Cox J.E., Cardon C.M., Van Vranken J.G., Dephoure N., Redin C., Boudina S., Gygi S.P., Brivet M., Thummel C.S., Rutter J. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 2012;337(6090):96-100. doi:10.1126/science.1218099
^ Herzig S., Raemy E., Montessuit S., Veuthey J.L., Zamboni N., Westermann B., Kunji E.R., Martinou J.C. Identification and functional expression of the mitochondrial pyruvate carrier. Science 2012;337(6090):93-6. doi:10.1126/science.1218530
^ Zara V., Assalve G., Ferramosca A. Multiple roles played by the mitochondrial citrate carrier in cellular metabolism and physiology. Cell Mol Life Sci 2022;79(8):428. doi:10.1007/s00018-022-04466-0
La lattato deidrogenasi o LDH (EC 1.1.1.27) è una famiglia di ossidoreduttasi che catalizza la conversione reversibile del piruvato a lattato, con la concomitante interconversione di NADH e NAD+, i quali agiscono da cofattori.
Sono enzimi a struttura tetramerica, dove ogni subunità è dotata di attività catalitica. Le subunità, codificate da geni distinti, si possono combinare in proporzioni differenti a dare isoenzimi con proprietà cinetiche e regolatorie specifiche.[1]
La lattato deidrogenasi è presente in quasi tutti i tessuti animali e nelle piante, ma anche nei microorganismi. Sebbene sia presente per la maggior parte nel citosol, la sua presenza è stata dimostrata anche nei mitocondri, dove catalizza l’ossidazione del lattato a piruvato, e nei perossisomi.[2][3]
Nell’uomo in tessuti differenti prevalgono isoenzimi differenti, sulla base dello specifico fenotipo metabolico del tessuto.
La lattato deidrogenasi è un enzima cruciale nel metabolismo cellulare, essendo coinvolta nella produzione di energia dai carboidrati in condizioni anaerobiche, nella sintesi del glucosio dal lattato e nell’utilizzo dello scheletro carbonioso del lattato a fini energetici in condizioni aerobiche.[1][4]
Nei mammiferi esistono diversi geni che codificano per le subunità della lattato deidrogenasi, indicati come LDHA, LDHB, LDHC, LDHx e LDHD.
I primi quattro codificano enzimi che riconoscono come substrato gli L-isomeri dell’acido lattico, la principale forma enantiomerica della molecola presente nei vertebrati, e hanno come cofattore il NAD, mentre LDHD codifica per un enzima che riconosce come substrato l’isomero D dell’acido lattico e ha come cofattore il FAD.[5][6]
LDHA, presente sul cromosoma 11p15.4, codifica per subunità M, dall’inglese muscle, mentre LDHB, presente sul cromosoma 12p12.2-p12.1, codifica per la subunità H, dall’inglese heart.[2][7]
LDHC, presente sul cromosoma 11p15.5-p15.3 e probabilmente derivante dalla duplicazione del gene LDHA, codifica per una subunità indicata come C.[8]
Infine, LDHx codifica per la subunità specifica dei perossisomi.[2] LDHx è la forma readthrough del gene LDHB; infatti quello che succede è che il ribosoma, nel momento in cui arriva al codone di stop dell’mRNA per la subunità H inserisce un aminoacido anziché terminare la traduzione, che procede aggiungendo altri sette aminoacidi che costituiscono il targeting perossisomiale di quella che sarebbe una normale subunità H.[3]
Struttura
Gli enzimi appartenenti alla famiglia della lattato deidrogenasi hanno struttura oligomerica, nello specifico sono tetrameri derivanti dall’assemblaggio di subunità, di circa 35 kD, che possono essere di uno stesso tipo o due tipi differenti. Ognuna subunità è dotata di attività catalitica, quindi il tetramero presenta quattro siti attivi. Tuttavia le subunità, prese singolarmente, risultano cataliticamente inattive.[7][9]
Considerando le subunità H e M, le loro strutture primarie sono per circa il 75% identiche, e di conseguenza anche la loro struttura tridimensionale risulta molto simile, ma con piccole differenze a livello del sito di legame per il substrato che portano a grosse differenze nelle proprietà cinetiche delle proteine.[1][10] Altra conseguenza delle differenze nella struttura primaria riguarda la carica netta di superficie che risulta essere di -6 per la subunità M e +1 per la H.[11]
La struttura secondaria è costituita per circa un 40% da alfa-eliche e un 23% da foglietti beta, a formare strutture beta-alfa-beta, con i foglietti beta paralleli come componente principale.[12]
Nell’uomo gli isoenzimi prevalenti sono quelli formati dalle subunità H e M. L’assemblaggio, in proporzioni differenti, delle due subunità porta alla formazione di cinque isoenzimi, indicati come:
La subunità C va a formare una sesta isoforma, omotetramerica, indicata come LDH-6 o C4.[8]
Una settima isoforma deriva dall’assemblaggio della subunità LDHx.[3][8]
Reazione
La lattato deidrogenasi catalizza l’interconversione del piruvato e lattato, che le forme ionizzate rispettivamente dell’acido piruvico e dell’acido lattico, e del NADH e NAD+. In entrambe i casi si verifica la rimozione, da quello che è l’agente riducente, di due atomi di idrogeno, e il successivo trasferimento di uno ione idruro, ossia un protone e due elettroni, all’agente ossidante, mentre il protone rimanente viene rilasciato nel mezzo acquoso. L’enzima è in grado di aumentare la velocità di reazione di 14 volte.[5]
Quando la reazione procede da piruvato a lattato, il primo passaggio è il legame del NADH, l’agente riducente, all’enzima, legame che è seguito da cambiamenti conformazionali che facilitano la formazione di legami idrogeno tra specifici residui delimitanti il sito attivo e il carbonile del piruvato, ossia il C2, e la successiva interazione tra NADH e il piruvato.[13] A questo punto si verifica il trasferimento di uno ione idruro dall’anello nicotinamidico del NADH al C2 del piruvato, che viene quindi ridotto a lattato. Ciò provoca l’ossidazione del coenzima e la neutralizzazione della carica positiva dell’azoto dell’anello nicotinamidico, e la riduzione del piruvato a lattato. Pertanto, in questo caso il C2 del piruvato viene ridotto da chetone ad alcol.
Quando la reazione procede in direzione opposta lo ione idruro viene trasferito dal C2 del lattato, che in questo caso è l’agente riducente, al C-4 dell’anello nicotinamidico. In questo caso il C2 del lattato è ossidato da alcool a chetone.[14][15]
Sito attivo
Il sito attivo delle subunità H e M ha una struttura ben conservata nelle differenti specie, con gli stessi aminoacidi che partecipano alla reazione.[16] Tra questi, non solo per la lattato deidrogenasi umana, ma anche per quella di molte altre specie, c’è His-193, aminoacido che si trova vicino al sito di legame per il coenzima.
Tuttavia, le piccole differenze nella struttura primaria delle due subunità, determinano differenze nelle proprietà biochimiche. Tra le differenze, una in particolare risulta cruciale per le proprietà catalitiche: una alanina della subunità M è sostituita, nella subunità H, da una glutammina, ossia un aminoacido non polare è sostituito da uno polare.[11] Di seguito una breve rassegna delle differenza catalitiche tra le due subunità.
La subunità H lega il substrato più velocemente della subunità M, ma ha, rispetto alla M un’attività catalitica circa cinque volte inferiore.
La subunità M ha una maggiore affinità per il piruvato, favorendo quindi la sintesi di lattato e NAD+, e non è inibita da alte concentrazioni di piruvato. Di contro, la subunità H ha un’affinità maggiore per il lattato, il che favorisce la sintesi di piruvato e NADH, mentre è inibita da elevate concentrazioni di piruvato.[17][18]
Da tutto ciò consegue che le proprietà cinetiche/catalitiche dei differenti isoenzimi dipendono dalla prevalenza di una delle due subunità.[19]
Regolazione ad opera del piruvato e lattato
Gli isoenzimi della lattato deidrogenasi sono soggetti a inibizione da parte del piruvato e lattato.
Gli isoenzimi dove predomina la subunità H sono inibiti da concentrazioni di piruvato più basse rispetto a quelli dove predomina la subunità M. Ad esempio, H4 è inibito da concentrazioni di piruvato pari a circa 0,2 mM, mentre M4 è debolmente inibito dal piruvato fino a concentrazioni di 5 mM.
Di contro, H4 è inibito dal lattato in concentrazione superiori a 20-40 mM mentre M4 è inibito in misura minore da elevate concentrazioni di lattato.[20]
Distribuzione tissutale
Nell’uomo i differenti isoenzimi della lattato deidrogenasi hanno localizzazione tissutali preferenziali, che in genere riflettono il fenotipo metabolico del tessuto.
Infatti, tessuti con un fenotipo metabolico prevalentemente o esclusivamente aerobico, come il cuore, producono in misura maggiore isoenzimi a prevalenza della subunità H.[21]
Al contrario, nei tessuti dove il metabolismo anaerobico è importante, come fibre muscolari scheletriche nel corso di un esercizio inteso, o alcune zone scarsamente ossigenate nei tumori solidi, sono prodotti isoenzimi con prevalenza della subunità M.[5]
Esistono tuttavia eccezioni, come ad esempio il fegato, organo con fenotipo metabolico aerobico dove predomina LDH-5, ma dove l’ossidazione del lattato a piruvato, che può essere considerata come parte del ramo epatico del ciclo di Cori, è favorita dal basso rapporto NADH/NAD+ presente nel citosolico dell’epatocita.[13]
Di seguito una breve panoramica.
LDH-1 o H4: muscolo cardiaco, rene e globuli rossi;
LDH-2 o M1H3 ha una distribuzione simile a LDH-1;
LDH-3 o M2H2: milza, cervello, globuli bianchi, rene e polmone;
LDH-4 o M3H1: milza, polmone, muscolo scheletrico, globuli rossi e rene;
LDH-5 o M4: fegato, muscolo scheletrico e polmone.
LDH-6: sperma e testicolo
Nei singoli organi, LDH-1 e LDH-2 predominano nel cuore, nei reni e nei globuli rossi, LDH-3 nei polmoni, LDH-4 e LDH-5 nel muscolo scheletrico e LDH-5 nel fegato.
Gli isoenzimi presenti nel siero, e il cui dosaggio viene utilizzato a fini diagnostici, sono sempre di derivazione tissutale.[5]
Infine, in uno stesso tessuto/organo, i differenti isoenzimi possono essere presenti in quantità significative in differenti tipi cellulari, come ad esempio accade nel muscolo scheletrico, nel cervello, ma anche nei tumori solidi.
Ruolo
La lattato deidrogenasi è un enzima essenziale sia per la produzione che l’utilizzazione/rimozione del lattato.[4]
In condizioni ipossiche la cellula ricava l’ATP dall’ossidazione anaerobica del glucosio. A mezzo della via glicolitica il monosaccaride viene ossidato a due molecole di piruvato, ricavando da ciò 2 molecole di ATP e due di NADH. Tuttavia, affinché la glicolisi possa procedere, il NADH prodotto dovrà essere riossidato a NAD+, essendo il coenzima ossidato necessario alla reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12). Poiché in condizioni anaerobiche il complesso della piruvato deidrogenasi inibito e la fosforilazione ossidativa bloccata, l’ossidazione del NADH è portata a termine dalla reazione catalizzata dalla lattato deidrogenasi, che quindi consente alla glicolisi di procedere. Questa via metabolica porta quindi alla produzione di lattato dal glucosio, ed è nota come fermentazione lattica.[13][22]
L’acido lattico è spesso considerato un prodotto finale del metabolismo dei glucosio, e il suo accumulo nel corpo risulta essere pericoloso, potendo essere causa di acidosi lattica.[22] Ne consegue che la molecola deve essere rapidamente rimossa dai tessuti e dal circolo. Considerando ad esempio il lattosio prodotto dai globuli rossi, o quello prodotto dalle fibre muscolari che lavorano in condizioni di scarso apporto di ossigeno, come durante un esercizio intenso, questi può raggiungere, trasportato dal circolo ematico, tra gli altri, il fegato e il cuore. Nel fegato, sebbene LDH-5 favorisca la riduzione del lattato a piruvato, il basso rapporto NADH/NAD+ citosolico sposta l’equilibrio della reazione verso la formazione del piruvato, che potrà essere utilizzato per la produzione di energia, o entrare nella gluconeogenesi.[23] Nel cardiomiocita invece, l’isoenzima prevalente, LDH-1, ossida il lattato a piruvato che sarà utilizzato a fini energetici.
Bibliografia
^ abc Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002
^ abc Markert C.L., Shaklee J.B., Whitt G.S. Evolution of a gene. Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science 1975;189(4197):102-14. doi:10.1126/science.1138367
^ abc Schueren F., Lingner T., George R., Hofhuis J., Dickel C., Gärtner J., Thoms S. Peroxisomal lactate dehydrogenase is generated by translational readthrough in mammals. Elife 2014;3:e03640. doi:10.7554/eLife.03640
^ abc Laughton J.D., Charnay Y., Belloir B., Pellerin L., Magistretti P.J., Bouras C. Differential messenger RNA distribution of lactate dehydrogenase LDH-1 and LDH-5 isoforms in the rat brain. Neuroscience 2000;96(3):619-25. doi:10.1016/s0306-4522(99)00580-1
^ abcd Farhana A., Lappin S.L. Biochemistry, lactate dehydrogenase. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557536/
^ Jin S., Chen X., Yang J., Ding J. Lactate dehydrogenase D is a general dehydrogenase for D-2-hydroxyacids and is associated with D-lactic acidosis. Nat Commun 2023;14(1):6638. doi:10.1038/s41467-023-42456-3
^ ab Krieg A.F., Rosenblum L.J., Henry J.B. Lactate dehydrogenase isozymes a comparison of pyruvate-to-lactate and lactate-to-pyruvate assays. Clin Chem 1967;13(3):196-203. doi:10.1093/clinchem/13.3.196
^ abc Zinkham W.H., Blanco A., Clowry L.J. Jr. An unusual isozyme of lactic dehydrogenase in mature testes: localization, ontogeny, and kinetic properties. Ann N Y Acad Sci 1964;121:571-88. doi:10.1111/j.1749-6632.1964.tb14227.x
^ Bellamacina C.R. The nicotinamide dinucleotide binding motif: a comparison of nucleotide binding proteins. FASEB J 1996;10(11):1257-69. doi:10.1096/fasebj.10.11.8836039
^ ab Read J.A., Winter V.J., Eszes C.M., Sessions R.B., Brady R.L. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins 2001;43(2):175-85.
^ Auerbach G., Ostendorp R., Prade L., Korndörfer I., Dams T., Huber R., Jaenicke R. Lactate dehydrogenase from the hyperthermophilic bacterium thermotoga maritima: the crystal structure at 2.1 A resolution reveals strategies for intrinsic protein stabilization. Structure 1998;6(6):769-81. doi:10.1016/s0969-2126(98)00078-1
^ abc Forkasiewicz A., Dorociak M., Stach K., Szelachowski P., Tabola R., Augoff K. The usefulness of lactate dehydrogenase measurements in current oncological practice. Cell Mol Biol Lett 2020;25:35. doi:10.1186/s11658-020-00228-7
^ Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
^ Nelson D.L., M. M. Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ Holmes R.S., Goldberg E. Computational analyses of mammalian lactate dehydrogenases: human, mouse, opossum and platypus LDHs. Comput Biol Chem 2009;33(5):379-85. doi:10.1016/j.compbiolchem.2009.07.006
^ Feng Y., Xiong Y., Qiao T., Li X., Jia L., Han Y. Lactate dehydrogenase A: a key player in carcinogenesis and potential target in cancer therapy. Cancer Med 2018;7(12):6124-6136. doi:10.1002/cam4.1820
^ Gray L.R., Tompkins S.C., Taylor E.B. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-604. doi:10.1007/s00018-013-1539-2
^ Rogatzki M.J., Ferguson B.S., Goodwin M.L., Gladden L.B. Lactate is always the end product of glycolysis. Front Neurosci 2015;9:22. doi:10.3389/fnins.2015.00022
^ Stambaugh R., Post D. Substrate and product inhibition of rabbit muscle lactic dehydrogenase heart (H4) and muscle (M4) isozymes. J Biol Chem 1966;241(7):1462-7. doi:10.1016/S0021-9258(18)96733-5
^ Wroblewski F, Gregory KF. Lactic dehydrogenase isozymes and their distribution in normal tissues and plasma and in disease states. Ann N Y Acad Sci 1961;94:912-32. doi:10.1111/j.1749-6632.1961.tb35584.x
^ ab Li X., Yang Y., Zhang B., Lin X., Fu X., An Y., Zou Y., Wang J.X., Wang Z., Yu T. Lactate metabolism in human health and disease. Signal Transduct Target Ther 2022;7(1):305. doi:10.1038/s41392-022-01151-3
^ Gleeson T.T. Post-exercise lactate metabolism: a comparative review of sites, pathways, and regulation. Annu Rev Physiol 1996;58:565-81. doi:10.1146/annurev.ph.58.030196.003025
I carbanioni sono ioni contenenti un atomo di carbonio con carica negativa.
Si formano a seguito della rottura eterolitica di un legame covalente tra un atomo di carbonio e un altro atomo o gruppo.[1]
Avendo una coppia di elettroni non condivisa, sono nucleofili potenti e basi forti e attaccano, al fine di formare un legame covalente, un protone o un centro elettrofilo, come un centro polarizzato o carico positivamente.[2]
I carbanioni sono estremamente reattivi. Pertanto, per consentire loro l’attacco ai centri nucleofili, devono essere stabilizzati.[3] La stabilizzazione può avvenire per effetto induttivo, per risonanza, e può anche dipendere dall’ibridazione dell’atomo di carbonio portatore di carica negativa.[1][2]
Sono intermedi in molte reazioni catalizzate da enzimi.
Considerando due atomi o gruppi, indicati come A e B, uniti da legame covalente, esistono due modi per rompere suddetto legame: l’eterolisi e l’omolisi.
Nell’eterolisi, la rottura del legame covalente porta alla formazione di due atomi dotati di carica, ossia due ioni, un catione e un anione, poiché entrambi gli elettroni di legame vengono presi da uno dei due atomi precedentemente legati, quello più elettronegativo.[4] La reazione può procedere in due modi:
A:B → :A– + B+, se A è più elettronegativo di B;
A:B → A+ + :B–, se B è più elettronegativo di A.
Nell’eterolisi di un legame covalente che coinvolge un atomo di carbonio, se entrambi gli elettroni di legame vengono presi dall’atomo di carbonio, l’atomo avrà una carica negativa, quindi è un anione, ed è definito carbanione. Se invece il carbonio perde entrambi gli elettroni di legame avrà carica positiva, quindi è un catione, ed è definito carbocatione.[5]
Nell’omolisi, la rottura del legame covalente tra A e B porta alla formazione di due radicali liberi, poiché ciascun atomo o gruppo prende uno dei due elettroni di legame.[6] Anche i radicali liberi, che sono elettricamente neutri, sono molecole molto instabili. Si noti che la scissione omolitica è meno comune della scissione eterolitica.[7]
Stabilizzazione dei carbanioni
I carbanioni sono specie chimiche estremamente reattive e, come i carbocationi e i radicali liberi, sono quasi sempre intermedi transienti in alcune reazioni organiche. Per permettere il loro attacco ai centri elettrofili è necessario che siano stabilizzati. La loro stabilizzazione dipende dalla dispersione della carica negativa, dispersione che può avvenire per effetto induttivo, per risonanza, e può anche dipendere dal carattere s degli orbitali ibridi dell’atomo di carbonio con carica negativa.
L’effetto induttivo è dovuto alla presenza nella molecola di uno o più dipoli permanenti in uno o più legami, dipoli a loro volta derivanti dalla differenza di elettronegatività tra due gruppi che determina una distribuzione non uniforme degli elettroni di legame. L’effetto induttivo può essere positivo, detto anche effetto +I, caratteristico di atomi o gruppi che tendono a respingere gli elettroni, oppure negativo, detto anche effetto –I, caratteristico di atomi o gruppi che tendono ad attrarre gli elettroni. Gli atomi o gruppi con effetto +I tendono a diminuire la stabilità dei carbanioni, mentre quelli con effetto –I, quindi più elettronegativi, tendono a stabilizzarli.[1]
La stabilità dei carbanioni aumenta quando sono legati a una struttura elettrofila dove la coppia di elettroni non condivisi può delocalizzarsi per risonanza, quindi una struttura che agisca da trappola per gli elettroni. Le strutture aromatiche, come il gruppo fenile, sono particolarmente efficaci.[2]
Infine la stabilità è anche una funzione del carattere s degli orbitali ibridi dell’atomo di carbonio caricato negativamente, aumentando all’aumentare della percentuale del suddetto carattere s. Pertanto aumenterà passando dall’ibridazione sp3, che ha il 25% di carattere s, a sp2, con il 33% di carattere s, a sp, con il 50% di carattere s.[1]
R-CH2– < R1R2C=CH– < RC≡C–
I carbanioni nelle reazioni enzimatiche
Esempi di reazioni enzimatiche che hanno carbanioni tra gli intermedi sono quelle catalizzata da tre complessi multienzimatici appartenenti alla famiglia delle 2-ossiacido deidrogenasi o alfa-chetoacido deidrogenasi, i quali sono coinvolti nelle decarbossilazioni ossidative di chetoacidi, in particolare degli alfa-chetoacidi, di seguito brevemente descritti.
Il complesso della piruvato deidrogenasi, che catalizza la decarbossilazione ossidativa del piruvato, la base coniugata dell’acido piruvico, in acetil-CoA, agendo così da ponte di collegamento tra glicolisi e ciclo dell’acido conversione citrico;
il complesso della ossoglutarato deidrogenasi o alfa-chetoglutarato deidrogenasi, che catalizza la decarbossilazione ossidativa dell’alfa-chetoglutarato a succinil-CoA nel corso del ciclo dell’acido citrico;
il complesso della alfa-chetoacido deidrogenasi a catena ramificata, che catalizza la decarbossilazione ossidativa degli aminoacidi ramificati valina, leucina e isoleucina in acetil-CoA e succinil-CoA, il cui scheletro carbonioso rimanente può quindi entrare nel ciclo dell’acido citrico.[3]
I tre complessi multienzimatici hanno strutture e meccanismi di reazione molto simili. Nello specifico, sono le loro subunità E1, che hanno come cofattore la tiamina pirofosfato, che catalizzano una reazione in cui si viene a formare un intermedio carbanionico, la cui formazione e stabilizzazione per risonanza coinvolge direttamente la tiamina.[8]
Anche la transchetolasi (EC 2.2.1.1) catalizza una reazione che comporta la formazione di un intermedio carbanionico. Questo enzima, che catalizza le tappe 6 e 8 della via del pentoso fosfato, richiede come cofattore la tiamina pirofosfato e ha un meccanismo di reazione simile a quello delle subunità E1 dei complessi multienzimatici visti in precedenza.[7]
Altro esempio di enzima che catalizza una reazione che vede la formazione di un intermedio carbanionico è l’acetil-CoA carbossilasi (EC 6.4.1.2), che catalizza la tappa di comando della sintesi degli acidi grassi, ossia la carbossilazione dell’acetil-CoA a malonil-CoA.[9]
Bibliografia
^ abcd Soderberg T. Organic chemistry with a biological emphasis. Volume I. Chemistry Publications. 2019
^ abc Solomons T. W.G., Fryhle C.B., Snyder S.A. Solomons’ organic chemistry. 12th Edition. John Wiley & Sons Incorporated, 2017
^ ab Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011
^ Heterolysis, in IUPAC Compendium of Chemical Terminology, 3rd ed. International Union of Pure and Applied Chemistry; 2006. Online version 3.0.1, 2019. doi:1351/goldbook.H02809
^ Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
^ Homolysis, in IUPAC Compendium of Chemical Terminology, 3rd ed. International Union of Pure and Applied Chemistry; 2006. Online version 3.0.1, 2019. doi:1351/goldbook.H02851
^ ab Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. H. Freeman and Company, 2012
^ Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002
I chetoacidi sono composti organici contenenti due gruppi funzionali: un gruppo carbossilico (−COOH) e uno carbonilico (˂C=O).
In base alla posizione del gruppo carbonilico rispetto a gruppo carbossilico, cui le regole della nomenclatura IUPAC assegnano la priorità più alta, sono suddivisi in alfa-chetoacidi, beta-chetoacidi, e gamma-chetoacidi.[1]
I chetoacidi, e in particolare gli alfa-chetoacidi, sono molto importanti in biochimica, essendo coinvolti in molte vie metaboliche.
Alfa-chetoacidi
Negli alfa-chetoacidi il gruppo carbonilico è adiacente al carbonio carbossilico. Molti di questi composti, in forma delle rispettive basi coniugate, svolgono importanti funzioni biologiche. Di seguito alcuni esempi.[2]
L’acido piruvico, il più semplice alfa-chetoacido, è il prodotto finale della glicolisi.
L’acido ossalacetico e l’acido alfa-chetoglutarico sono intermedi del ciclo dell’acido citrico.
Gli alfa-chetoacidi possono derivare da reazioni di transaminazione e di deaminazione ossidativa di aminoacidi. Nelle reazioni di transaminazione il gruppo amminico in alfa dell’amminoacido viene trasferito a un alfa-chetoacido, di solito l’alfa-chetoglutarato, con formazione di un nuovo amminoacido e di un alfa-chetoacido. Queste reazioni sono catalizzate da enzimi detti transaminasi o aminotransferasi (EC 6.1.-).
alfa-Chetoacido + Aminoacido ⇄ Nuovo aminoacido + Nuovo alfa-chetoacido
Nelle reazioni di deaminazione ossidativa gli amminoacidi sono convertiti nei corrispettivi alfa-chetoacidi a mezzo dell’eliminazione del gruppo amminico, che è convertito in ammoniaca e sostituito da un gruppo carbonilico. Essendo le reazione reversibile gli alfa-chetoacidi sono anche precursori degli aminoacidi.
Nota: l’ammoniaca è un composto tossico e, nel fegato, viene convertita in urea attraverso il ciclo dell’urea.[2]
Piruvato, ossalacetato e alfa-chetoglutarato, quest’ultimo via ossalacetato, sono i punti di ingresso nella gluconeogenesi degli scheletri carboniosi di molti amminoacidi glucogenici.[3]
E’ stato inoltre osservato che, in vitro, linee cellulari tumorali murine e umane rilasciano nel microambiente tumorale 2-chetoacidi quali α-chetoisocaproato, l’α-cheto-β-metilvalerato e l’α-chetoisovalerato, molecole in grado di influenzare l’attività antitumore dei macrofagi.[4]
Beta-chetoacidi
Nei beta-chetoacidi il gruppo carbonilico è sul secondo carbonio dopo il carbonio carbossilico.
Esempi sono l’acido acetoacetico, il più semplice 3-chetoacido, e l’acido β-idrossibutirrico, i quali sono due dei tre corpi chetonici, assieme all’acetone, prodotti dall’epatocita quando l’acetil-CoA è prodotto in eccesso rispetto alla capacità della citrato sintasi (EC 2.3.3.1 ), quindi del ciclo dell’acido citrico, di ossidarlo, situazione che si presenta durante il digiuno prolungato o diete molto povere in carboidrati.
Da notare che acetoacetil-CoA e beta-idrossibutirril-CoA, ossia le due forme attivate dei rispettivi beta-chetoacidi, sono intermedi della via di sintesi dell’acido butirrico seguita dalla maggior parte dei batteri del microbiota intestinale produttori dell’acido grasso.[5][6][7]
Gamma-chetoacidi
Nei gamma-chetoacidi il gruppo carbonilico è sul terzo carbonio dopo il carbonio carbossilico. Un esempio è l’acido levulinico, il più semplice gamma-chetoacido, che deriva dal catabolismo della cellulosa.
^ ab Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ D’Andrea G. Classifying amino acids as gluco(glyco)genic, ketogenic, or both. Biochem Educ 2000;28(1):27-28. doi:10.1016/s0307-4412(98)00271-4
^ Cai Z., Li W., Brenner M., Bahiraii S., Heiss E.H., Weckwerth W. Branched-chain ketoacids derived from cancer cells modulate macrophage polarization and metabolic reprogramming. Front Immunol 2022;13:966158. doi:10.3389/fimmu.2022.966158
^ Miller T.L., Wolin M.J. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl Environ Microbiol 1996;62(5):1589-92. doi:10.1128/aem.62.5.1589-1592
^ Portincasa P., Bonfrate L.,Vacca M., De Angelis M., Farella I., Lanza E., Khalil M.,Wang D.Q.-H., Sperandio M., Di Ciaula A. Gut microbiota and short chain fatty acids: implications in glucose homeostasis. Int J Mol Sci 2022;23:1105. doi:10.3390/ijms23031105
^ Pryde S.E., Duncan S.H., Hold G.L., Stewart C.S., Flint H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett 2002;217(2):133-9. doi:10.1111/j.1574-6968.2002.tb11467.x
Un ciclo futile, o ciclo del substrato, si instaura quando due reazioni opposte, non all’equilibrio e catalizzate da enzimi differenti, o due vie metaboliche opposte avvengono simultaneamente senza altro effetto complessivo se non un dispendio di energia.[1]
Il nome di ciclo futile fu coniato in quanto all’apparenza questi cicli sembravano non portare alcun beneficio alla cellula, una sorta di imperfezione metabolica.[2]
Tuttavia, recentemente, sono stati riconosciuti come importanti per la produzione di calore, l’amplificazione dei segnali metabolici, la redistribuzione di energia, in forma di trigliceridi, tra tessuto adiposo e fegato, e per le modificazioni a carico degli acidi grassi immagazzinati nei trigliceridi del tessuto adiposo.[3][4][5]
Per evitare un dispendio incontrollato di energia, i cicli futili sono strettamente controllati.
Esempi di cicli futili sono la glicolisi e la gluconeogenesi quando procedono simultaneamente a elevata velocità nella stessa cellula, il ciclo di Cori, e il ciclo trigliceridi/acidi grassi.[6]
Consideriamo una via ciclica che da A porta a B e viceversa. Supponiamo che il tasso di conversione di A in B sia 100, e quella di B in A 90. Ne risulta un flusso netto di A verso B di 10. Si immagini ora un effettore che aumenti la conversione di A in B del 30%, portandola a 130, e contemporaneamente riduca del 30% la conversione di B in A, portandola a 63. Il flusso netto risultante è pari a 130-63 = 67. Ne risulta un aumento del flusso netto pari a 570% a seguito di una variazione del 30% nella velocità delle reazioni opposte. Modifiche di questo tipo potrebbero, almeno in parte, spiegare l’aumento anche di 1000 volte del flusso di carbonio lungo la glicolisi nella fase iniziale di un esercizio intenso.[2]
Regolazione
Nel corso dell’evoluzione, la selezione di enzimi differenti per la catalisi di reazioni irreversibili e contrapposte ha permesso di evitare l’instaurarsi di cicli futili o di metterli sotto stretto controllo. In che modo? La selezione di un enzima che catalizza la conversione di A in B, e di un secondo enzima che catalizza la conversione di B in A, le cui attività sono regolate separatamente, permette il controllo della direzione netta del flusso metabolico.[7]
Il controlla delle attività enzimatiche può avvenire mediante:
meccanismi allosterici;
modificazioni covalenti;
modificazioni nella concentrazione degli enzimi coinvolti, conseguenti a variazioni del rapporto tra la loro sintesi e degradazione. Un meccanismo differente regola la glucochinasi epatica (EC 2.7.1.2). Durante il digiuno, l’enzima si lega in modo reversibile alla GKPR, una delle proteine specifiche del fegato, che lo ancora all’interno del nucleo, separandolo dagli altri enzimi glicolitici, e impedendo così l’instaurarsi di un ciclo futile tra glicolisi e gluconeogenesi.
Grazie a questi meccanismi di controllo è così possibile ottenere una regolazione coordinata delle due vie contrapposte, evitando un ciclo futile incontrollato. Ovviamente, una regolazione così fine non potrebbe essere ottenuta se un singolo enzima operasse in entrambe le direzioni.[8]
Glicolisi e gluconeogenesi
Considerando la glicolisi e la gluconeogenesi, la prima via porta alla conversione del glucosio in piruvato, la base coniugata dell’acido piruvico, con produzione di ATP, mentre la seconda converte il piruvato in glucosio, con consumo di ATP. Se le due vie procedessero simultaneamente ad alta velocità nella stessa cellula si avrebbe solo un consumo netto di ATP, quindi un ciclo futile. Ciò è evitato attraverso il controllo delle tappe irreversibili dei due processi, in particolare delle reazioni catalizzate dalla fosfofruttochinasi-1 o PFK-1 (EC 2.7.1.11) e dalla fruttosio-1,6-bisfosfatasi o FBPasi (EC 3.1.3.11), principalmente ad opera dall’effettore allosterico fruttosio 2,6-bifosfato.[7]
Da notare che mentre nella glicolisi il controllo allosterico coinvolge tutte e tre le reazioni irreversibili, nella gluconeogenesi i punti di regolazione cruciali sono le reazioni catalizzate dalla piruvato carbossilasi (EC 6.4.1.1) e dalla fruttosio-1,6-bisfosfatasi.[8]
Ciclo di Cori
Con il ciclo di Cori una parte dell’acido lattico prodotto dal muscolo e da altri tessuti extraepatici raggiunge il fegato, dove è convertito in glucosio che, rilasciato in circolo, torna al muscolo e ad altri tessuti extraepatici chiudendo il ciclo. Dal punto di vista energetico il ciclo di Cori può essere considerato un ciclo futile perché per ogni ciclo si ha come unico effetto apparente un consumo netto di 4 ATP, come per il ciclo glicolisi/gluconeogenesi. Tuttavia, questo ciclo permette il funzionamento di molti differenti tipi di cellule extraepatiche a spese del fegato.[6]
Ciclo trigliceridi/acidi grassi
Nel ciclo trigliceridi/acidi grassi, i trigliceridi nel tessuto adiposo vengono parzialmente o completamente idrolizzati in acidi grassi liberi e glicerolo, in un processo chiamato lipolisi; quindi, gli acidi grassi rilasciati vengono utilizzati per sintetizzare nuove molecole di trigliceridi.[3][6][7] Per ogni mole di trigliceridi che completa il ciclo vengono consumate 4 moli di ATP.
Questo ciclo futile può avvenire:
tra il tessuto adiposo, che rilascia acidi grassi, e il fegato, che li riesterifica in trigliceridi, portando a una ridistribuzione dell’energia immagazzinata;[4]
negli adipociti, dove può contribuire alla termogenesi e alla modifica degli acidi grassi immagazzinati.
Per quanto riguarda le modifiche degli acidi grassi immagazzinati, il ciclo li rende accessibili a reazioni di allungamento e desaturazione, che consentono la conversione degli acidi grassi saturi in acidi grassi insaturi. Tuttavia, sembra che l’efficienza del processo dipenda dal tipo di acidi grassi. Ad esempio, il metabolismo degli acidi grassi a catena media è più rapido della conversione dell’acido palmitico, uno degli acidi grassi a catena lunga, negli acidi palmitoleico e oleico, e poi, negli epatociti, in acido arachidonico. Si noti che la conversione degli acidi grassi a catena media e dell’acido palmitico in acidi grassi insaturi a catena lunga riduce il rischio per la salute associato al loro accumulo nei trigliceridi immagazzinati.[5]
Generazione di calore
In alcuni casi un ciclo futile ha la sola funzione di produrre calore a mezzo dell’idrolisi di ATP.
Questo si verifica ad esempio nei muscoli alari dei bombi, i quali, per volare devono mantenere una temperatura toracica di circa 30 °C, anche quando la temperatura esterna è bassa, fino anche a 10 °C. La temperatura toracica è mantenuta ai livelli ottimali per consentire il volo grazie al ciclo futile che si instaura tra le reazioni catalizzate dagli enzimi PFK-1 e FBPasi. Infatti, la FBAasi dei muscoli alari non è inibita dall’AMP, il che sembra suggerire che nel corso dell’evoluzione questa proteina sia stata selezionata appositamente per la produzione di calore.
Al contrario dei bombi, nei muscoli alari delle api mellifere non è quasi presente la FBPasi, e pertanto questi insetti non possono volare quando la temperatura esterna è bassa.[2]
Bibliografia
^ Newsholme E.A. Substrate cycles: their metabolic, energetic and thermic consequences in man. Biochem Soc Symp. 1978;43:183–205.
^ abc Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002
^ ab Reshef L., Olswang Y., Cassuto H., Blum B., Croniger C.M., Kalhan S.C., Tilghman S.M., Hanson R.W. Glyceroneogenesis and the triglyceride/fatty acid cycle. J Biol Chem 2003;278(33):30413-6. doi:10.1074/jbc.R300017200
^ ab Sharma A.K., Wolfrum C. Lipid cycling isn’t all futile. Nat Metab 2023;5(4):540-541. doi:10.1038/s42255-023-00779-x
^ ab Wunderling K., Zurkovic J., Zink F., Kuerschner L., Thiele C. Triglyceride cycling enables modification of stored fatty acids. Nat Metab 2023;5(4):699-709. doi:10.1038/s42255-023-00769-z
^ abc Brownstein A.J., Veliova M., Acin-Perez R., Liesa M., Shirihai O.S. ATP-consuming futile cycles as energy dissipating mechanisms to counteract obesity. Rev Endocr Metab Disord 2022;23(1):121-131. doi:10.1007/s11154-021-09690-w
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
Gli acidi grassi a catena corta o SCFAs sono acidi grassi saturi con catena carboniosa lineare o ramificata composta da un numero di atomi di carbonio compreso tra 2 e 5, e sono l’acido acetico, l’acido propionico, l’acido butirrico, l’acido isobutirrico, l’acido valerico, l’acido isovalerico, e l’acido 2-metilbutirrico.[1]
Nell’uomo sono, con i sali biliari secondari, i principali metaboliti prodotti dai batteri del microbiota intestinale nel cieco e nel colon, e derivano quasi interamene dalla fermentazione anerobica di carboidrati non digeribili.[11] I più abbondanti sono l’acido acetico, l’acido propionico e l’acido butirrico, che costituiscono circa il 95% del totale degli acidi grassi a catena corta prodotti.[6] La percentuale rimanente è costituita da quelli a catena ramificata.
Sono i principali anioni presenti nel colon. La loro concentrazione è maggiore nel cieco e nella parte prossimale del colon rispetto a quella distale, dove i substrati per la loro sintesi vanno esaurendosi.[1][2][4] Sono in grado di ridurre il valore del pH del lume intestinale e quindi acidificare le feci.
Circa il 90-95% viene assorbito nel cieco e nel colon, mentre il restante 5-10% viene escreto con le feci.[13]
Si ritiene forniscano circa il 70% del fabbisogno energetico necessario ai colonociti.[4]
Gli acidi grassi a catena corta sono in grado di modulare sia la fisiologia che la composizione del microbiota intestinale.[7] Inoltre, un numero crescente di ricerche suggerisce che svolgano un ruolo importante nel concorrere al mantenimento della salute dell’uomo.[4]
Al pari degli acidi grassi a catena media e degli acidi grassi a catena lunga, gli acidi grassi catena corta sono presenti nei tessuti animali e vegetali principalmente in forma di trigliceridi, sebbene in quantità molto inferiori rispetto a quelli a catena lunga.
Nell’adulto la principale fonte alimentare è costituita da latte e derivati, dove l’acido butirrico è quello presente in concentrazione maggiore. Altre fonti sono alcuni oli vegetali, come l’olio di cocco e l’olio di palmisti.
Nei neonati allattati al seno la principale fonte è il latte materno.[10]
Tuttavia, per l’uomo, e per la maggior dei mammiferi, la fonte più importante è costituita dalla fermentazione anaerobica delle fibre e dell’amido resistente, ossia di carboidrati indigeribili, ad opera dei batteri del microbiota intestinale.[6] Questa via porta alla produzione di circa 500-600 mM di SCFAs al giorno. Gli acidi acetico, propionico e butirrico sono presenti in un rapporto molare rispettivamente di circa 60:20:20, sebbene la proporzione relativa di ciascuno essi dipenda dal substrato, dalla composizione del microbiota, e dal tempo di transito intestinale.[7][11]
Proprietà
Gli acidi grassi a catena corta hanno catene carboniose composte da un numero di atomi di carbonio compreso tra 2 e 5, caratteristica che influenza fortemente le loro proprietà fisiche.[9]
L’acido acetico, l’acido butirrico, l’acido propionico e l’acido valerico sono acidi grassi a catena lineare, mentre l’acido isobutirrico, l’acido isovalerico e l’acido 2-metil butirrico sono acidi grassi a catena ramificata.
Sono molecole di piccole dimensioni, e tra i lipidi sono i più piccoli.
A temperatura ambiente sono liquidi, e sono solubili in solventi polari come l’acqua, differendo in questo dagli acidi grassi con catene carboniose più lunghe, la cui solubilità in solventi polari, considerando quelli a catena lineare, va diminuendo all’aumentare della lunghezza della catena stessa, essendo questa la parte della molecola dotata di carattere non polare, a differenza del gruppo carbossilico che è polare.[10]
Va infine notato che l’acido butirrico e l’acido isobutirrico, che hanno formula molecolare C4H8O2, sono un esempio di isomeria di catena, al pari dell’acido valerico, dell’acido isovalerico e dell’acido 2-metilbutirrico, i quali hanno formula molecolare C5H10O2.
Effetti sulla salute
Gli acidi grassi a catena corta sono ritenuti svolgere un ruolo cruciale nel mantenimento della salute dell’uomo.[4] La loro azione sembra esplicarsi attraverso effetti diretti e indiretti su processi cellulari come la proliferazione, la differenziazione e l’espressione genica, concorrendo in questo modo alla regolazione di processi quali l’omeostasi del glucosio, la funzionalità intestinale e immunitaria, e la regolazione dell’asse microbiota-intestino-cervello.[6] Tutto ciò sembra essere confermato anche da studi che mostrano come una disbiosi intestinale appaia essere implicata in patologie metaboliche, come disturbi che coinvolgono l’omeostasi del glucosio, e comportamentali e neurologiche, come l’Alzheimer, il Parkinson, e la depressione.[11]
Sintesi
Nell’uomo, il corredo enzimatico responsabile della digestione dei carboidrati è privo degli enzimi in grado di digerire le fibre e l’amido resistente, quest’ultimo così chiamato proprio perché resiste all’azione idrolitica della alfa-amilasi. Al contrario, i batteri del microbiota intestinale codificano per un gran numero di differenti glicoside idrolasi, oltre 260, grazie alle quali sono in grado di liberare monosaccaridi dalle fibre e dall’amido resistente.[7] Gli esosi e i desossiesosi entrano nella glicolisi, e i pentosi nella via del pentoso fosfato, a dare il piruvato, la base coniugata dell’acido piruvico, che è il principale precursore per la sintesi degli acidi grassi a catena corta.[2][4][5]
La sintesi degli SCFAs è influenzata da diversi fattori, alcuni dei quali sono di seguito elencati.[4][11][13]
Il contenuto di fibre della dieta. Ad esempio, una dieta ricca di fibre, come la dieta mediterranea, può influenzarne la sintesi.
La composizione del microbiota intestinale.
Il pH del lume intestinale, in quanto per valori di circa 5,5 dominano i batteri che producono acido butirrico, mentre per valori di circa 6,5 dominano i batteri che producono acido acetico e acido propionico.
La velocità di transito intestinale.
La quantità di ossigeno nel lume intestinale.
L’acido acetico e l’acido propionico sono prodotti principalmente da specie appartenenti al phylum Bacteroides, mentre l’acido butirrico, per la cui sintesi è particolarmente importante l’amido resistente, da specie batteriche appartenenti al phylum Firmicutes.[6]
Sintesi dell’acido acetico
L’acido acetico, il più abbondante tra gli SCFAs presenti nel colon, può essere sintetizzato attraverso la via di Wood-Ljungdahl nella direzione riduttiva, a mezzo della riduzione della CO2 ad acetato, o dall’acetil-CoA, la via metabolica più importante essendo responsabile della produzione di circa due terzi dell’acido grasso presente nel lume intestinale.[4]
Sintesi dell’acido propionico
L’acido propionico può essere sintetizzato attraverso tre differenti vie metaboliche: le vie dell’acrilato e del succinato, che utilizzano come precursore l’acido lattico prodotto da altri batteri, e la via del propandiolo, nella quale i precursori sono desossiesosi.[2][3][10]
Nella via dell’acrilato, l’acido lattico viene convertito in propionil-Coa via lattoil-CoA. Nel passaggio finale il propionil-Coa è idrolizzato a dare acido propionico.
Nella via del succinato, il lattato viene ridotto a piruvato, che a sua volta viene carbossilato a ossalacetato, che è convertito in propionil-CoA attraverso una via metabolica che ha come intermedi il malato, il fumarato, il succinato e il metilmalonil-CoA. Infine il propionil-CoA viene idrolizzato a coenzima A e acido propionico. La via del succinato è considerata la via dominante per la sintesi dell’acido propionico nell’intestino.
Nella via del propandiolo, alcuni desossiesosi, come il fucosio e il ramnosio, sono convertiti, via 1,2-propandiolo, in propionil-Coa e quindi in acido propionico.
Sintesi dell’acido butirrico
La sintesi dell’acido butirrico può seguire due vie.[7][10]
Nella maggior parte dei batteri produttori di acido butirrico, l’acido grasso a catena corta è prodotto attraverso una via metabolica che ha inizio con la condensazione di due molecole di acetil-CoA a dare acetoacetil-CoA. Di seguito l’acetoacetil-CoA viene convertito in beta-idrossibutirril-CoA, crotonil-CoA, e butirril-CoA. Nell’ultima tappa si ha la liberazione di acido butirrico dal butirril-CoA.[8]
In un numero ridotto di specie batteriche il butirril-CoA viene convertito in butirrilfosfato, da cui viene infine liberato acido butirrico.[2]
Sintesi dagli aminoacidi
L’acido acetico, l’acido propionico e l’acido butirrico possono essere prodotti anche a partire da aminoacidi derivanti dalla degradazione di peptidi e proteine, sebbene la quantità prodotta da queste vie sia minima.[7]
Questi processi si verificano nella parte distale del colon, spesso ad opera di batteri non commensali.[3] Dal metabolismo di aminoacidi differenti sono prodotti acidi grassi a catena corta diversi; di seguito alcuni esempi.[4]
Dall’acido glutammico sono prodotti principalmente acido acetico e acido butirrico.
Dall’acido aspartico derivano principalmente acido acetico e acido propionico.
Dagli aminoacidi basici lisina, arginina e istidina sono prodotti acido acetico e acido butirrico.
Dalla cisteina derivano gli acidi acetico, propionico e butirrico.
Dal metabolismo della metionina derivano principalmente l’acido propionico e l’acido butirrico.
Dagli aminoacidi ramificati leucina, isoleucina e valina si originano acidi grassi a catena corta ramificati.
Il pH del lume intestinale influenza il metabolismo delle proteine da parte del microbiota intestinale, a partire dalla loro degradazione negli aminoacidi costituenti, più probabile a pH neutro o debolmente alcalino.
Si noti che dal metabolismo intestinale degli aminoacidi sono prodotti anche composti potenzialmente tossici, quali ammoniaca, solfiti e fenoli.
Sintesi endogena
L’uomo, e più in generale i mammiferi, hanno il corredo enzimatico per la sintesi endogena di acidi grassi a catena corta. La sintesi, prevalentemente epatica, avviene a mezzo di cicli di reazioni di beta-ossidazione che portano alla formazione di acil-CoA con catena carboniosa via via più corta dell’acido grasso di partenza. L’acido grasso legato al coenzima A è quindi rilasciato a seguito dell’idrolisi operata da una acil-CoA tioesterasi (EC 3.1.2.20).[10][12]
Recettori di membrana
Gli acidi grassi a catena corta sono in grado di legarsi a specifici recettori presenti sulla membrana plasmatica, i recettori accoppiati alle proteine G, tra cui GPR41, GPR43 e GPR109A.[6]
Gli effetti del legame ai recettori dipendono dal tipo di cellula. Ad esempio, il legame ai recettori presenti sulle cellule L dell’intestino è associato al rilascio di GLP-1, o glucagon-like peptide-1, e del peptide YY, ormoni che hanno effetto sull’appetito e sull’assunzione di cibo. Il legame alle cellule enterocromaffini induce il rilascio di serotonina, il che potrebbe avere effetto sulla motilità intestinale. Infine il legame ai recettori presenti sulle cellule beta del pancreas aumenta il rilascio di insulina.[11]
I diversi acidi grassi a catena corta hanno capacità differenti di attivare i recettori: GPR43 è più probabile sia attivato dall’acido acetico e dall’acido propionico, GPR41 dall’acido propionico e dall’acido butirrico, mentre GPR109A dall’acido butirrico.[4]
Assorbimento
Circa il 90% degli acidi grassi a catena corta presenti nel lume intestinale è assorbito dai colonociti. Il passaggio attraverso la membrana plasmatica può avvenire per diffusione passiva o a mezzo di un trasporto attivo mediato da due tipi di trasportatori di membrana: i trasportatori per i monocarbossilati H+-dipendenti e quelli Na+-dipendenti, indicati rispettivamente come MCTs e SMCTs.[6][7]
Il trasporto passivo interessa le forme protonate degli SCFAs, quindi è influenzato dal pH presente nel colon. Una debole acidificazione del lume intestinale, che può essere conseguente all’attività metabolica dei microrganismi presenti, aumenta la prevalenza della forma protonata e quindi del trasporto passivo.[10]
Funzioni nel colonocita
Nei colonociti, gli acidi grassi a catena corta possono essere utilizzati a scopi energetici e regolatori.
Quando utilizzati a fini energetici, l’acido acetico e l’acido butirrico sono convertiti in acetil-CoA, e l’acido propionico in propionil-CoA. Attraverso la produzione di ATP contribuiscono al mantenimento dell’omeostasi cellulare, ma anche, ad esempio, al mantenimento dell’integrità delle giunzioni strette presenti agli apici cellulari, e quindi dell’integrità della barriera intestinale.[11] Dei tre principali acidi grassi a catena corta prodotti nell’intestino, l’acido butirrico è la principale fonte di energia per i colonociti, mentre gli acidi acetico e propionico sono scarsamente metabolizzati e per la maggior parte drenati dalla vena porta.[2][13]
A fini regolatori gli SCFAs sono ad esempio in grado di inibire l’attività delle istone deacetilasi (EC 3.5.1.98), enzimi che catalizza la rimozione dei gruppi acetilici da residui di lisina delle proteine istoniche, gruppi acetilici precedentemente inseriti dalla istone acetiltransferasi (EC 2.3.1.48).[7] I gruppi R delle lisine deacetilate portano cariche positive, il che fa si che le proteine istoniche avvolgano più strettamente il DNA, che porta cariche negative. Ciò rende il nucleosoma più compatto, e di conseguenza più difficile eseguire la trascrizione e l’espressione genica. I diversi acidi grassi a catena corta hanno differenti capacità di inibire l’attività delle istone deacetilasi:
fino all’80% per l’acido butirrico;
fino al 60% per l’acido propionico;
l’acido acetico ha la minor capacità inibitoria.[4]
Questa modalità d’azione sulle istone deacetilasi è stata osservata non solo nell’intestino e nel tessuto immunitario associato, ma anche nel sistema nervoso centrale e periferico.[11]
Trasporto
Gli acidi grassi a catena corta che non sono stati utilizzati nel colonocita lasciano la cellula per diffusione passiva e trasporto attivo, a livello della membrana basolaterale, per entrare nella circolazione portale, dove la concentrazione maggiore è raggiunta dall’acido acetico, circa 260 mM/L, mentre gli acidi propionico e butirrico raggiungono concentrazione di circa 30 mM/L.[2]
Nel retto, una piccola quantità di questi piccoli lipidi può passare direttamente nella circolazione sistemica, bypassando il fegato, a mezzo della vena iliaca interna.[13]
A differenza degli acidi grassi a catena lunga, quelli a catena corta e media viaggiano in circolo in forma libera, come acidi grassi non esterificati, e, legati all’albumina, raggiungono il fegato. Il successivo uptake e trasporto citosolico non richiede l’intervento delle proteine leganti gli acidi grassi, di traslocasi per gli acidi grassi nella membrana plasmatica, o di proteine leganti gli acidi grassi nel citosol. Pertanto, muovendosi liberamente, la loro ossidazione potrebbe essere assai più veloce rispetto a quella degli acidi grassi a catena lunga e ai membri più lunghi degli acidi grassi a catena media, ossia quelli con catene carboniose con più di 8 atomi.[10]
Metabolismo epatico ed extraepatico
Il fegato è un sito importante per il metabolismo degli acidi grassi a catena corta.[13] Infatti l’organo è in grado di assorbire circa il 40% dell’acido acetico e l’80% del dell’acido propionico presenti nella vena porta. L’acido propionico è per la maggior parte metabolizzato nel fegato, dove può essere utilizzato anche come substrato per la gluconeogenesi.[7]
Una piccola quantità degli SCFAs prodotti nell’intestino, circa il 36% per l’acido acetico, il 9% per l’acido propionico e solo il 2% per l’acido butirrico, raggiunge, attraverso la circolazione sistemica, i tessuti periferici. Nel muscolo, l’acido acetico può essere utilizzato per la sintesi dei lipidi o essere ossidato per la produzione di energia. Inoltre, si ritiene che le concentrazioni presenti nella circolazione sistemica, seppure piccole, siano in grado di influenzare il metabolismo e la fisiologia delle cellule dei tessuti periferici.
Bibliografia
^ ab Abdul Rahim M.B.H., Chilloux J., Martinez-Gili L., Neves A.L., Myridakis A., Gooderham N., Dumas M.-E. Diet-induced metabolic changes of the human gut microbiome: importance of short-chain fatty acids, methylamines and indoles. Acta Diabetol 2019;56:493-500. doi:10.1007/s00592-019-01312-x
^ abcde Deleu S., Machiels K., Raes J., Verbeke K., Vermeire S. Short chain fatty acids and its producing organisms: an overlooked therapy for IBD? EBioMedicine 2021;66:103293. doi:10.1016/j.ebiom.2021.103293
^ ab Koh A., De Vadder F., Kovatcheva-Datchary P., Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 2016;165(6):1332-1345. doi:10.1016/j.cell.2016.05.041
^ abcdefghij Liu X.F., Shao J.H., Liao Y.T., Wang L.N., Jia Y., Dong P.J., Liu Z.Z., He D.D., Li C., Zhang X. Regulation of short-chain fatty acids in the immune system. Front Immunol 2023;14:1186892. doi:10.3389/fimmu.2023.1186892
^ Miller T.L., Wolin M.J. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl Environ Microbiol 1996;62(5):1589-92. doi:10.1128/aem.62.5.1589-1592
^ abcdef Parada Venegas D., De la Fuente M.K., Landskron G., González M.J., Quera R., Dijkstra G., Harmsen H.J.M., Faber K.N., Hermoso M.A. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol 2019;10:1486. doi:10.3389/fimmu.2019.00277
^ abcdefgh Portincasa P., Bonfrate L., Vacca M., De Angelis M., Farella I., Lanza E., Khalil M.,Wang D.Q.-H., Sperandio M., Di Ciaula A. Gut microbiota and short chain fatty acids: implications in glucose homeostasis. Int J Mol Sci 2022;23:1105. doi:10.3390/ijms23031105
^ Pryde S.E., Duncan S.H., Hold G.L., Stewart C.S., Flint H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett 2002;217(2):133-9. doi:10.1111/j.1574-6968.2002.tb11467.x
^ abcdefg Schönfeld P., Wojtczak L. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res 2016;57(6):943-54. doi:10.1194/jlr.R067629
^ abcdefg Silva Y.P., Bernardi A., Frozza R.L. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front Endocrinol (Lausanne) 2020;11:25. doi:10.3389/fendo.2020.00025
^ abcde Xiong R.G., Zhou D.D., Wu S.X., Huang S.Y., Saimaiti A., Yang Z.J., Shang A., Zhao C.N., Gan R.Y., Li H.B. Health benefits and side effects of short-chain fatty acids. Foods 2022;11(18):2863. doi: 10.3390/foods11182863
L’amido fosforilasi o alfa-glucano fosforilasi (EC 2.4.1.1) è una proteina multimerica, dotata di attività enzimatica e regolatoria, che svolge un ruolo importante nel metabolismo dei carboidrati, sia nei procarioti che negli eucarioti.[10]
L’enzima catalizza il trasferimento di un’unità glucosidica dal glucosio-1-fosfato all’estremità non riducente di un α-(1→4)-glucano nascente, a cui viene legata per mezzo di un legame glicosidico α-(1→4).[6][8] La reazione è reversibile e la direzione dipende dal rapporto fosfato/glucosio-1-fosfato presente in vivo.[2] L’enzima, al pari delle amido sintasi (EC 2.4.1.21), che intervengono nella sintesi dell’amilosio e dell’amilopectina, i polisaccaridi che compongono i granuli di amido, della glicogeno fosforilasi (EC 2.4.1.1), enzima che interviene nella glicogenolisi, e della glicogeno sintasi (EC 2.4.1.11) che interviene nella sintesi del glicogeno, appartiene alla famiglia delle glucosiltransferasi (EC 2.4).[6]
Si noti che il substrato donatore dell’unità glucosidica nella reazione catalizzata dalla amido fosforilasi non è ne l’ADP-glucosio, utilizzato dalle amido sintasi, ne l’UDP-glucosio utilizzato dalla glicogeno sintasi, ma il glucosio-1-fosfato.[2]
L’amido fosforilasi sembra intervenire sia nella sintesi che nella degradazione dell’amilosio e amilopectina.[9]
A livello industriale l’azione fosforolitica della fosforilasi è utilizzata nella produzione del glucosio-1-fosfato e nella preparazione di carboidrati quali glucani e amidi modificati.[6]
Nelle piante sono presenti almeno due isoforme dell’amido fosforilasi, una nello stroma dei plastidi, indicata come Pho1, e una a localizzazione citosolica, denominata Pho2. Entrambe gli isoenzimi svolgono un ruolo fondamentale nella sintesi e degradazione dell’amido.[6][8]
Degradazione dell’amido
Sebbene il primo enzima a intervenire nella degradazione del polisaccaride sia l’alfa-amilasi (EC 3.2.1.1) nelle prime fasi della germinazione, e la beta-amilasi (EC 3.2.1.2) nel caso della degradazione dell’amido transitorio nei cloroplasti, anche altre attività enzimatiche sono state chiamate in causa, quali l’alfa-glucano acqua dichinasi (EC 2.7.9.4) e la fosfoglucano acqua dichinasi (EC 2.7.9.5), e l’enzima deramificante.[10]
Dei due isoenzimi dell’amido fosforilasi, Pho1 sembra avere un’azione indiretta o regolatrice in grado di influenzare l’attività degli altri enzimi che intervengono nella degradazione dell’amido, mentre Pho2 è in grado di degradare i granuli di amido e altri glucani ramificati.
Sintesi dell’amido
Nel corso della sintesi dell’amido, l’amido fosforilasi, in particolare Pho1, sembra svolgere sia un’azione enzimatica che regolatrice.
Pho1, sembra essere coinvolta nei passaggi iniziali della sintesi dell’amido, concorrendo all’allungamento della catena nascente.[1][8]
La sintesi dell’amido coinvolge un set di almeno cinque classi di enzimi, ossia la ADP-glucosio pirofosforilasi (EC 2.7.7.27), le amido sintasi, gli enzimi ramificanti l’amido (EC 2.4.1.18), gli enzimi deramificanti l’amido (EC2.1.41) e le amido fosforilasi.[5][10] A queste vanno aggiunte proteine prive di attività catalitica ma necessarie alla corretta sintesi del granulo di amido. Nell’endosperma dei cereali la formazione di complessi multienzimatici tra le amido sintasi e gli enzimi ramificanti dipendono, oltre che da specifiche fosforilazioni delle proteine stesse, anche dalla presenza di Pho1.[1][3]
L’amido fosforilasi è in grado di formare anche un complesso con l’enzima disproporzionante (EC 2.4.1.25).[1][10] Questo complesso sembra in grado di sintetizzare brevi malto-oligosaccaridi o MOS, che sono α-(1→4)-glucani con un grado di polimerizzazione compreso tra 2 e 7.[7] I MOS agiscono come primer per l’amido sintasi IV e l’amido sintasi legata ai granuli nelle fasi iniziali della sintesi rispettivamente dell’amilopectina e dell’amilosio, una funzione questa analoga a quella svolta dalla glicogenina nella della sintesi del glicogeno.[4] I MOS possono derivare anche dall’azione degli enzimi deramificanti l’amido nel corso del rifinitura di molecole di amilopectina vicine.
Bibliografia
^ abc Crofts N., Abe N., Oitome N.F., Matsushima R., Hayashi M., Tetlow I.J., Emes M.J., Nakamura Y., Fujita N. Amylopectin biosynthetic enzymes from developing rice seed form enzymatically active protein complexes. J Exp Bot 2015;66(15):4469-82. doi:10.1093/jxb/erv212
^ ab Orzechowski S. Starch metabolism in leaves. Acta Biochim Pol 2008;55(3):435-45. doi:10.18388/abp.2008_3049
^ Pfister B., Zeeman S.C., Rugen M.D., Field R.A., Ebenhöh O., Raguin A. Theoretical and experimental approaches to understand the biosynthesis of starch granules in a physiological context. Photosynth Res 2020;145:55-70. doi:10.1007/s11120-019-00704-y
^ Qu J., Xu S., Zhang Z., Chen G., Zhong Y., Liu L., Zhang R., Xue J., Guo D. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci Rep 2018;8(1):12736. doi:10.1038/s41598-018-30411-y
^ abcd Rathore R.S., Garg N., Garg S., Kumar A. Starch phosphorylase: role in starch metabolism and biotechnological applications. Crit Rev Biotechnol 2009;29(3):214-24. doi:10.1080/07388550902926063
^ Tetlow I.J., Bertoft E. A review of starch biosynthesis in relation to the building block-backbone model. Int J Mol Sci 2020;21(19):7011. doi:10.3390/ijms21197011
^ abc Tickle P., Burrell M.M., Coates S.A., Emes M.J., Tetlow I.J., Bowsher C.G. Characterization of plastidial starch phosphorylase in Triticum aestivum L. endosperm. J Plant Physiol 2009;166(14):1465-78. doi:10.1016/j.jplph.2009.05.004
^ Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011
^ abcd Yu G., Shoaib N., Xie Y., Liu L., Mughal N., Li Y., Huang H., Zhang N., Zhang J., Liu Y., Hu Y., Liu H., Huang Y. Comparative study of starch phosphorylase genes and encoded proteins in various Monocots and Dicots with emphasis on maize. Int J Mol Sci 2022;23:4518. doi:doi.org/10.3390/ijms23094518
L’amilopectina è un polisaccaride altamente ramificato formato da unità di alfa-D-glucosio. Insieme all’amilosio è uno dei due principali costituenti dei granuli di amido, il mezzo attraverso cui le piante immagazzinano energia, e la più diffusa e abbondante forma di accumulo di carboidrati presente sulla Terra.[12]
I residui glucosidici sono legati da legami glicosidici α-(1→4) a formare catene da cui si dipartono ramificazioni a mezzo di legami glicosidici α-(1→6).[2]
La sua sintesi richiede l’intervento coordinato di almeno quattro distinte classi di enzimi: le amido sintasi, gli enzimi ramificanti l’amido (EC 2.4.1.18), gli enzimi deramificanti l’amido, e l’amido fosforilasi (EC 2.4.1.1).[17]
Nel granulo di amido l’amilopectina è presente in quantità maggiore rispetto all’amilosio, e forma una matrice semicristallina all’interno della quale si ritiene venga depositato l’amilosio.
Il rapporto amilosio/amilopectina influenza fortemente le proprietà chimico-fisiche dell’amido, e di conseguenza sia i suoi usi industriali, come la produzione di additivi alimentari, che i possibili effetti sulla salute.[3]
L’amilopectina è un polisaccaride altamente ramificato con un peso molecolare compreso tra i 107 e 108 Dalton, quindi molto più grande dell’amilosio. Contiene 104-105 molecole di glucosio organizzate a formare molte catene relativamente corte, composte in media da 18-25 molecole di glucosio legate da legami glicosidici α-1,4.[17] La lunghezza delle catene glucosidiche varia a seconda della fonte dell’amido nonché delle condizioni ambientali e nutritive prevalenti nel corso della crescita della pianta e della formazione del seme.
Le catene sono legate tra di loro attraverso legami glicosidici α-1,6, a formare numerose ramificazioni che creano una struttura ad albero, con le catene vicine organizzate in strutture simili a grappoli o cluster.[1]
Nella maggior parte degli amidi i legami glicosidici α-1,6 rappresentano circa il 5% di tutti i legami glicosidici, una percentuale più bassa rispetto al 9% che si osserva nella molecola del glicogeno, dove le ramificazioni sono anche più uniformemente distribuite. La lunghezza e la posizione della ramificazioni influenzano direttamente le proprietà fisico-chimiche dell’amilopectina, quali la solubilità, la viscosità, la facilità di retrogradazione e la temperatura di gelificazione e incollaggi.[3] Ad esempio, il glicogeno è solubile in acqua mentre l’amilopectina non lo è.
Catene dell’amilopectina
Le catene glucosidiche dell’amilopectina possono essere classificate sulla base della loro lunghezza o della presenza o assenza di ramificazioni.
La classificazione basata sulla lunghezza individua due tipi principali di catene: le catene corte e lunghe. Le catene corte hanno un grado di polimerizzazione compreso tra 6 e 36, sebbene il limite superiore dipenda dalla fonte di amilopectina, mentre le catene lunghe hanno un grado di polimerizzazione maggiore o uguale a 36. La distribuzione molare delle catene corte e lunghe è dell’ordine di 6-19 nella maggior parte degli amidi, ed è di solito più bassa negli amidi B-cristallini, come quelli delle patate, rispetto agli amidi A-cristallini, come gli amidi di deposito dell’endosperma dei cereali.[17]
La classificazione basata sulle connessioni con altre catene individua tre categorie: catene A, catene B e catene C.[7]
Le catene A non hanno ramificazioni, sono corte, con un grado di polimerizzazione di circa 13, e sono le catene esterne.
Le catene B hanno almeno una ramificazione,ossia legano una catena A o B, sono più lunghe delle catene A, e si trovano nella parte più interna della molecola.
Le catene B a loro volta sono state suddivisi in catene B1, che hanno un grado di polimerizzazione di circa 22, le catene B2, con un grado di polimerizzazione di circa 42, le B3, con un grado di polimerizzazione di circa 69, B4 e così via.
La catene C è la catena B che porta l’estremità libera riducente.
Le catene A e le catene B1 partecipano alla formazione dei cluster, mentre si ritiene che le catene B2, B3, e B4 si estendano rispettivamente in due, tre e quattro cluster.[7][11]
Polimorfi di tipo A, B e C
All’interno dei cluster, i segmenti di catena lineari vicini formano doppie eliche, parallele tra di loro, con un periodo di 2,1 nm, e dove ogni giro conta sei unità glucosidiche per catena.[10] Le doppie eliche si dispongono a formare due tipi di strutture cristalline indicate come polimorfo di tipo A, più denso e tipico dell’amido dei cereali, e polimorfo di tipo B, a struttura esagonale, meno denso, più idratato e tipico degli amidi dei tuberi e degli amidi ad alto contenuto di amilosio. In alcuni tipi di amido, come quello presente nelle radici, legumi e alcuni frutti, sono presenti polimorfi miscele dei due precedenti, chiamati polimorfi di tipo C.[2]
Questo tipo di organizzazione è alla base della natura semicristallina dei granuli di amido.
Anelli di crescita
Sebbene i granuli di amido abbiamo forme molto differenti, la loro architettura interna è notevolmente conservata tra le differenti specie. Infatti, quando osservati al microscopio, la maggior parte degli amidi presenta uno schema regolare di strutture ad anello più scure e più chiare, strutture note come anelli di crescita, così chiamati in quanto ricordano gli anelli di crescita delle piante.[10]
Gli anelli di crescita circondano l’ilo del granulo, ossia, il centro del granulo, la cui struttura non è nota, sebbene sembri essere formata da una struttura relativamente disordinata di alfa-glucani. Gli anelli hanno uno spessore di 200-400 nm, e sono formati da regioni amorfe e semicristalline che si alternano, meno dense le prime e più dense le seconde.[10]
Secondo il modello a cluster le regioni semicristalline sono formate dall’alternanza di lamelle cristalline e lamelle amorfe, impilate con una periodicità di circa 9-10 nm.[11] Le lamelle cristalline sono formate dalle catene lineari dell’amilopectina disposte a dare i polimorfi di tipo A, B o C, e si estendono per circa 6-7 nm, mentre le lamelle amorfe presentano la maggior parte dei punti di ramificazione e si estendono per circa 3 nm.[5]
Sintesi dell’amilopectina
La sintesi dell’amilopectina si ritiene avvenga a partire dall’ilo.[22]
La sintesi richiede l’intervento coordinato di almeno quattro distinte classi di enzimi: le amido sintasi, l’amido fosforilasi, gli enzimi ramificanti l’amido e gli enzimi deramificanti l’amido.[12] Ciascuna classe si compone di diverse isoforme con proprietà biochimiche distinte.
Al pari della sintesi dell’amilosio, anche per la sintesi dell’amilopectina sono necessari corti malto-oligosaccaridi o MOS, α-(1→4)-glucani lunghi da 2 a 7 residui glucosidici, che agiscono come primer e sono allungati dalle amido sintasi, una funzione analoga a quella svolta dalla proteina glicogenina nelle fasi iniziali della sintesi del glicogeno.[16]
I MOS sembra possano avere diverse origini, che chiamano tutte in causa enzimi coinvolti nella sintesi del granulo di amido, ossia:
amido sintasi III e amido fosforilasi, quest’ultima in combinazione con l’enzima disproporzionate (EC 2.4.1.25), che utilizzano come substrati rispettivamente l’ADP-glucosio e il glucosio-1-fosfato;
gli enzimi deramificanti l’amido, nel corso del rifinitura di altre molecole di amilopectina.[16]
Grazie alla loro scarsa solubilità in ambiente acquoso, i MOS sembrano in grado di eludere l’azione idrolitica delle alfa-amilasi (EC 3.2.1.1) e delle beta-amilasi (EC 3.2.1.2).
La coordinazione, sia spaziale che temporale, degli enzimi coinvolti, che in molti casi interagiscono anche fisicamente a formare complessi multienzimatici, è essenziale per permettere la conversione dei prodotti della fotosintesi nella struttura organizzata e insolubile del polisaccaride. E, come nel caso della sintesi dell’amilosio e del glicogeno, la polimerizzazione del glucosio in amilopectina, e più in generale di monosaccaridi osmoticamente attivi in polisaccaridi osmoticamente inerti, permette il deposito di grandi quantità di monosaccaridi all’interno della cellula senza alcun aumento sostanziale della pressione osmotica.
Amido sintasi
Sono note sei isoforme dell’amido sintasi, tutte strutturalmente correlate, di cui cinque sono coinvolte nella sintesi dell’amilopectina, isoforme indicate come amido sintasi I, II, III, IV e V o, rispettivamente, SSI, SSII, SSIII, SSIV e SSV (EC 2.4.1.21), presenti nello stroma dei plastidi o suddivise tra stroma e granuli, mentre la sesta, la amido sintasi legata ai granuli o GBSS (EC 2.4.1.242), è quasi esclusivamente legata ai granuli ed è coinvolta nella sintesi dell’amilosio.[10]
Le prime quattro isoforme sono dotate di attività catalitica e appartengono, come GBSS, la glicogeno fosforilasi (EC 2.4.1.1) e la glicogeno sintasi (EC 2.4.1.11), enzimi che intervengono rispettivamente nella glicogenolisi e nella sintesi del glicogeno, alla famiglia delle glicosiltransferasi (EC 2.4). La amido sintasi V è invece priva di attività catalitica.
Nel corso della sintesi dell’amilopectina, l’amido sintasi catalizza il trasferimento di un’unità di glucosio all’estremità non riducente di un α-(1→4)-glucano, cui il monosaccaride è legato per mezzo di un legame glicosidico α-(1→4).[5]
Si noti che, a differenza della glicogeno sintasi, le amido sintasi utilizzano come donatore dell’unità glucosidica l’ADP-glucosio e non l’UDP glucosio.
La modalità d’azione dell’amido sintasi I, II, III, e IV è differente rispetto a quella della GBSS in quanto sono in grado di catalizzare l’aggiunta di una sola unità di glucosio per incontro con il substrato, modalità d’azione definita distributiva, mentre l’amido sintasi legata ai granuli è in grado di catalizzare l’aggiunta di più di un’unità glucosidica per incontro con l’α-(1→4)-glucano nascente, modalità d’azione definita processuale.[17]
Ruolo delle amido sintasi
Le fasi iniziali della sintesi dell’amilopectina, nonché la formazione di un granulo di amido normale, richiedono la presenza di SSIV, sebbene anche SSIII sembra svolgere un ruolo, sovrapponendo la sua azione a quella di SSIV.[16]
Al pari della GBSS, SSIV richiede la presenza di PTST2, una proteina della famiglia PTST. PTST2 è priva di attività catalitica, ma è in grado, grazie alla presenza di uno specifico dominio che lega i carboidrati, di facilitare il legame dell’enzima agli alfa-glucani. SSIV è anche capace di formare dimeri, una caratteristica importante sia per l’attività catalitica che per la capacità di interagire con altre proteine.
Secondo un modello d’azione di PTST2, la proteina, a mezzo del dominio che lega i carboidrati, riconosce e forma un complesso con MOS dotati di una specifica forma tridimensionale elicoidale. In complesso proteina-MOS a sua volta interagisce con un dimero SSIV, che è ora in grado di catalizzare l’allungamento dell’alfa-glucano, mentre PTST2 viene rilasciata così da permetterle di legare un altro malto-oligosaccaride e facilitarne una successiva interazione con un altro dimero di SSIV.[15]
L’azione di SSIV è seguita da quella delle altre isoforme dell’amido sintasi. SSI catalizza l’allungamento di malto-oligosaccaridi lunghi da 6 a 7 residui glucosidici, a dare oligosaccaridi con un grado di polimerizzazione compreso tra 8 e 12, che a loro volta sono ottimi substrati per SSII, che porta il grado di polimerizzazione tra 12 e 30. Questi alfa-glucani sono ulteriormente allungati dall’azione catalitica di SSIII, a dare catene lineari con un grado di polimerizzazione superiore a 30.[12] Quindi SSIII sembra agire non solo nelle fasi iniziali della sintesi del granulo di amido, ma anche nelle fasi successive.
SSIV e SSV sembrano necessarie per la sintesi di un numero regolare di granuli di amido di normale morfologia.[1][16]
Enzimi ramificanti l’amido
Gli enzimi ramificanti l’amido catalizzano la formazione di legami glicosidici α-(1→6), creando quindi punti di ramificazione nella catena lineare di alfa-glucani, di cui i principali sono il glicogeno e l’amilopectina.[20] La loro azione aumenta il numero delle estremità non riducenti, che possono servire come accettori di unità glucosidiche nelle reazioni di allungamento.[14]
SBE catalizza dapprima la scissione idrolitica di un legame α-(1→4) all’interno di una catena di alfa-glucano, liberando un oligosaccaride la cui estremità riducente viene legata al gruppo idrossilico in posizione C6 di un’unità glucosidica di un alfa-glucano. Quindi le due catene sono legate a mezzo di un legame glicosidico α-(1→6).
La catena cui l’oligosaccaride viene legato può essere la stessa da cui è stato staccato, e in questo caso di parla di trasferimento intracatena, o una catena differente, e in questo caso si parla di trasferimento intercatena.
Tra i fattori determinanti il tipo di trasferimento sembra esserci la concentrazione relativa della catene lineari α-(1→4). In particolare sembra che le catena strettamente associate, come nelle doppie eliche che si formano nei cluster, promuovano il trasferimento inter-catena.[19] Infine sembra che l’interazione tra l’amido sintasi I e l’enzima ramificante sia cruciale nel determinare la distribuzione bimodale della lunghezza delle catene osservata negli amidi delle piante.
Isoforme degli enzimi ramificanti l’amido
Nelle piante sono presenti due isoforme dell’enzima ramificante l’amido, indicate come SBEI e SBEII.
Codificate da geni distinti, hanno proprietà biochimiche distinte, il che suggerisce che svolgano ruoli differenti nella determinazione della struttura dell’amilopectina e dell’amilosio.[17]
SBEI sembra essere espresso maggiormente nei tessuti di deposito, il che suggerisce un suo ruolo importante nel determinare le proprietà strutturali degli amidi di deposito, mostra una preferenza di substrato per l’amilosio, e in grado di trasferire oligosaccaridi con un grado di polimerizzazione superiore ai 30, sebbene la maggior parte sia tra 10 e 13.[6] Sembra inoltre intervenire nella formazione delle catene superlunghe ed extralunghe dell’amilopectina, mentre sembrano meno importanti altri suoi contributi alla struttura dell’amilopectina. Non tutte le piante esprimono SBEI; ad esempio Arabidopsis e Canola (Brassica napus L.), che immagazzinano olio, e dove l’amido è presente solo nei tessuti fotosintetici, hanno solo SBEII.
SBEII è maggiormente espresso nelle graminacee, nei cereali, e in molte altre piante. La sua perdita determina importanti alterazioni nell’architettura dell’amilopectina e influenza anche la quantità totale di amido, riducendone il contenuto. L’enzima mostra una preferenza di substrato per l’amilopectina e trasferisce oligosaccaridi relativamente corti, formati da 6-14 unità di glucosio. Nei cereali e nelle graminacee SBEII è presente con due isoforme tessuto-specifiche e codificate da geni distinti, indicate come SBEIIa, presente principalmente nelle foglie, e SBEIIb, presente prevalentemente nell’endosperma.[17]
Enzimi deramificanti l’amido
Gli enzimi deramificanti l’amido catalizzano l’idrolisi dei legami glicosidici α-(1→6), e appartengono alla superfamiglia delle alfa-amilasi.
Nelle piante sono presenti due tipi di enzimi deramificanti: le isoamilasi (EC 3.2.1.68) e le pullulanasi (EC 3.2.1.41).[8] Le isoamilasi deramificano l’amilopectina e altri poliglucani, mentre le pullulanasi l’amilopectina e il pullulano, un polisaccaride fungino. Isoamilasi e pullulanasi differiscono anche riguardo alla specificità di substrato in quanto le isoamilasi richiedono che le ramificazioni siano formate da almeno tre residui glucosidici, mentre le pullulanasi sono in grado di agire su ramificazioni con non meno di due residui glucosidici.[18]
Nel corso della formazione del granulo di amido gli enzimi deramificanti svolgono un ruolo cruciale nel determinare le proprietà insolubili e la struttura fine dell’amilopectina. Si ritiene infatti che le idrolisi operate permettano il raggruppamento delle ramificazioni rimanenti, promuovendo in questo modo le interazioni tra catene adiacenti e la formazione delle alfa-eliche, che a loro volta sono importanti nella formazione delle strutture semicristalline insolubili in acqua dell’amilopectina, e quindi dell’amido.[17] Nella struttura semicristallina le ramificazioni sono presumibilmente inaccessibili all’azione della alfa-amilasi e beta-amilasi e dell’enzima deramificante stesso.
Gli enzimi deramificanti l’amido sono utilizzati industrialmente nella preparazione dell’amido resistente e delle ciclodestrine, che sono oligosaccaridi ciclici.
Amido fosforilasi
L’amido fosforilasi appartiene, al pari delle amido sintasi, alla famiglia delle glicosiltransferasi, ed è una fosforilasi simile alla glicogeno fosforilasi. Nelle piante è presente con almeno due forme isoenzimatiche, Pho1, che si ritrova nello stroma dei plastidi, ed è considerata la vera amido fosforilasi che si ritiene agisca nella sintesi dell’amido, e Pho2, isoforma a localizzazione citosolica.[4]
L’amido fosforilasi sembra essere coinvolta nei passaggi iniziali della sintesi dell’amido, catalizzando il trasferimento, reversibile, di unità glucosidiche a un alfa-glucano, unità che sono legate attraverso un legame glicosidico α-(1→4).[3] A differenza delle amido sintasi, il substrato donatore dell’unità glucosidica è il glucosio-1-fosfato e non l’ADP-glucosio.[4]
Fosforilazione dell’amilopectina
L’amilopectina, analogamente al glicogeno, lega covalentemente gruppi fosfato in quantità variabili a seconda dell’origine botanica dell’amido. Ad esempio, l’amido delle patate ha un contenuto di gruppi fosfato relativamente alto, con un grado di sostituzione di circa lo 0,1-0,3%, mentre negli amidi di deposito dell’endosperma dei cereali è generalmente inferiore allo 0,01%.[9]
La fosforilazione dell’amilopectina è catalizzata da due dichinasi presenti nei plastidi: la alfa-glucano acqua dichinasi (EC 2.7.9.4) e la fosfoglucano acqua dichinasi (EC 2.7.9.5). I due enzimi catalizzano il trasferimento del gruppo fosfato beta dell’ATP a un residuo glucosidico di una di catena di alfa-glucano, mentre il gruppo fosfato gamma è trasferito all’acqua. Nello specifico, la alfa-glucano acqua dichinasi catalizza la fosforilazione del gruppo ossidrilico in posizione C6, mentre la fosfoglucano acqua dichinasi del gruppo ossidrilico in posizione C3, di solito di un alfa-glucano prefosforilato.[13] La posizione C6 raccoglie circa i due terzi dei gruppi fosfato, mentre la posizione C3 circa il 20-30%. Gruppi fosfato si ritrovano anche in posizione C2, sebbene in percentuale molto ridotta rispetto alle altre posizioni. Non è noto quale enzima catalizzi questa fosforilazione.
Riguardo alla specificità di substrato sembra che le fosforilazioni si accumulino più facilmente sulle catene più lunghe. Inoltre sembra esistere anche una correlazione inversa tra il contenuto totale di fosfato e la frequenza delle ramificazioni dell’amilopectina.
Le cariche negative portate dai gruppi fosfato determinano modificazioni localizzate della struttura del polisaccaride, in quanto tendono a far allontanare le une dalle altre le catene fosforilate adiacenti. Queste repulsioni sembra permettano l’apertura e l’idratazione delle catene, influenzando in questo modo l’attività degli enzimi della biosintesi e rendendo anche più suscettibili le catene stesse all’attacco delle amilasi.[21]
Rapporto amilosio/amilopectina
I granuli di amido sono costituiti per la maggior da amilosio e amilopectina.[12] I due polisaccaridi sono presenti in percentuali variabili, con l’amilosio che non costituisce più del 35% del peso secco del granulo.[5] Esistono tuttavia piante nelle quali i granuli di amido sono costituiti per la maggior parte, o quasi interamente, da amilopectina, amidi definiti cerosi, e altre dove, al contrario, l’amilosio costituisce la maggior parte, e in alcuni casi, la quasi totalità della componente polisaccaridica del granulo.[19]
Il rapporto amilosio/amilopectina influenza le proprietà fisico-chimiche dell’amido, quali la capacità di assorbire acqua, la gelatinizzazione e la retrogradazione, o la resistenza all’idrolisi enzimatica, quest’ultima importante nel determinare la velocità con cui, nel corso della digestione dei carboidrati, amilosio e amilopectina sono idrolizzati a maltosio e maltotriosio ad opera della alfa-amilasi.[3] Pertanto, il rapporto amilosio/amilopectina influenza gli effetti dei differenti tipi di amido sulla salute nonché i possibili usi industriali.
Bibliografia
^ ab Abt M.R., Pfister B., Sharma M., Eicke S., Bürgy L., Neale I., Seung D., Zeeman S.C. STARCH SYNTHASE5, a noncanonical starch synthase-like protein, promotes starch granule initiation in Arabidopsis. Plant Cell 2020;32(8):2543-2565. doi:10.1105/tpc.19.00946
^ ab Cornejo-Ramírez Y.I., Martínez-Cruz O., Del Toro-Sánchez C.L., Wong-Corral F.J., Borboa-Flores J. & Cinco-Moroyoqui F.J. The structural characteristics of starches and their functional properties. CYTA J Food 2018;16(1):1003-1017. doi:10.1080/19476337.2018.1518343
^ abcd Crofts N., Abe N., Oitome N.F., Matsushima R., Hayashi M., Tetlow I.J., Emes M.J., Nakamura Y., Fujita N. Amylopectin biosynthetic enzymes from developing rice seed form enzymatically active protein complexes. J Exp Bot 2015;66(15):4469-82. doi:10.1093/jxb/erv212
^ ab Cuesta-Seijo J.A., Ruzanski C., Krucewicz K., Meier S., Hägglund P., Svensson B., Palcic M.M. Functional and structural characterization of plastidic starch phosphorylase during barley endosperm development. PLoS One 2017;12(4):e0175488. doi:10.1371/journal.pone
^ abc Gous P.W., Fox G.P. Review: Amylopectin synthesis and hydrolysis – Understanding isoamylase and limit dextrinase and their impact on starch structure on barley (Hordeum vulgare) quality. Trends Food Sci Technol 2017;62:23-32. doi:10.1016/j.tifs.2016.11.013
^ Guan H.P., Preiss J. Differentiation of the properties of the branching isozymes from maize (Zea mays). Plant Physiol 1993;102(4):1269-1273. doi:10.1104/pp.102.4.1269
^ ab Li G., Yacine Y., Zhu F. Relationships between supramolecular organization and amylopectin fine structure of quinoa starch. Food Hydrocoll 2021;117:106685. doi:10.1016/j.foodhyd.2021.106685
^ Møller M.S., Henriksen A., Svensson B. Structure and function of α-glucan debranching enzymes. Cell Mol Life Sci. 2016;73(14):2619-41. doi:10.1007/s00018-016-2241-y
^ Nitschke F., Wang P., Schmieder P., Girard J.M., Awrey D.E., Wang T., Israelian J., Zhao X., Turnbull J., Heydenreich M., Kleinpeter E., Steup M., Minassian B.A. Hyperphosphorylation of glucosyl C6 carbons and altered structure of glycogen in the neurodegenerative epilepsy Lafora disease. Cell Metab 2013;17(5):756-67. doi:10.1016/j.cmet.2013.04.006
^ abcd Pfister B., Zeeman S.C. Formation of starch in plant cells. Cell Mol Life Sci 2016;73(14):2781-807. doi:10.1007/s00018-016-2250-x
^ ab Pfister B., Zeeman S.C., Rugen M.D., Field R.A., Ebenhöh O., Raguin A. Theoretical and experimental approaches to understand the biosynthesis of starch granules in a physiological context. Photosynth Res 2020;145:55-70. doi:10.1007/s11120-019-00704-y
^ abcd Qu J., Xu S., Zhang Z., Chen G., Zhong Y., Liu L., Zhang R., Xue J., Guo D. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci Rep 2018;8(1):12736. doi:10.1038/s41598-018-30411-y
^ Ritte G., Heydenreich M., Mahlow S., Haebel S., Kötting O., Steup M. Phosphorylation of C6- and C3-positions of glucosyl residues in starch is catalysed by distinct dikinases. FEBS Lett 2006;580(20):4872-6. doi:10.1016/j.febslet.2006.07.085
^ Sawada T., Nakamura Y., Ohdan T., Saitoh A., Francisco P.B. Jr, Suzuki E., Fujita N., Shimonaga T., Fujiwara S., Tsuzuki M., Colleoni C., Ball S. Diversity of reaction characteristics of glucan branching enzymes and the fine structure of α-glucan from various sources. Arch Biochem Biophys 2014;562:9-21. doi:10.1016/j.abb.2014.07.032
^ Seung D., Boudet J., Monroe J., Schreier T.B., David L.C., Abt M., Lu K.J., Zanella M., Zeeman S.C. Homologs of PROTEIN TARGETING TO STARCH control starch granule initiation in Arabidopsis leaves. Plant Cell 2017;29(7):1657-1677. doi:10.1105/tpc.17.00222
^ abcd Szydlowski N., Ragel P., Raynaud S., Lucas M.M., Roldán I., Montero M., Muñoz F.J., Ovecka M., Bahaji A., Planchot V., Pozueta-Romero J., D’Hulst C., Mérida A. Starch granule initiation in Arabidopsis requires the presence of either class IV or class III starch synthases. Plant Cell 2009;21(8):2443-57. doi:10.1105/tpc.109.066522
^ abcdefg Tetlow I.J., Bertoft E. A review of starch biosynthesis in relation to the building block-backbone model. Int J Mol Sci 2020;21(19):7011. doi:10.3390/ijms21197011
^ Xia W., Zhang K., Su L., Wu J. Microbial starch debranching enzymes: developments and applications. Biotechnol Adv 2021;50(3):107786. doi:10.1016/j.biotechadv.2021.107786
^ ab Wang J., Hu P., Lin L., Chen Z., Liu Q., Wei C. Gradually decreasing starch branching enzyme expression is responsible for the formation of heterogeneous starch granules. Plant Physiol 2018;176(1):582-595. doi:10.1104/pp.17.01013
^ Wilkens C., Svensson B., Møller M.S. Functional roles of starch binding domains and surface binding sites in enzymes involved in starch biosynthesis. Front Plant Sci 2018;9:1652. doi:10.3389/fpls.2018.01652
^ Zhou W., He S., Naconsie M., Ma Q., Zeeman S.C., Gruissem W. & Zhang P. Alpha-glucan, water dikinase 1 affects starch metabolism and storage root growth in Cassava (Manihot esculenta Crantz). Sci Rep 2017;7:9863 doi:10.1038/s41598-017-10594-6
^ Ziegler G.R., Creek J.A., Runt J. Spherulitic crystallization in starch as a model for starch granule initiation. Biomacromolecules 2005;6(3):1547-54. doi:10.1021/bm049214p
L’ipertensione arteriosa viene definita come una pressione arteriosa media a riposo di 140/90 o maggiore e/o l’uso corrente di medicinali antiipertensivi.
E’ il problema di salute pubblica più comune nei paesi industrializzati. Spesso definita “killer silenzioso”, in quanto le persone affette possono essere asintomatiche per molti anni e poi andare incontro a un attacco di cuore fatale, è uno tra i maggiori fattori di rischio per lo sviluppo di malattia coronarica, infarto del miocardio, insufficienza cardiaca, ictus, e una delle principali cause di morbilità e mortalità. Tuttavia, tra i fattori di rischio di malattie cardiovascolari, è quello maggiormente modificabile.
L’ipertensione può essere primaria o essenziale e secondaria.
L’ipertensione primaria, responsabile di circa il 95% dei casi, è probabilmente conseguenza di fattori ambientali, fattori genetici, e della loro interazione. Tra i fattori ambientali la dieta ha un ruolo centrale. Tra i fattori genetici l’interesse si è focalizzato sui fattori che influenzano la risposta pressoria all’assunzione di sale, e sono stati identificati diversi genotipi, molti dei quali influenzano l’asse renina-angiotensina-aldosterone o il trattamento del sale a livello renale.
L’ipertensione secondaria è causata da altre patologie, solitamente endocrine, ad esempio l’ipertiroidismo, l’iperaldosteronismo, e la sindrome di Cushing.
Una pressione arteriosa al di sopra dei valori ottimali, anche se non ancora nell’intervallo ipertensivo o pre-ipertensivo, conferisce un aumento del rischio di malattie cardiovascolari, come mostrato dal fatto che quasi un terzo delle morti da malattia coronarica correlate alla pressione sono stimate accadere in soggetti non ipertesi, con pressione sistolica di 120-139 mm Hg o diastolica di 80-89 mm Hg. Ciò significa che il rischio di malattie cardiovascolari aumenta per tutto l’intervallo pressorio a partire da 115/75 mm Hg.
Categoria
Pressione arteriosa (mm Hg)
Sistolica
Diastolica
Ottimale
< 120
< 80
Normale
< 130
< 85
Normale – Alta
130 – 139
85 – 89
Ipertensione di grado 1
140 – 159
90 – 99
Ipertensione di grado 2
160 – 179
100 – 109
Ipertensione di grado 3
≥180
≥110
Ipertensione sistolica isolata
≥140
≤ 90
Infine, i soggetti pre-ipertesi hanno un elevato rischio, pari a circa il 90%, di sviluppare nel tempo l’ipertensione, sebbene il passaggio non sia inevitabile.
Ipertensione nella terza età
La prevalenza dell’ipertensione aumenta con l’aumentare dell’età, come mostrato dal fatto che più della metà della popolazione adulta oltre i 60 anni è ipertesa.
Il rischio correlato all’età è funzione di variabili legate allo stile di vita quali l’aumento di peso, la scarsa attività fisica, l’uso eccessivo di sale, grassi e acidi grassi saturi, alcool e ipercolesterolemia, e un basso apporto di frutta e verdura, piuttosto che all’invecchiamento in se. Ad esempio, studi condotti su vegetariani che vivono in paesi industrializzati hanno mostrato che tali abitudini alimentari sono associate con un minor aumento della pressione con l’aumentare dell’età, e con una pressione marcatamente più bassa se comparata a quella dei non vegetariani.
Ipertensione ed età pediatrica
Secondo uno studio condotto da un team di ricercatori della Johns Hopkins University la prevenzione dell’ipertensione ha inizio nell’infanzia.
Inoltre, una meta-analisi su studi condotti su diverse popolazioni, pubblicati tra gennaio 1970 e luglio 2006, ha esaminato il cammino della pressione dall’infanzia all’età adulta dimostrando che la pressione dell’infanzia è associata con quella nei periodi successivi della vita, e che un valore elevato nell’infanzia è probabilmente di aiuto nel predire l’ipertensione in età avanzata.
Infine, altri studi hanno anche dimostrano che l’aumentata pressione arteriosa tra i bambini è correlata alla crescente epidemia di obesità.
Come prevenire l’ipertensione
Nelle ultime due decadi negli USA è stato documentato un trend discendente della pressione arteriosa, cui ha contribuito l’adozione di uno stile di vita più salutare.
Le modificazioni dello stile di vita che riducono efficacemente la pressione sono:
limitare l’apporto di sale e di altre forme di sodio;
seguire un’alimentazione ricca di frutta fresca, verdura, carboidrati complessi e prodotti caseari a basso contenuto di grassi;
aumentare l’apporto di potassio consumando frutta, verdura e legumi;
perdere peso corporeo se in sovrappeso, o prevenirne l’aumento tra coloro che sono magri;
aumentare l’attività fisica di bassa o moderata intensità;
smettere di fumare;
Questi cambiamenti rappresentano la prima linea di difesa nella lotta contro la pressione alta, e non dovrebbero essere fatti uno alla volta: i migliori risultati si ottengono quando i cambiamenti avvengono contemporaneamente, come dimostrato da due studi in cui interventi “multicomponent” hanno ridotto la pressione arteriosa nei partecipanti ipertesi e non ipertesi.
Infine, è stato dimostrato una relazione anche tra assunzione di alcool e ipertensione.
Ruolo del potassio
Il potassio, un nutriente essenziale per l’uomo, è il più abbondante catione presente nei fluidi intracellulari. E’ quindi ampiamente diffuso nei cibi che derivino da tessuti animali e vegetali, ma che non abbiano subito processi di salatura e/o essicazione. Anche i metodi di cottura tendono a ridurne la quantità. Considerando le verdure, il peggior metodo di cottura è la bollitura in abbondante acqua, prolungata per più di un’ora, mentre il migliore è la cottura a microonde.
Potassio
Contenuto
>250 mg/100 g
Legumi
Legumi secchi, quali ceci, fagioli, lenticchie, piselli e soia, e fagioli freschi.
Carne/prodotti della pesca, sia freschi che conservati. Questi ultimi sono da limitare a causa dell’elevato contenuto in sodio.
Un’elevata assunzione di potassiocon gli alimenti e la pressione arteriosa sono inversamente correlate, come dimostrato da studi condotti su animali, studi epidemiologici osservazionali, sperimentazioni cliniche, studi di alimentazione controllata, come il DASH Study e l’OmniHeart trial, e meta-analisi. Inoltre, un elevato apporto di potassio aumenta anche l’escrezione urinaria di sodio.
La strategia ottimale per aumentare l’apporto di potassio è quella di consumare alimenti naturalmente ricchi del minerale quali frutta e verdura di stagione, e legumi, alimenti tipici della dieta mediterranea. Non è quindi difficile raggiungere il livello di assunzione giornaliera raccomandata, per la popolazione sana, che è pari a 4,7 g/die.
Ruolo del sodio
A differenza del potassio, il sodio è il principale catione dei liquidi extracellulari, di cui ne condiziona fortemente il valore della pressione osmotica.
Le principali fonti alimentari sono tre.
La più intuitiva è il sale utilizzato a tavola, che rappresenta fino al 20% dell’apporto giornaliero. Si noti che, sebbene molto spesso i termini sale e sodio siano usati in modo interscambiabile, ciò non è corretto. Sulla base del peso, il sale è costituito per il 40% da sodio e per il restante 60% da cloro.
Una seconda fonte è il sale o i composti del sodio aggiunti durante la preparazione o la trasformazione dei cibi. Tra il 35 e 80% del sodio assunto giornalmente proviene dai cibi trasformati, quali:
carne e pesce affumicati, sott’olio o sotto sale;
estratti di carne e salse, snack salati, la salsa di soia, e la salsa piccante;
Trascurabile è invece la terza fonte, il sodio naturalmente presente negli alimenti, in genere basso negli alimenti freschi.
Un elevata assunzione di sodio contribuisce all’aumento della pressione arteriosa e allo sviluppo dell’ipertensione. Ciò è supportato da molti studi epidemiologici, su animali, di migrazione delle popolazioni, e meta-analisi, con la prova finale derivante da studi accuratamente controllati dose-risposta. Inoltre, nelle società primitive, dove l’apporto di sodio è molto basso, le persone raramente vanno incontro a ipertensione, e con l’avanzare dell’età non si verifica l’aumento della pressione arteriosa.
Pertanto, per prevenire lo sviluppo dell’ipertensione si raccomanda una riduzione del suo apporto. In ragione degli alimenti a disposizione e dell’elevato consumo corrente, una raccomandazione ragionevole può essere di porre come limite superiore un apporto di sodio di 2,3 g/die, pari a 5,8 g/die di sale.
Come può essere raggiunto questo valore?
Usando meno sale possibile durante la preparazione dei cibi.
Evitando di aggiungere sale una volta che i cibi sono in tavola.
Limitando il consumo di cibi trasformati molto salati.
Studi clinici hanno documentato che una ridotta assunzione di sodio è in grado di abbassare la pressione arteriosa anche in un quadro di assunzione di farmaci antipertensivi, e può facilitare il controllo dell’ipertensione stessa.
Alcuni componenti della dieta possono modificare la risposta pressoria al sodio. Un elevato apporto dietetico di cibi ricchi di potassio e calcio può prevenire o attenuare l’aumento della pressione a seguito di un dato aumento dell’apporto di sodio. Al contrario, alcuni dati, osservati principalmente in modelli animali, suggeriscono che una elevata assunzione di saccarosio potrebbe potenziare la sensibilità della pressione arteriosa verso sodio.
Nota: elevati apporti di sodio possono contribuire allo sviluppo dell’osteoporosi, in quanto determinano un aumento dell’escrezione renale di calcio, in modo particolare se l’assunzione giornaliera di calcio è bassa.
Ruolo del peso corporeo
Il peso corporeo, in particolare il sovrappeso e l’obesità, sono un fattore determinante della pressione arteriosa a ogni età. Infatti:
è stato stimato che il rischio di sviluppare una pressione elevata è da due a sei volte maggiore nelle persone in sovrappeso rispetto ai normopeso;
c’è una correlazione lineare tra la pressione arteriosa e il peso corporeo o l’indice di massa corporea, che, se maggiore di 27, è correlato con un aumento della pressione;
anche quando l’assunzione di sodio è mantenuta costante, la correlazione tra variazione di peso e pressione arteriosa è lineare;
il 60% dei soggetti affetti da ipertensione è più del 20% in sovrappeso;
la distribuzione centrale della massa grassa, come fattore determinante l’aumento della pressione arteriosa, con una circonferenza addominale superiore a 88 cm nelle donne e 102 negli uomini, è più importante della localizzazione periferica del grasso stesso, sia negli uomini che nelle donne;
la perdita di peso, sia nei soggetti affetti da ipertensione che nei normotesi, può ridurre la pressione arteriosa, e la riduzione si verifica prima, e senza, il raggiungimento di un peso corporeo desiderabile.
Ruolo dell’attività fisica
L’attività fisica produce un calo della pressione sistolica e diastolica. Quindi, per la prevenzione primaria dell’ipertensione è importante aumentare l’attività fisica di bassa o moderata intensità per 30-45 minuti per 3-4 volte alla settimana fino a un’ora quasi tutti i giorni, come raccomandato dalla Organizzazione Mondiale della Sanità. Di contro, le persone meno attive hanno dal 30 al 50% di probabilità in più di sviluppare ipertensione rispetto a quelle attive.
Bibliografia
Appel L.J., Brands M.W., Daniels S.R., Karanja N., Elmer P.J. and Sacks F.M. Dietary approaches to prevent and treat HTN: a scientific statement from the American Heart Association. Hypertension 2006;47:296-08. doi:10.1161/01.HYP.0000202568.01167.B6
Bibbins-Domingo K., Chertow G.M., Coxson P.G., Moran A., Lightwood J.M., Pletcher M.J., and Goldman L. Projected effect of dietary salt reductions on future cardiovascular disease. N Engl J Med 2010;362:590-9. doi:10.1056/NEJMoa0907355
Cappuccio FP. Overview and evaluation of national policies, dietary recommendtions and programmes around the world aiming at reducing salt intake in the population. World Health Organization. Reducing salt intake in populations: report of a WHO forum and technical meeting. WHO Geneva 2007;1-60.
Chen J, Gu D., Jaquish C.E., Chen C., Rao D.C., Liu D., Hixson J.E., Lee Hamm L., Gu C.C., Whelton P.K. and He J. for the GenSalt Collaborative Research Group. Association between blood pressure responses to the cold pressor test and dietary sodium intervention in a chinese population. Arch Intern Med. 2008;168:1740-1746. doi:10.1001/archinte.168.16.1740
Chen X. and Wang Y. Tracking of blood pressure from childhood to adulthood. A systematic review and meta-regression analysis. Circulation 2008;117:3171-80. doi:10.1161/CIRCULATIONAHA.107.730366
Denton D., Weisinger R., Mundy N.I., Wickings E.J., Dixson A., Moisson P., Pingard A.M., Shade R., Carey D., Ardaillou R., Paillard F., Chapman J., Thillet J. & Michel J.B. The effect of increased salt intake on blood pressure of chimpanzees. Nature Med 1995;10:1009-1016. doi:10.1038/nm1095-1009
Ford E.S., Ajani U.A., Croft J.B., Critchley J.A., Labarthe D.R., Kottke T.E., Giles W.H, and Capewell S. Explaining the decrease in U.S. deaths from coronary disease, 1980-2000. N Engl J Med 2007;356:2388-98. doi:10.1056/NEJMsa053935
Geleijnse J.M., Witteman J.C., den Breeijen J.H., Hofman A., de Jong P., Pols H.A. and Grobbee D.E. Dietary electrolyte intake and blood pressure in older subjects: the Rotterdam Study. J Hyperten 1996;14:73741. doi:10.1097/00004872-199606000-00009
Gutiérrez O.M. Sodium- and phosphorus-based food additives: persistent but surmountable hurdles in the management of nutrition in chronic kidney disease. Adv Chronic Kidney Dis 2013;20(2):150-6. doi:10.1053/j.ackd.2012.10.008
Harlan W.R. and Harlan L.C. Blood pressure and calcium and magnesium intake. In: Laragh J.H., Brenner B.M., eds. Hypertension: pathophysiology, diagnosis and management. 2end ed. New York: Raven Press 1995;1143-1154
He F.J., Tan M., Ma Y., MacGregor G.A. Salt reduction to prevent hypertension and cardiovascular disease: JACC state-of-the-art review. J Am Coll Cardiol 2020;75(6):632-647. doi:10.1016/j.jacc.2019.11.055
Holmes E., Loo R.L., Stamler J., Bictash M., Yap I.K.S., Chan Q., Ebbels T., De Iorio M., Brown I.J., Veselkov K.A., Daviglus M.L., Kesteloot H., Ueshima H., Zhao L., Nicholson J.K. and Elliott P. Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 2008;453:396-400. doi:10.1038/nature06882
Nugroho P., Andrew H., Kohar K., Noor C.A., Sutranto A.L. Comparison between the world health organization (WHO) and international society of hypertension (ISH) guidelines for hypertension. Ann Med 2022;54(1):837-845. doi:10.1080/07853890.2022.2044510
Sesso H.D., Cook N.R., Buring J.E., Manson J.E. and Gaziano J.M. Alcohol consumption and the risk of hypertension in women and men. Hypertension 2008;51:1080-1087. doi:10.1161/HYPERTENSIONAHA.107.104968
Simpson F.O. Blood pressure and sodium intake. In: Laragh J.H., Brenner B.M. eds. Hypertension: pathophysiology, diagnosis and management. 2end ed. New York: Raven Press 1995;273-281
Stone M.S., Martyn L., Weaver C.M. Potassium intake, bioavailability, hypertension, and glucose control. Nutrients 2016;8(7):444. doi:10.3390/nu8070444
Strazzullo P., D’Elia L., Kandala N. and Cappuccio F.P. Salt intake, stroke, and cardiovascular disease: meta-analysis of prospective studies. BMJ 2009;339:b4567. doi:10.1136/bmj.b4567
Tzoulaki I., Brown I.J., Chan Q., Van Horn L., Ueshima H., Zhao L., Stamler J., Elliott P., for the International Collaborative Research Group on Macro-/Micronutrients and Blood Pressure. Relation of iron and red meat intake to blood pressure: cross sectional epidemiological study. BMJ 2008;337:a258. doi:10.1136/bmj.a258
Unger T., Borghi C., Charchar F., Khan N.A., Poulter N.R., Prabhakaran D., Ramirez A., Schlaich M., Stergiou G.S., Tomaszewski M., Wainford R.D., Williams B., Schutte A.E. 2020 International society of hypertension global hypertension practice guidelines. hypertension 2020;75(6):1334-1357. doi:10.1161/HYPERTENSIONAHA.120.15026
Weinberger M.H. The effects of sodium on blood pressure in humans. In: Laragh J.H., Brenner B.M., eds. Hypertension: pathophysiology, diagnosis and management. 2end ed. New York: Raven Press 1995;2703-2714.
Writing Group of the PREMIER Collaborative Research Group. Effects of comprehensive lifestyle modification on blood pressure control: main results of the PREMIER Clinical Trial. JAMA 2003;289:2083-2093. doi:10.1001/jama.289.16.2083
World Health Organization, International Society of Hypertension Writing Group. 2003 World Health Organization (WHO)/ISH statement on management of HTN. Guidelines and recommendations. J Hyperten 2003;21:1983-92. doi:10.1097/00004872-200311000-00002
E il colesterolo assunto con gli alimenti?
Non esiste una correlazione diretta tra i livelli di colesterolo plasmatico e l’assunzione di colesterolo con gli alimenti. Tuttavia, il colesterolo alimentare può aumentare i livelli del colesterolo plasmatico se assunto in associazione agli acidi grassi saturi e agli acidi grassi trans.
Se comunque vuoi ridurre l’assunzione di colesterolo è consigliabile diminuire l’uso di prodotti di origine animale e/o consumare latte parzialmente scremato o scremato, formaggi con basso contenuto di grassi, yogurt magro, e carne magra.
Un fattore di rischio per l’ipercolesterolemia è rappresentato da un’elevata assunzione di acidi grassi saturi, un gruppo di lipidi che possono essere facilmente utilizzati per la produzione endogena di colesterolo.
Questi acidi grassi sono presenti nella carne, nei latticini, e in elevata quantità nei grassi e oli vegetali, come la margarina, l’olio di palma, l’olio di semi di palma, e l’olio di cocco, molto utilizzati nell’industria dolciaria.
Cosa fare:
eliminare il grasso visibile dalla carne o acquistarne tagli magri;
sostituire il latte intero, il burro, i formaggi grassi, le creme, e i gelati con prodotti con un contenuto in grassi più basso, come lo yogurt magro, il latte parzialmente scremato o scremato, e i formaggi magri;
evitare i prodotti di pasticceria.
Acidi grassi trans
Gli acidi grassi trans o grassi trans costituiscono un fattore di rischio molto elevato, e non solo per l’ipercolesterolemia.
Studi condotti su volontari ne hanno evidenziato una grande capacità aterogenica dovuta a modificazione a carico delle lipoproteine plasmatiche, dove si osserva una diminuzione dei livelli delle HDL e un aumento dei livelli delle LDL e dei trigliceridi.
Dove si possono trovare?
In molti alimenti per bambini.
Nei prodotti da forno industriali come cracker, grissini, biscotti, pane confezionato, e snack.
In prodotti come zuppe, piatti pronti, freschi o surgelati, e in paste di base per torte e pizze casalinghe.
Nei dadi da brodo.
Nelle caramelle morbide.
In alcuni cereali per la prima colazione.
Nei gelati, nei surrogati vegetali della panna, e nella margarina.
In molte conserve vegetali, incluse le marmellate.
Per quanto riguarda il contenuto in acidi grassi saturi e trans, molto spesso non ci sono differenze tra i prodotti classici e quelli “naturali” o “biologici”.
Cosa fare?
Evitare di acquistare prodotti che nella composizione presentino grassi/oli vegetali idrogenati o parzialmente idrogenati, ed evitare di acquistare prodotti fritti.
Sovrappeso
Un significativo aumento della massa grassa contribuisce all’ipercolesterolemia.
Per molte persone la riduzione dell’assunzione degli acidi grassi saturi e trans non si accompagna a una riduzione della colesterolemia fino a che non c’è un concomitante calo del peso corporeo.
Cosa fare per tenere sotto controllo il peso?
Ridurre il consumo di grassi di origine animale e vegetale.
Ridurre i cibi ricchi di zuccheri semplici come dolci, soft drinks, dessert, caramelle e biscotti.
Non reintrodurre le calorie eliminate nei due punti precedenti con un eccessivo consumo di olio di oliva e amido, ossia pasta, patate, riso, pane.
Aumentare l’attività fisica.
Aumentare il consumo di frutta e verdura.
Cause genetiche
In questo caso è necessario un intervento farmacologico, che dovrà essere prescritto dal proprio medico curante, ma che deve essere comunque associato a una alimentazione adeguata.
Bibliografia
Ascherio A., Katan M.B., Zock P.L., Stampfer M.J., Willett W.C. Trans fatty acids and coronary heart disease. N Engl J Med 1999;340:1994-1998. doi:10.1056/NEJM199906243402511
Benito-Vicente A., Uribe K.B., Jebari S., Galicia-Garcia U., Ostolaza H., Martin C. Familial hypercholesterolemia: the most frequent cholesterol metabolism disorder caused disease. Int J Mol Sci 2018;19(11):3426. doi:10.3390/ijms19113426
Fernandez M.L., Murillo A.G. Is there a correlation between dietary and blood cholesterol? Evidence from epidemiological data and clinical interventions. Nutrients 2022;14(10):2168. doi:10.3390/nu14102168
Hu F.B., Willett W.C. Optimal diet for prevention of coronary heart disease JAMA 2002;288:2569-2578. doi:10.1001/jama.288.20.2569
Lichtenstein A.H. Dietary fat, carbohydrate, and protein: effects on plasma lipoprotein patterns J. Lipid Res. 2006;47:1661-1667. doi:10.1194/jlr.R600019-JLR200
Lichtenstein A.H., Ausman L., Jalbert S.M., Schaefer E.J. Effect of different forms of dietary hydrogenated fats on serum lipoprotein cholesterol levels. N Engl J Med 1999;340:1933-1940. doi:10.1056/NEJM199906243402501
Mensink R.P., Katan M.B. Effect of dietary trans fatty acids on high-density and low-density lipoprotein cholesterol levels in healthy subjects. N Engl J Med 1990;323:439-445. doi:10.1056/NEJM199008163230703
Mozaffarian D., Katan M.B., Ascherio A., Stampfer M.J., Willett W.C. Trans fatty acids and cardiovascular disease. N Engl J Med 2006;354:1601-1613. doi:10.1056/NEJMra054035
Shils M.E., Olson J.A., Shike M., Ross A.C.: “Modern nutrition in health and disease” 9th ed., by Lippincott, Williams & Wilkins, 1999
L’amido sintasi (EC 2.4.1.21) è un enzima che catalizza il trasferimento di molecole di glucosio dall’ADP-glucosio all’estremità non riducente di un α-(1→4)-glucano preesistente, cui i monosaccaridi sono legati attraverso un legame glicosidico α-(1→4).[4]
L’enzima è coinvolto nella sintesi dell’amilosio e dell’amilopectina, i due polisaccaridi che costituiscono la quasi totalità del granulo di amido, la più abbondante forma di accumulo di carboidrati ed energia presente nelle piante.[7]
L’amido sintasi appartiene alla famiglia delle glicosiltransferasi (EC 2.4), al pari dell’amido fosforilasi (EC 2.4.1.1), un altro enzima coinvolto nel metabolismo dell’amido, e della glicogeno sintasi (EC 2.4.1.11) e glicogeno fosforilasi (EC 2.4.1.1), enzimi coinvolti rispettivamente nella sintesi del glicogeno e nella degradazione del glicogeno o glicogenolisi.[6] Tuttavia, mentre la glicogeno sintasi utilizza come donatore dell’unità glucosidica l’UDP-glucosio, e l’amido fosforilasi il glucosio-1-fosfato, l’amido sintasi utilizza l’ADP-glucosio.[2][3]
Nelle piante sono note sei isoforme di amido sintasi. Sono proteine strutturalmente correlate, cinque coinvolte nella sintesi dell’amilopectina, indicate come amido sintasi I, II, III, IV e V o, rispettivamente, SSI, SSII, SSIII, SSIV e SSV, e una coinvolta nella sintesi dell’amilosio, l’amido sintasi legata ai granuli o GBSS (EC 2.4.1.242).[6] GBSS, SSI, SSII, SSIII, e SSIV sono dotate di attività catalitica, mentre la SSV ne è priva.[1]
GBSS è quasi esclusivamente legata ai granuli, sembra per la maggior parte posizionata al suo interno, come evidenziato dal trattamento della superficie dei granuli con proteasi. Le altre isoforme dell’amido sintasi sono presenti o solamente nello stroma dei plastidi o suddivise tra stroma e granuli di amido, e sono dette solubili.[8]
Le amido sintasi I, II, III, e IV catalizzano l’aggiunta di una sola unità di glucosio per incontro con l’α-(1→4)-glucano nascente, modalità d’azione definita distributiva, mentre GBSS è in grado di catalizzare l’aggiunta di più unità glucosidiche per incontro con il substrato, modalità d’azione definita processuale.[11]
Amido sintasi e MOS
Le amido sintasi coinvolte nelle prime fasi della sintesi di amilosio e amilopectina necessitano della presenza di corti malto-oligosaccaridi o MOS per iniziare la sintesi de novo dei due polisaccaridi.[11]
Questi piccoli oligosaccaridi, ossia α-(1→4)-glucani lunghi da 2 a 7 residui glucosidici, fungono da primer e sono allungati, funzione analoga a quella svolta dalla glicogenina nella sintesi del glicogeno.
I MOS possono derivare dall’azione dell’amido sintasi III, dell’amido fosforilasi e degli enzimi deramificanti l’amido, in quest’ultimo caso nel corso del rifinitura di molecole di amilopectina.
Essendo scarsamente solubili, i MOS sembrano in grado di evitare l’azione idrolitica delle alfa-amilasi (EC 3.2.1.1) e delle beta-amilasi.[8]
Amido sintasi e sintesi dell’amilopectina
La sintesi dell’amilopectina richiede l’azione temporalmente coordinata di almeno quattro classi di enzimi, ossia gli isoenzimi dell’amido sintasi, l’amido fosforilasi, gli enzimi ramificanti l’amido (EC 2.4.1.18), e gli enzimi deramificanti l’amido.[2][7] Si ritiene inoltre che, in molti casi, questi enzimi interagiscono fisicamente tra loro a dare complessi multienzimatici, strutture in grado di aumentare l’efficienza di una via metabolica.[11]
E’ generalmente accettato che la crescita del granulo di amido avvenga a partire da una zona centrale detta ilo, la cui esatta struttura non è nota, sebbene sembra sia formata da una struttura relativamente disordinata di α-glucani.[13] L’iniziazione dell’ilo, la formazione di un granulo di amido strutturalmente normale, e il grado di accumulo dell’amido richiedono la presenza di SSIV, sebbene sia stato suggerito che anche SSIII e SSV possano svolgere un ruolo, sovrapponendo la loro azione con quella della SSIV.[10]
GBSS e sintesi dell’amilosio
La sintasi legata ai granuli è coinvolta nella sintesi dell’amilosio.
Questo enzima fu scoperto nei primi anni sessanta del secolo scorso dal gruppo di Luis Federico Leloir, lo stesso ricercatore che nel 1948 aveva scoperto la principale via per il metabolismo del galattosio, la via di Leloir.[5]
La sua azione catalitica non è del tutto contemporanea a quella delle altre amido sintasi in quanto necessita della presenza di un’impalcatura di amilopectina per essere indirizzata verso i granuli nascenti.[6]
Nelle Graminacee si conoscono due isoforme dell’enzima, codificate da geni distinti, e indicate come GBSSI e GBSSII.[12]
GBSS richiede, per la sua azione catalitica, la presenza di una proteina della famiglia PTST, la PTST1 che permette il suo legame al granulo di amido, e la cui azione sembra essere più importante nei cloroplasti rispetto agli amiloplasti.[9]
La proteina sembra si associ, nello stoma del plastidio, a GBSS. Il complesso formato si lega al granulo nascente, la proteina si dissocia dall’enzima che inizia a catalizzare l’allungamento dei malto-oligosaccaridi. PTST1 torna quindi nello stroma dove recluta un altro enzima.
Bibliografia
^ Abt M.R., Pfister B., Sharma M., Eicke S., Bürgy L., Neale I., Seung D., Zeeman S.C. STARCH SYNTHASE5, a noncanonical starch synthase-like protein, promotes starch granule initiation in Arabidopsis. Plant Cell 2020;32(8):2543-2565. doi:10.1105/tpc.19.00946
^ ab Crofts N., Abe N., Oitome N.F., Matsushima R., Hayashi M., Tetlow I.J., Emes M.J., Nakamura Y., Fujita N. Amylopectin biosynthetic enzymes from developing rice seed form enzymatically active protein complexes. J Exp Bot 2015;66(15):4469-82. doi:10.1093/jxb/erv212
^ Cuesta-Seijo J.A., Ruzanski C., Krucewicz K., Meier S., Hägglund P., Svensson B., Palcic M.M. Functional and structural characterization of plastidic starch phosphorylase during barley endosperm development. PLoS One 2017;12(4):e0175488. doi:10.1371/journal.pone
^ Gous P.W., Fox G.P. Review: Amylopectin synthesis and hydrolysis – Understanding isoamylase and limit dextrinase and their impact on starch structure on barley (Hordeum vulgare) quality. Trends Food Sci Technol 2017;62:23-32. doi:10.1016/j.tifs.2016.11.013
^ Leloir L.F., de Fekete M.A., Cardini C.E. Starch and oligosaccharide synthesis from uridine diphosphate glucose. J Biol Chem 1961;236:636-41. doi:10.1016/S0021-9258(18)64280-2
^ abc Pfister B., Zeeman S.C. Formation of starch in plant cells. Cell Mol Life Sci 2016;73(14):2781-807. doi: 10.1007/s00018-016-2250-x
^ ab Qu J., Xu S., Zhang Z., Chen G., Zhong Y., Liu L., Zhang R., Xue J., Guo D. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci Rep 2018;8(1):12736. doi:10.1038/s41598-018-30411-y
^ ab Seung D., Boudet J., Monroe J., Schreier T.B., David L.C., Abt M., Lu K.J., Zanella M., Zeeman S.C. Homologs of PROTEIN TARGETING TO STARCH control starch granule initiation in Arabidopsis leaves. Plant Cell 2017;29(7):1657-1677. doi:10.1105/tpc.17.00222
^ Seung D., Soyk S., Coiro M., Maier B.A., Eicke S., and Zeeman S.C. PROTEIN TARGETING TO STARCH is required for localising GRANULE-BOUND STARCH SYNTHASE to starch granules and for normal amylose synthesis in Arabidopsis. PLoS Biol 2015;13:e1002080. doi:10.1371/journal.pbio.1002080
^ Szydlowski N., Ragel P., Raynaud S., Lucas M.M., Roldán I., Montero M., Muñoz F.J., Ovecka M., Bahaji A., Planchot V., Pozueta-Romero J., D’Hulst C., Mérida A. Starch granule initiation in Arabidopsis requires the presence of either class IV or class III starch synthases. Plant Cell 2009;21(8):2443-57. doi:10.1105/tpc.109.066522
^ abc Tetlow I.J., Bertoft E. A review of starch biosynthesis in relation to the building block-backbone model. Int J Mol Sci 2020;21(19):7011. doi:10.3390/ijms21197011
^ Vrinten P.L., Nakamura T. Wheat granule-bound starch synthase I and II are encoded by separate genes that are expressed in different tissues. Plant Physiol 2000;122(1):255-64. doi:10.1104/pp.122.1.255
^ Ziegler G.R., Creek J.A., Runt J. Spherulitic crystallization in starch as a model for starch granule initiation. Biomacromolecules 2005;6(3):1547-54. doi:10.1021/bm049214p
Il concetto di dieta mediterranea è stato introdotto negli anni ’50 dello scorso secolo dal fisiologo americano Ancel Benjamin Keys.
Keys per primo identificò la relazione tra dieta e insorgenza delle malattie cardiovascolari o CVD, in particolare grazie allo studio epidemiologico noto come Seven Countries Study, il primo studio a evidenziare questo modello alimentare.
La dieta mediterranea, che è riconosciuta come uno dei modelli alimentari più sani, è ricca di alimenti vegetali minimamente lavorati, quali verdure, legumi, cereali, meglio se integrali, con l’olio di oliva extravergine come principale fonte di lipidi. Quindi è un modello nutrizionale ricco in sostanze antiossidanti e con azione antiinfiammatoria.
Dopo lo Studio di Keys, molti altri gruppi di ricerca hanno evidenziato, in popolazioni appartenenti a paesi industrializzati e non, il ruolo protettivo di questo tipo di alimentazione non solo nei confronti della CVD, ma anche di malattie cronico-degenerative, dei disturbi depressivi, nonché una correlazione con miglioramenti sulla capacità di apprendimento. Ed è stato dimostrato che una maggiore aderenza alla dieta mediterranea è associata al miglioramento dello stato di salute e alla riduzione della mortalità in generale
Per tutto questo, non esiste associazione scientifica che sostenga la tesi secondo cui sia dannosa per la nostra salute.
In aggiunta, grazie al suo ridotto consumo di carni, è in grado di migliorare la salute pubblica anche concorrendo alla riduzione delle emissione dei gas serra.
In definitiva, la dieta mediterranea rappresenta un tipo di alimentazione che deve essere salvaguardata e promossa, in contrapposizione al trend mondiale verso una uniformità dietetica che sta avanzando anche nei paesi del bacino mediterraneo.
Keys individuò la correlazione tra dieta e il rischio di sviluppare malattie cardiovascolari nei primi anni ’50 del secolo scorso confrontando i tassi di insorgenza di CVD tra dirigenti aziendali americani e popolazioni europee appena uscite dalla seconda guerra mondiali. Mentre tra i primi, soggetti ben nutriti, i tassi di insorgenza erano elevati, nei secondi, che invece si trovavano in una fase di indigenza alimentare, i tassi di insorgenza di CVD erano bassi. Queste osservazioni portarono Keys a ipotizzare una correlazione tra il contenuto di grassi nella dieta e i decessi per malattie cardiovascolari.
La successiva osservazione di una frequenza estremamente bassa di malattie coronariche e certi tipi di tumore nella popolazione dell’isola di Creta, in molta della restante popolazione greca e in quella del sud dell’Italia rispetto a quanto osservato negli USA, portò Keys a ipotizzare che l’alimentazione di quelle popolazioni, caratterizzata da un basso contenuto di grassi di origine animale, costituisse un fattore di protezione.
Il passo successivo fu l’avvio di quello che sarà conosciuto come il Seven Countries Study, uno studio epidemiologico osservazionale a lungo termine, ben 25 anni, e lo studio più conosciuto sulla dieta mediterranea, nel quale fu osservata una correlazione inversa tra la dieta e il rischio di morte generale e legata a malattie cardiovascolari. Lo Studio mostrò che i grassi saturi sono il principale fattore di rischio dietetico e che seguire un’alimentazione di tipo mediterraneo portava a un riduzione del rischio di insorgenza di malattie cardiovascolari.
Caratteristiche della dieta mediterranea
La dieta mediterranea è un modello nutrizionale caratterizzato dal consumo prevalente di cibi di origine vegetale quali legumi, verdure, cereali integrali e olio extravergine d’oliva, il che assicura un buon apporto di fibre, antiossidanti, fitosteroli, polifenoli e acidi grassi insaturi.
Per quanto riguarda i prodotti di origine animale, il consumo di carne, in particolare carne rossa e prodotti derivati, nonché di prodotti caseari ad alto contenuto di grassi dovrebbe essere limitato, mentre dovrebbero essere presenti pesci e frutti di mare.
Il consumo di etanolo dovrebbe essere moderato, da assumersi con il vino rosso durante i pasti.
E tra i greci che hanno partecipato allo studio EPIC, l’olio extravergine di oliva, le verdure, i legumi, un modesto apporto di etanolo, assieme a un basso consumo di carne e derivati sono risultati predittori di una minore mortalità.
Pietra angolare della dieta mediterranea è l’olio extravergine di oliva. Da utilizzarsi preferibilmente crudo, è un’ottima fonte di acidi grassi monoinsaturi e contiene oltre 2000 diverse molecole, molte con attività antiossidante.
Rappresentando la principale fonte di lipidi, quando associato a un basso consumo di prodotti di origine animale ad alto contenuto di grassi, assicura un elevato rapporto tra acidi grassi monoinsaturi e acidi grassi saturi, il che concorre al miglioramento del profilo lipidico e del controllo glicemico nei diabetici.
E’ tuttavia fuorviante focalizzare l’attenzione su un singolo componente della dieta mediterranea; non esiste il “proiettile magico”, come mostrato da studi che si sono concentrati su un unico nutriente. Le persone non mangiano un singolo e isolato nutriente, ma un complesso di nutrienti e, cosa ancora più importante, i nutrienti interagiscono tra di loro in modo sinergistico o antagonistico. Quindi, i benefici per la salute apportati dalla dieta mediterranea sono dovuti a tutti i suoi componenti.
Dieta mediterranea e malattie croniche
Dopo il Seven Countries Study numerosissimi studi hanno dimostrato l’efficacia di questo modello nutrizionale nella prevenzione sia primaria che secondaria delle principali malattie croniche, dalle malattie cardiovascolari ai disturbi depressivi, nonché una riduzione della mortalità generale.
Di seguito alcuni esempi tra i moltissimi disponibili tra le pubblicazioni scientifiche.
Una meta-analisi ha analizzato l’associazione tra l’aderenza alla dieta mediterranea, mortalità, e incidenza di malattie dimostrando che “la maggiore aderenza alla dieta mediterranea è associata in modo significativo con una riduzione della mortalità generale, mortalità cardiovascolare, incidenza e mortalità per cancro, e incidenza del morbo di Parkinson.”
Uno studio multicentrico randomizzato ha dimostrato l’efficacia nella prevenzione primaria degli eventi cardiovascolari in soggetti ad alto rischio cardiovascolare.
E’ correlata sia a un minor rischio di morbo di Alzheimer che con il suo decorso e i suoi esiti: la maggior aderenza è associata con una minore mortalità e si suggerisce un effetto dose-risposta.
Ci sono prove crescenti che indicano un effetto protettivo sull’aumento di peso.
E’ stata riporta un’associazione inversa tra l’aderenza a queste abitudini dietetiche e l’incidenza del diabete di tipo 2 tra soggetti inizialmente in salute e in pazienti sopravvissuti a un infarto miocardico.
E’ associata a una minor prevalenza della sindrome metabolica.
Studi epidemiologici e di intervento hanno rivelato un effetto protettivo nei confronti dell’infiammazione cronica lieve e delle sue complicanze metaboliche.
Ci sono prove che l’aderenza al modello della dieta mediterranea possa avere un potenziale ruolo protettivo per quanto riguarda la prevenzione dei disturbi depressivi.
Ruolo nella riduzione dei gas serra
La dieta mediterranea è in grado di migliorare la salute pubblica anche concorrendo alla riduzione dell’emissione dei gas serra, ossia anidride carbonica o CO2, metano, ossido di azoto e simili, provenienti dal settore zootecnico, responsabile dei 4/5 delle emissioni legate all’agricoltura. Queste emissioni sono maggiori di quelle dovute ai trasporti, e seconde solo a quelle derivanti dalla produzione di energia. Se a questo si aggiunge che la popolazione mondiale sta crescendo, e tale crescita è accompagnata da un aumento del consumo pro capite di carne, con stime secondo cui entro il 2030 si avrà un incremento della produzione di carne dell’85% rispetto al 2000, il ruolo della dieta mediterranea nei confronti della riduzione dell’emissione dei gas serra diviene ancora più evidente.
Allevamento bovino e gas serra
Analizzando nel dettaglio i gas serra derivanti dall’allevamento bovino, il maggiore responsabile dell’emissione nel comparto zootecnico, questi derivano come di seguito indicato:
40% dalla perdita di piante annuali, erbe e alberi che ricoprirebbero il terreno dove si coltiva il foraggio;
32% dalle emissioni di metano da parte dei rifiuti animali e dagli animali stessi;
14% dai fertilizzanti per la coltivazione dei cereali da foraggio, di cui ne sono necessari 16 chili per chilo di carne consumata;
14% dalla produzione agricola generale.
Nella tabella sono confrontate le emissioni di CO2 dovute alla produzione di diversi cibi, considerando porzioni da 225 g, con quelle rilasciate da una macchina a benzina che fa circa 12 chilometri con un litro.
Alimenti
Distanza
Grammi di CO2 equivalente
Patate
300 metri
59
Mele
320 metri
68
Asparagi
440 metri
91
Pollo
1,17 chilometri
249
Maiale
4,1 chilometri
862
Manzo
15,8 chilometri
3360
Quindi produrre 225 grammi di carne di manzo porta al rilascio di una quantità di gas serra quasi 13 volte maggiore di quella rilasciata a seguito della produzione di una eguale quantità di pollo e addirittura 57 volte maggiore se consideriamo le patate.
Per fare un altro esempio produrre i 41 chili di carne bovina consumata annualmente dallo statunitense medio genera la stessa quantità di CO2 di un’automobile che percorra 3000 chilometri.
Bibliografia
Di Daniele N., Noce A., Vidiri M., Moriconi E., Marrone G., Annicchiarico-Petruzzelli M., D’Urso G., Tesauro M., Rovella V., De Lorenzo A. Impact of Mediterranean diet on metabolic syndrome, cancer and longevity. Oncotarget 2017;8:8947-8979. doi:10.18632/oncotarget
Estruch R., Ros E., Salas-Salvadó J., et al. Primary prevention of cardiovascular disease with a Mediterranean Diet. N Engl J Med 2013;368:1279-1290. doi:10.1056/NEJMoa1200303
Friel S., Dangour A.D., Garnett T., Lock K., Chalabi Z., Roberts I., Butler A., Butler C.D., Waage J., McMichael A.J. and Haines A. Public health benefits of strategies to reduce greenhouse-gas emissions: food and agriculture. Lancet 2009;374:2016-2025. doi:10.1016/S0140-6736(09)61753-0
Giugliano D. and Esposito K. Mediterranean Diet and Cardiovascular Health. Annals NY Acad Sci 2005;1056(1):253-260. doi:10.1196/annals.1352.012
Giugliano D. and Esposito K. Mediterranean diet and metabolic diseases. Curr Opin Lipidol 2008;19:63-68. doi:10.1097/MOL.0b013e3282f2fa4d
Keys A. Mediterranean diet and public health: personal reflections. Am J Clin Nutr 1995;61:1321S-1323S doi:10.1093/ajcn/61.6.1321S
Keys A., Aravanis C., Blackburn H., Buzina R., Djordjevic B.S., Dontas A.S., Fidanza F., Karvonen M.J., Kimura N., Menotti A., Mohacek I., Nedeljkovic S., Puddu V., Punsar S., Taylor H.L., Van Buchem F.S.P. Seven Countries: A Multivariate Analysis of Death and Coronary Heart Disease. Harvard University Press, Cambridge, Harvard University Press, ISBN: 0-674-80237-3, 1980. 381 pp.
Martínez-González M.Á., de la Fuente-Arrillaga C., Nunez-Cordoba J.M., Basterra-Gortari F.J., Beunza J.J., Vazquez Z., Benito S., Tortosa A., Bes-Rastrollo M. Adherence to Mediterranean diet and risk of developing diabetes: prospective cohort study. BMJ 2008;336:1348-1351. doi:10.11bmj.39561.501007.BE
Martín-Peláez S., Fito M., Castaner O. Mediterranean diet effects on type 2 diabetes prevention, disease progression, and related mechanisms. A review. Nutrients 2020;12(8):2236. doi:10.3390/nu12082236
Mentella M.C., Scaldaferri F., Ricci C., Gasbarrini A., Miggiano G.A.D. Cancer and Mediterranean Diet: a review. Nutrients 2019; 11(9):2059. doi:10.3390/nu11092059
Nestle M. Mediterranean diets: historical and research overview. Am J Clin Nutr 1995;61:1313S-1320S doi:10.1093/ajcn/61.6.1313S
Samieri C., Okereke O.I., E. Devore E.E. and Grodstein F. Long-term adherence to the Mediterranean Diet is associated with overall cognitive status, but not cognitive decline, in women. J Nutr 2013;143:493-499. doi:10.3945/jn.112.169896
Sánchez-Villegas A., Delgado-Rodríguez M., Alonso A., Schlatter J., Lahortiga F., Serra Majem L., Martínez-González M.A. Association of the Mediterranean Dietary pattern with the incidence of depression: The Seguimiento Universidad de Navarra/University of Navarra Follow-up (SUN) Cohort. Arch Gen Psychiatry. 2009;66:1090-1098 doi:10.1001/archgenpsychiatry.2009.129
Scarmeas N., Luchsinger J.A., Mayeux R. and Stern Y. Mediterranean diet and Alzheimer disease mortality. Neurology 2007;69(11):1084-1093 doi:10.1212/01.wnl.0000277320.50685.7c
Schröder H. Protective mechanisms of the Mediterranean diet in obesity and type 2 diabetes. J Nutr Biochem 2007;18:149-160. doi:10.1016/j.jnutbio.2006.05.006
Sofi F., Cesari F., Abbate R., Gensini G.F. and Casini A. Adherence to Mediterranean diet and health status: meta-analysis. BMJ 2008;337:a1344. doi:10.1136/bmj.a1344
Subak S. Global environmental costs of beef production. Ecol. Econom. 1999;30:79-91. doi:10.1016/S0921-8009(98)00100-1
Trichopoulou A., Bamia C. and Trichopoulos D. Anatomy of health effects of Mediterranean diet: Greek EPIC prospective cohort study. BMJ 2009;338:b2337. doi:10.1136/bmj.b2337
Trichopoulou A., Costacou T., Bamia C., Trichopoulos D. Adherence to a Mediterranean Diet and Survival in a Greek Population. N Engl J Med 2003;348:2599-2608. doi:10.1056/NEJMoa025039
L’amilosio è un polisaccaride formato da unità di α-D-glucosio legate da legami glicosidici α-(1→4), con poche ramificazioni connesse alla catena principale da legami glicosidici α-(1→6).[17]
Insieme all’amilopectina è uno dei due principali costituenti dell’amido, la più diffusa e abbondante forma di accumulo di energia e carboidrati presente nella Biosfera.[12]
La sua sintesi è catalizzata dall’enzima amido sintasi legata ai granuli o GBSS (EC 2.4.1.242), e necessita della presenza di una seconda proteina, la PTST1, che è priva di attività catalitica.[15]
Nel granulo di amido l’amilosio viene depositato all’interno della matrice semicristallina formata dall’amilopectina.[13] Sebbene a differenza dell’amilopectina l’amilosio non sia necessario per la formazione dei granuli semicristallini di amido, dove è presente in quantità inferiore rispetto all’amilopectina, l’amilosio ha una grande influenza sulle proprietà fisico-chimiche dell’amido.[7]
Sono state selezionate piante dove il granulo di amido può contenere quantità molto piccole di amilosio, e altre dove invece i granuli sono formati per la quasi totalità dal polisaccaride. Questi fenotipi possono avere vantaggi sia dal punto di vista della salute che industriale.[4][20]
Le molecole di amilosio hanno un peso molecolare di circa 106 daltons, sono per la maggior parte lineari e formate da residui di α-D-glucosio, di seguito indicato come glucosio, legati a mezzo di legami glicosidici α-(1→4), ossia legami in cui ciascuna unità glucosidica è legata alla successiva a mezzo di un legame covalente tra il C-1 di una unità e il gruppo ossidrilico sul C-4 dell’unità successiva.[1] Le catene lineari sono costituite da un numero di monosaccaridi variabile da poche centinaia a diverse migliaia, risultando quindi molto più lunghe di quelle dell’amilopectina.[17]
Le poche ramificazioni sono connesse alla catena lineare da legami glicosidici α-(1→6), al pari di quanto accade con l’amilopectina e il glicogeno, la forma di accumulo dei carboidrati presente negli animali. Nel caso dei legami glicosidici α-(1→6), le due unità di glucosio sono unite da un legame covalente tra il C-1 di un’unità e il gruppo ossidrilico sul C-6 dell’altra unità. Il numero di ramificazioni è compreso tra 5 e 20, a seconda dell’origine botanica dell’amido, ramificazioni che, rispetto all’amilopectina, non sono raggruppate.[6] Studi sulla lunghezza delle ramificazioni dell’amilosio hanno evidenziato una loro distribuzione bimodale, con le due frazioni indicate come:
AM1, che raggruppa le catene più corte, con un grado di polimerizzazione compreso tra i 100 e i 700 daltons;
AM2, dove si ritrovano le catene più lunghe, con un grado di polimerizzazione compreso tra i 700 e i 40.000 daltons.[19]
Analoga distribuzione bimodale della lunghezza delle ramificazioni si osserva nell’amilopectina, le cui frazioni sono indicate come AP1, più corte e abbondanti, e AP2.
La variazione intraspecie della distribuzione delle frazioni AM1 e AM2 è relativamente piccola, mentre è grande tra specie differenti, variazioni che hanno una base genetica.[19]
Localizzazione dell’amilosio nel granulo di amido
L’esatta localizzazione dell’amilosio all’interno del granulo di amido non è nota, sebbene si ritenga che la maggior parte si trovi nelle regioni amorfe. Tuttavia alcuni studi hanno suggerito che la sua localizzazione non sia limitata alle regioni amorfe, ma sia anche presente tra le catene di amilopectina e sulla superficie dei granuli.[13] Ne consegue che l’amilosio potrebbe avere diverse localizzazione all’interno del granulo di amido.
Sintesi
Nelle piante la sintesi dell’amilosio è catalizzata dalla GBSS, una selle sei isoforme dell’amido sintasi.[11] L’enzima, la cui azione è la principale determinante del contenuto in amilosio del granulo di amido, necessita della presenza della proteina PTST1.[15] L’inserzione delle ramificazioni sembra essere catalizzata dall’enzima ramificante l’amido o SBE (EC 2.4.1.18).
Come nel caso della sintesi dell’amilopectina, si ritiene che gli enzimi coinvolti interagiscano fisicamente a formare complessi multienzimatici, strutture in grado di ottimizzare l’efficienza dell’intero processo.[17]
Poiché la sintesi dell’amilosio necessita della presenza di un’impalcatura di amilopectina per poter indirizzare l’amido sintasi legata ai granuli verso i granuli di amido, la sintesi dei due polisaccaridi non è del tutto contemporanea.[11]
Nelle fasi iniziali, l’amido sintasi legata ai granuli, al pari delle altre amido sintasi, necessita della presenza di corti malto-oligosaccaridi o MOS, α-(1→4)-glucani lunghi da 2 a 7 residui glucosidici, che fungono da primer e sono allungati.[17] I MOS possono originare da diverse fonti, tra cui il processo di rifinitura di molecole di amilopectina nascenti per opera dell’enzima deramificante, o dall’attività dell’amido fosforilasi (EC 2.4.1.1), un altro enzima coinvolto nel metabolismo dell’amido.[16] Essendo scarsamente solubili in acqua, i MOS sembrano in grado di eludere l’azione idrolitica delle alfa-amilasi (EC 3.2.1.1) e delle beta-amilasi (EC 3.2.1.2), e diffondono all’interno della matrice dei granuli di amido in formazione dove sono allungati per azione della GBSS.[14] Una volta superati i sette residui glucosidici non riescono a diffondere facilmente al di fuori del granulo e sono ulteriormente allungati.
La fase iniziale della sintesi dell’amilosio e dell’amilopectina, richiedendo la presenza di un primer, somiglia a quella del glicogeno. Tuttavia, il primer richiesto nel corso della sintesi del glicogeno non è un oligosaccaride ma una proteina auto-glicosilante, la glicogenina.
Da notare che la polimerizzazione del glucosio in amilopectina, amilosio, o glicogeno, e più in generale di monosaccaridi osmoticamente attivi in polisaccaridi osmoticamente inerti, permette il deposito di grandi quantità di molecole osmoticamente attive all’interno della cellula senza che si verifichi un aumento della pressione osmotica.
Amido sintasi legata ai granuli
GBSS e le altre isoforme dell’amido sintasi appartengono alla famiglia delle glicosiltransferasi (EC 2.4), al pari dell’amido fosforilasi.[11]
L’enzima, scoperto dal gruppo di Louis Federico Leloir, che in precedenza aveva scoperto la principale via per il metabolismo del galattosio, la via di Leloir, è la più abbondante tra le proteine associate ai granuli di amido.[9] E’ presente quasi esclusivamente legata ai granuli, a differenza delle altre isoforme della amido sintasi che sono per la maggior parte presenti nello stroma del plastidio, o suddivise tra stroma e granuli. Inoltre, il trattamento della superficie dei granuli con proteasi ha evidenziato che la maggior parte di GBSS è presente all’interno del granulo piuttosto che sulla sua superficie, posizionamento coerente con la sintesi dell’amilosio all’interno dei granuli nascenti.[15]
Nelle graminacee la sintasi è presente in due isoforme, codificate da geni distinti, indicate come GBSSI e GBSSII, presenti rispettivamente negli amiloplasti dei tessuti di deposito, quindi non fotosintetici, e nei cloroplasti, quindi nei tessuti fotosintetici dove interviene nella sintesi dell’amido transiente.[18]
GBSS catalizza il trasferimento di una molecola di glucosio dall’ADP-glucosio all’estremità non riducente di un α-(1→4)-glucano, cui l’unità glucosidica viene legata attraverso un legame glicosidico α-(1→4).[5]
Si noti che, mentre le amido sintasi utilizzano come donatore dell’unità glucosidica l’ADP-glucosio, la glicogeno sintasi, che interviene nella sintesi del glicogeno, utilizza l’UDP-glucosio.[3]
L’amido sintasi legata ai granuli è in grado di aggiungere più di un’unità di glucosio per incontro con il substrato, modalità d’azione definita processiva, a differenza delle altre isoforme di amido sintasi che aggiungono una sola unità di glucosio per incontro con la catena di glucano nascente, azione definita distributiva. L’azione processiva permette la sintesi di lunghe catene lineari, e sembra essere fortemente incrementata dalla presenza di amilopectina.[17]
PTST1
La sintesi dell’amilosio richiede la presenza di una proteina della famiglia PTST, ossia PTST1, scoperta cinquanta anni dopo GBSS.[15]
PTST1 non ha attività catalitica, ma permette il legame di GBSS al granulo di amido, azione che sembra essere più importante nei cloroplasti, e quindi per la sintesi dell’amido transiente, che negli amiloplasti. Secondo un modello di azione di PTST1, la proteina si associa a GBSS nello stroma del plastidio, il complesso si lega al granulo di amido nascente, PTST1 si dissocia dall’enzima che inizia la sintesi dell’amilosio, mentre la proteina torna nello stroma per reclutare un’altra molecola di enzima.
L’importanza di PTST1 è sottolineata dal fatto che è conservata in tutto il regno vegetale e la sua perdita causa il distacco dell’enzima dal granulo di amido nascente.
Enzima ramificante l’amido
Non è noto l’enzima che catalizza l’inserzione delle poche ramificazioni presenti nelle molecole di amilosio, sebbene sembra possa essere coinvolta un’isoforma dell’enzima ramificante l’amido, la SBEI.[13]
Presente per la maggior parte nello stroma dei plastidi, e in piccola parte legato al granulo, è principalmente espressa nei tessuti di deposito. La bassa frequenza con cui sono presenti le ramificazioni nell’amilosio potrebbe essere il risultato della sintesi del polisaccaride all’interno dei granuli, dove la molecola nascente sarebbe protetta dall’azione dell’enzima ramificante l’amido, ivi scarsamente presente.[8]
Rapporto amilosio/amilopectina
Nei granuli di amido delle piante terrestri l’amilosio è quasi sempre presente, ma in percentuali variabili, in genere comprese tra il 5 e il 35%.[5] La variabilità si osserva non solo tra specie differenti, ma anche all’interno di una stessa specie in base all’organo o tessuto considerato, e, nei tuberi e semi, in base alla fase di sviluppo, essendo in genere il suo contenuto basso nelle prime fasi, per poi aumentare fino al raggiungimento del valore finale, una modalità coerente con la sua sintesi all’interno di una matrice di amilopectina.[13]
Esistono tuttavia piante il cui contenuto di amilosio è molto basso, o addirittura assente. I loro amidi sono detti cerosi, per l’aspetto simile a cera dell’endosperma dei chicchi crudi. Al contrario, ci sono piante i cui granuli di amido sono formati per la maggior parte, o in alcuni casi per la quasi totalità, da amilosio.[20]
Sebbene il suo ruolo biologico non sia ancora stato chiarito, la sua quasi costante presenza sembra indicare che questo polisaccaride svolga un ruolo strutturale importante nel granulo di amido, e conferisca alla pianta un qualche vantaggio nella fase di crescita e sviluppo.[13]
Il rapporto amilosio/amilopectina influenza fortemente le proprietà fisico-chimiche dell’amido, quali la sua capacità di assorbire acqua, che a sua volta influenza processi come la gelatinizzazione e la retrogradazione dell’amido, o la resistenza all’idrolisi enzimatica, quest’ultima importante ad esempio nel determinare la velocità con cui il maltosio e maltotriosio sono rilasciati per azione della alfa-amilasi e delle beta-amilasi.[10] Tutto ciò è in grado di influenzare gli utilizzi industriali dell’amido nonché i suoi effetti sulla salute.
Amidi ricchi di amilosio
I cerali ad alto contenuto di amilosio sono ottenuti attraverso l’aumento dell’espressione del gene per GBSSI, e dalla soppressione o eliminazione dei geni codificanti l’enzima ramificante l’amido, SSIIa, o altri enzimi e proteine coinvolti nella sintesi dell’amilopectina. Tuttavia, almeno per l’endosperma dei cereali, la metodica più efficace è risultata essere la soppressione o l’eliminazione di una o più isoforme dell’enzima ramificante l’amido.[4][20]
Gli amidi ad alto contenuto di amilosio hanno proprietà chimico-fisiche peculiari, tra cui una elevata forza gelificante, una maggior facilità di retrogradazione, e un’ottima capacità di formare film, caratteristiche che li rendono idonei ad applicazioni industriali come la produzione di plastica biodegradabile, di carta e di adesivi.[7][10]
Gli amidi ricchi in amilosio sono ricchi di amido resistente, un tipo di amido che resiste alla digestione intestinale ad opera della alfa-amilasi, una delle idrolasi coinvolte nella digestione dei carboidrati. Studi condotti utilizzando cibi arricchiti con amido resistente hanno evidenziato un miglioramento delle risposte insuliniche e glicemiche, nonché una riduzione del rischio di sviluppare malattie cardiovascolari, obesità e diabete mellito di tipo II.[2]
Quale potrebbe essere la loro modalità d’azione?
L’amido resistente, essendo in grado di eludere l’azione dell’alfa-amilasi, potrebbe ridurre l’indice glicemico degli alimenti in cui è presente, contribuendo così a una migliore regolazione della glicemia. Inoltre, una volta raggiunto il colon, l’amido resistente può essere fermentato dai batteri del microbiota intestinale, che è parte del più ampio microbiota umano, con produzione di acidi grassi a catena corta, in particolari l’acido butirrico, l’acido acetico e l’acido propionico, acidi grassi essenziali per la salute intestinale.
^ Birt D.F., Boylston T., Hendrich S., Jane J.L., Hollis J., Li L., McClelland J., Moore S., Phillips G.J., Rowling M., Schalinske K., Scott M.P., Whitley E.M. Resistant starch: promise for improving human health. Adv Nutr 2013;4(6):587-601. doi:10.3945/an.113.004325
^ Crofts N., Abe N., Oitome N.F., Matsushima R., Hayashi M., Tetlow I.J., Emes M.J., Nakamura Y., Fujita N. Amylopectin biosynthetic enzymes from developing rice seed form enzymatically active protein complexes. J Exp Bot 2015;66(15):4469-82. doi:10.1093/jxb/erv212
^ ab Funnell-Harris D.L., Sattler S.E., O’Neill P.M., Eskridge K.M., and Pedersen J.F. Effect of waxy (low amylose) on fungal infection of Sorghum grain. Phytopathology 2015;105(6):716-846. doi:10.1094/PHYTO-09-14-0255-R
^ ab Gous P.W., Fox G.P. Review: amylopectin synthesis and hydrolysis – Understanding isoamylase and limit dextrinase and their impact on starch structure on barley (Hordeum vulgare) quality. Trends Food Sci Technol 2016;62:23-32. doi:10.1016/j.tifs.2016.11.013
^ Hizukuri S., Takeda Y., Yasuda M., Suzuki A. Multi-branched nature of amylose and the action of debranching enzymes. Carbohydr Res 1981;94:205-213. doi:10.1016/S0008-6215(00)80718-1
^ ab Jobling S. Improving starch for food and industrial applications. Curr Opin Plant Biol 2004;7(2):210-8. doi:10.1016/j.pbi.2003.12.001
^ Kram A.M., Oostergetel G.T., Van Bruggen E. Localization of branching enzyme in potato tuber cells with the use of immunoelectron microscopy. Plant Physiol 1993;101(1):237-243. doi:10.1104/pp.101.1.237
^ Leloir L.F., de Fekete M.A., Cardini C.E. Starch and oligosaccharide synthesis from uridine diphosphate glucose. J Biol Chem 1961;236:636-41. doi:10.1016/S0021-9258(18)64280-29
^ ab Magallanes-Cruz P.A., Flores-Silva P.C., Bello-Perez L.A. Starch structure influences its digestibility: a review. J Food Sci 2017;82(9):2016-2023. doi:10.1111/1750-3841.13809
^ abc Pfister B., Zeeman S.C. Formation of starch in plant cells. Cell Mol Life Sci 2016;73(14):2781-807. doi:10.1007/s00018-016-2250-x
^ Qu J., Xu S., Zhang Z., Chen G., Zhong Y., Liu L., Zhang R., Xue J., Guo D. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci Rep 2018;8(1):12736. doi:10.1038/s41598-018-30411-y
^ abcde Seung D. Amylose in starch: towards an understanding of biosynthesis, structure and function. New Phytol 2020;228:1490-1504. doi:10.1111/nph.16858
^ Seung D., Boudet J., Monroe J., Schreier T.B., David L.C., Abt M., Lu K.J., Zanella M., Zeeman S.C. Homologs of PROTEIN TARGETING TO STARCH control starch granule initiation in Arabidopsis leaves. Plant Cell 2017;29(7):1657-1677. doi:10.1105/tpc.17.00222
^ abcd Seung D., Soyk S., Coiro M., Maier B.A., Eicke S., Zeeman S.C. PROTEIN TARGETING TO STARCH is required for localising GRANULE-BOUND STARCH SYNTHASE to starch granules and for normal amylose synthesis in Arabidopsis. PLOS Biol 2015;13(2):e1002080. doi:10.1371/journal.pbio.1002080
^ Szydlowski N., Ragel P., Raynaud S., Lucas M.M., Roldán I., Montero M., Muñoz F.J., Ovecka M., Bahaji A., Planchot V., Pozueta-Romero J., D’Hulst C., Mérida A. Starch granule initiation in Arabidopsis requires the presence of either class IV or class III starch synthases. Plant Cell 2009;21(8):2443-57. doi:10.1105/tpc.109.066522
^ abcde Tetlow I.J., Bertoft E. A review of starch biosynthesis in relation to the building block-backbone model. Int J Mol Sci 2020;21(19):7011. doi:10.3390/ijms21197011
^ Vrinten P.L., Nakamura T. Wheat granule-bound starch synthase I and II are encoded by separate genes that are expressed in different tissues. Plant Physiol 2000;122(1):255-64. doi:10.1104/pp.122.1.255
^ ab Wang K., Hasjim J., Wu A.C., Henry R.J., Gilbert R.G. Variation in amylose fine structure of starches from different botanical sources. J Agric Food Chem 2014;62(19):4443-53. doi:10.1021/jf5011676
^ abc Wang J., Hu P., Chen Z., Liu Q., Wei C. Progress in high-amylose cereal crops through inactivation of starch branching enzymes. Front Plant Sci 2017;8:469. doi:10.3389/fpls.2017.00469
La via di Leloir è la principale via per il metabolismo del galattosio.
Descritta per la prima volta da Leloir L.F. e colleghi nel 1948, porta alla conversione del galattosio in glucosio-1-fosfato, dunque all’inversione della configurazione del gruppo ossidrilico sul C4 del galattosio, carbonio che è uno dei centri di chiralità della molecola.[9]
Gli intermedi metabolici che portano alla isomerizzazione del galattosio a glucosio sono precursori utilizzati in vie metaboliche differenti, quali le reazioni di glicosilazione dei lipidi e proteine o la sintesi del glicogeno, a seconda della fase dello sviluppo, del tipo di tessuto e delle condizioni metaboliche presenti.[6]
Con l’esclusione della prima reazione, le altre reazioni della via di Leloir possono fluire in entrambe le direzioni, a seconda del livello dei substrati e delle richieste metaboliche del tessuto, il che permette l’interconversione di galattosio e glucosio.[4]
L’importanza della via di Leloir, e quindi del galattosio, è sottolineata dal fatto che è estremamente conservata in natura, essendo presente dai batteri fino alle piante e animali, e, nell’uomo, dalla gravità delle conseguenze dovute a mutazioni a carico di dei geni che codificano per gli enzimi che ne catalizzano le reazioni, mutazioni che provocano la malattia metabolica congenita nota come galattosemia.[2][8]
Il galattosio, con il glucosio e fruttosio, è uno dei monosaccaridi che può essere assorbito a livello intestinale. La principale fonte di galattosio è rappresentata dal lattosio che, con maltosio, trealosio e saccarosio, è uno dei disaccaridi presenti negli alimenti.
Essendo assenti trasportatori intestinali per i disaccaridi, nell’ultima tappa della digestione dei carboidrati i loro legami glicosidici sono idrolizzati con liberazione dei monosaccaridi costituenti, che per il lattosio sono glucosio e galattosio. Segue l’assorbimento dei monosaccaridi i quali, tramite il sistema portale, raggiungono il fegato, che è il sito principale per il metabolismo del galattosio e ne assorbe, attraverso diffusione passiva mediata dal trasportatore GLUT2, la maggior parte, circa l’88%.[3] La quantità residua circolante raggiunge altri organi e tessuti, come la ghiandola mammaria che, in fase di allattamento, lo utilizza per la produzione del lattosio e la glicosilazione delle proteine e dei lipidi del latte.[4]
Tappe della via di Leloir
La via di Leloir si compone di quattro reazioni catalizzate da altrettanti enzimi che sono la galattosio mutarotasi o aldoso-1-epimerasi (EC 5.1.3.3), la galattochinasi (EC 2.7.1.6), galattosio-1-fosfato uridiltransferasi (EC 2.7.7.12) e la galattosio 4-epimerasi (EC 5.1.3.2).[8]
La Via di Leloir
Reazione 1: galattosio mutarotasi
L’idrolisi del legame glicosidico β-(1→4) del lattosio porta alla liberazione di glucosio e beta-galattosio. Tuttavia il secondo enzima della via di Leloir, la galattochinasi, è specifica per l’alfa-galattosio. La conversione nell’anomero alfa è catalizzata dalla galattosio mutarotasi. L’enzima è in grado d’interconvertire le configurazioni anomeriche anche del glucosio, dello xilulosio, del maltosio e del lattosio, sebbene con efficienza variabile.[12]
Reazione 2: galattochinasi
Nel secondo passaggio l’alfa-galattosio viene fosforilato a galattosio-1-fosfato nella reazione catalizzata dalla galattochinasi.[8] La fosforilazione del galattosio è importante in quanto:
blocca il monosaccaride all’interno della cellula poiché è caricato negativamente e sono assenti sulla membrana plasmatica trasportatori per gli zuccheri fosforilati;
ne aumenta il contenuto in energia iniziandone la destabilizzazione il che faciliterà il successivo metabolismo;
ne mantiene bassa la concentrazione intracellulare, favorendone l’ulteriore passaggio all’interno della cellula.
La reazione catalizzata dalla galattochinasi è l’unica reazione irreversibile della via di Leloir.[4] Inoltre, a differenza della esochinasi e della glucochinasi (EC 2.7.1.1), che catalizzano la fosforilazione del gruppo ossidrilico sul C6 del glucosio, la galattochinasi e la fruttochinasi (EC 2.7.1.4) catalizzano la fosforilazione dei gruppi ossidrilici legati rispettivamente al C1 di galattosio e fruttosio.[11]
La conversione del galattosio-1-fosfato a glucosio-1-fosfato richiede due reazioni, rispettivamente la terza e la quarta della via di Leloir.
Nel terzo passaggio la galattosio-1-fosfato uridiltransferasi catalizza il trasferimento del gruppo UMD dall’UDP-glucosio al galattosio-1-fosfato, con formazione di UDP-galattosio e glucosio-1-fosfato. La reazione procede con un meccanismo a ping-pong con formazione di un intermedio covalente tra l’enzima e l’UMP.[7]
Reazione 4: UDP-galattosio 4-epimerasi
Nell’ultimo passaggio l’UDP-galattosio viene convertito in UDP-glucosio nella reazione catalizzata dalla UDP-galattosio 4-epimerasi.
L’enzima catalizza l’inversione della configurazione del gruppo ossidrilico sul C4 ed è il punto di interconversione tra UDP-galattosio e UDP-glucosio.[8]
L’enzima richiede come cofattore il NAD+ e la reazione procede attraverso la formazione di un intermedio chetonico sul C4, con contemporanea riduzione del NAD+ a NADH. Successivamente l’intermedio chetonico ruota presentando la faccia opposta dello zucchero al NADH. A questo punto uno ione idruro viene ritrasferito dal NADH al C4, ma in configurazione opposta.[13] Poiché il NAD nel corso della reazione viene prima ridotto e poi ossidato, non subisce alcuna ossidoriduzione netta per cui non compare nell’equazione di reazione. Sembra che la UDP-galattosio 4-epimerasi sia l’enzima limitante la via di Leloir.
L’UDP-glucosio prodotto viene riciclato nella reazione catalizzata dalla UDP-glucosio pirofosforilasi, con rilascio di glucosio-1-fosfato.[6]
Nei mammiferi la galattosio 4-epimerasi catalizza anche la interconversione tra UDP-N-acetilgalattosammina e UDP-N-acetilglucosammina.[3]
Si noti che UDP-galattosio e UDP-glucosio, o più in generale galattosio e glucosio, hanno la stessa formula molecolare. Sono quindi isomeri e differiscono solo per la configurazione del quarto atomo di carbonio. Pertanto sono un esempio di isomeria ottica.
Quali sono le funzioni della via di Leloir?
La via di Leloir permette alla cellula di utilizzare il galattosio o il glucosio derivato in diverse vie metaboliche, sia anaboliche che cataboliche, a seconda delle condizioni metaboliche della cellula o del tessuto. Inoltre, poiché la reazione catalizzata dalla UDP-galattosio 4-epimerasi è reversibile, è possibile la conversione del glucosio a galattosio e derivati nucleotidici.[4][6]
L’UDP-galattosio può essere utilizzato:
nelle reazioni di glicosilazioni di proteine e lipidi, come i galattocerebrosidi che sono i principali glicolipidi della mielina, ed è per questo che il galattosio fu inizialmente chiamato cerebrosio;
nella ghiandola mammaria in allattamento per la produzione del lattosio nella reazione catalizzata dal complesso della lattosio sintasi. Inoltre, essendo la reazione catalizzata dalla UDP-galattosio 4-epimerasi reversibile sarà anche possibile la conversione del glucosio, previa attivazione a UDP-glucosio nella reazione catalizzata dalla UDP-glucosio pirofosforilasi (EC 2.7.7.9), in UDP-galattosio per la sintesi del lattosio.
Nel fegato e nel muscolo scheletrico l’UDP-glucosio derivante dell’UDP-galattosio, può essere utilizzato:[4]
nelle reazioni di glicosilazione dei lipidi e delle proteine;
nella sintesi del glicogeno, quando la richiesta energetica della cellula è bassa; e, rispetto al glucosio e fruttosio, il galattosio è preferenzialmente incorporato nel glicogeno epatico piuttosto che essere indirizzato verso il metabolismo ossidativo;
a seguito della conversione in glucosio-1-fosfato, nella reazione catalizzata dalla UDP-glucosio pirofosforilasi, e isomerizzazione a glucosio-6-fosfato, nella reazione catalizzata dalla fosfoglucomutasi, può entrare in vie metaboliche differenti quali la glicolisi, la via del pentoso fosfato o la gluconeogenesi.[3]
Nota: l’UDP-galattosio è stato il primo zucchero nucleotidico a essere scoperto, scoperta avvenuta proprio nel corso degli studi sulla via di Leloir.[1]
Via di Leloir e galattosemia
Le glicosilazioni sono modificazioni post-traduzionali che hanno un ruolo importante nel rendere possibili e regolare differenti processi biologici. Difetti a loro carico sono stati correlati a molte condizioni patologiche come cancro, diabete ed errori congeniti del metabolismo quali i disturbi congeniti della glicosilazione, principalmente disordini monogenici autosomici recessivi.[5] Tra questi ultimi si annovera la galattosemia, descritta per la prima volta nel 1908 da von Reuss A.[14] La galattosemia è conseguente a mutazioni a carico di uno dei geni che codificano per i quattro enzimi della via di Leloir, e ne sono stati individuati quattro tipi:
tipo I, da carenza di galattosio-1-fosfato uridiltransferasi, la forma più comune, anche detta galattosemia classica;
A oggi, lo standard terapeutico per la cura della galattosemia è una dieta a ridotto contenuto di galattosio.
Galattosemia e cataratta
L’accumulo di galattosio ha quale effetto quello di attivare vie metaboliche alternative quali la sintesi del galattitolo e del galattonato.
Tra i sintomi della galattosemia si ritrova la comparsa precoce di cataratta, in genere entro i primi due anni di vita, e nei casi più severi danni al cervello, reni e fegato.[12]
Sembra che uno dei fattori che determinano l’insorgenza della cataratta sia la riduzione del galattosio, accumulatosi nella lente dell’occhio, a galattitolo, nella reazione catalizzata dalla aldoso reduttasi (EC 1.1.1.21).[10] Il galattitolo, un poliolo scarsamente metabolizzato, non diffonde attraverso la membrana plasmatica a causa della sua scarsa lipofilicità, ed essendo un composto osmoticamente molto attivo determina un aumento della pressione osmotica intracellulare, con conseguente richiamo d’acqua nella lente. Inoltre la sua sintesi, riducendo i livelli intracellulari di NADPH, può determinare una riduzione dell’attività della glutatione reduttasi (EC 1.8.1.7) causando un accumulo di radicali liberi.[3] L’effetto osmotico e l’accumulo di radicali liberi possono danneggiare l’integrità della cellula e causarne la morte. Inoltre, è stato riportato che il galattitolo è un inibitore della galattosio mutarotasi, per cui il suo accumulo potrebbe causare un ulteriore accumulo di galattosio non metabolizzato.[12]
Bibliografia
^ Cardini C.E., Paladini A.C., Caputto R., Leloir L.F. Uridine diphosphate glucose: the coenzyme of the galactose-glucose phosphate isomerization. Nature 1950;165:191-192. doi:10.1038/165191a0
^ Coelho A.I., Berry G.T., Rubio-Gozalbo M.E. Galactose metabolism and health. Curr Opin Clin Nutr Metab Care 2015;18(4):422-427. doi:10.1097/MCO.0000000000000189
^ abcd Coelho A.I., Rubio-Gozalbo M.E., Vicente J.B., Rivera I. Sweet and sour: an update on classic galactosemia. J Inherit Metab Dis 2017;40(3):325-342. doi:10.1007/s10545-017-0029-3
^ abcde Conte F., van Buuringen N., Voermans N.C., Lefeber D.J. Galactose in human metabolism, glycosylation and congenital metabolic diseases: time for a closer look. Biochim Biophys Acta Gen Subj 2021;1865(8):129898. doi:10.1016/j.bbagen.2021.129898
^ abc Frey P.A. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J 1996;10(4):461-70. doi:10.1096/fasebj.10.4.8647345
^ abcd Holden H.M., Rayment I., Thoden J.B. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem 2003;278(45):43885-43888. doi:10.1074/jbc.R300025200
^ Leloir L.F., de Fekete M.A., Cardini C.E. Starch and oligosaccharide synthesis from uridine diphosphate glucose. J Biol Chem 1961;236:636-41. doi:10.1016/S0021-9258(18)64280-2
^ Pintor J. Sugars, the crystalline lens and the development of cataracts. Biochem Pharmacol 2012;1:4. doi:10.4172/2167-0501.1000e1190
^ Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009
^ abc Thoden J.B., Timson D.J., Reece R.J., and M. Holden H.M. Molecular structure of human galactose mutarotase. J Biol Chem 2004;279(22):23431-23437. doi:10.1074/jbc.M402347200
^ Thoden J.B., Wohlers T.M., Fridovich-Keil J.L., Holden H.M. Human UDP-galactose 4-epimerase. Accommodation of UDP-N-acetylglucosamine within the active site. J Biol Chem 2001;276(18):15131-6. doi:10.1074/jbc.M100220200
^ Wada Y., Kikuchi A., Arai-Ichinoi N., Sakamoto O., Takezawa Y., Iwasawa S., Niihori T., Nyuzuki H., Nakajima Y., Ogawa E., Ishige M., Hirai H., Sasai H., Fujiki R., Shirota M., Funayama R., Yamamoto M., Ito T., Ohara O., Nakayama K., Aoki Y., Koshiba S., Fukao T., Kure S. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet Med 2019;21(6):1286-1294. doi:10.1038/s41436-018-0340-x
Nel 1906, il chimico russo-americano Martin André Rosanoff, allora alla New York University, scelse la gliceraldeide, un monosaccaride, come standard per descrivere la stereochimica dei carboidrati e di altre molecole con almeno un centro di chiralità, un sistema di nomenclatura noto come convenzione di Fischer-Rosanoff, o convenzione di Rosanoff, o sistema D-L.[7]
Non conoscendo la configurazione assoluta della gliceraldeide, Rosanoff assegnò in modo del tutto arbitrario:
la lettera D, dal latino dexter che significa “destra”, alla (+)-gliceraldeide, l’enantiomero destrorotatorio, ipotizzando che la configurazione, nelle proiezioni di Fischer, fosse quella con il gruppo idrossilico (–OH) legato al centro chirale disposto sul lato destro della molecola;
la lettera L, dal latino laevus che significa “sinistra”, alla (-)-gliceraldeide, l’enantiomero levorotatorio, ipotizzando che la configurazione, nelle proiezioni di Fischer, fosse quella con il gruppo idrossilico legato al centro chirale disposto sul lato sinistro della molecola.[4]
E, sebbene Fischer lo respinse, questo sistema di nomenclatura fu universalmente accettato e utilizzato per derivare le configurazioni relative delle molecole chirali.[3] In che modo? La configurazione attorno a un centro chirale viene messa in relazione con quella della gliceraldeide convertendo i gruppi della molecola in esame in quelli del monosaccaride attraverso reazioni che avvengano con ritenzione di configurazione, ossia reazioni che non comportano la rottura di nessuno dei legami del centro chirale. Grazie a ciò la disposizione spaziale dei gruppi attorno al centro chirale nel prodotto è la stessa che si ha nel reagente. La convenzione di Fischer-Rosanoff permette quindi di suddividere le molecole chirali, come gli amminoacidi e i monosaccaridi, in due gruppi, la serie D e la serie L, a seconda che la configurazione attorno al centro chirale sia correlata alla D-gliceraldeide o alla L-gliceraldeide.
Nota: non c’è correlazione tra ritenzione di configurazione e segno del potere rotatorio: il sistema D-L non specifica il segno della rotazione del piano della luce polarizzata provocato dalla molecola chirale, ma semplicemente correla la configurazione della molecola a quella della gliceraldeide.[6]
I monosaccaridi possono essere aldosi o chetosi. Gli aldosi, e i chetosi con più di tre atomi di carbonio hanno almeno un centro chirale, e, per convenzione, apparterranno alla serie D o alla serie L se la configurazione del carbonio chirale più lontano dal carbonio carbonilico, il carbonio più ossidato presente nella molecola, è rispettivamente la stessa della D-gliceraldeide o della L-gliceraldeide.
Nelle proiezioni di Fischer la catena di atomi di carbonio più lunga è orientata verticalmente, e gli atomi di carbonio sono numerati in modo che il carbonio carbonilico abbia il numero più basso possibile, quindi C-1 negli aldosi e C-2 nei chetosi.[8]
Nota: in Natura, i carboidrati della serie D sono molto più abbondanti di quelli della serie L.
Se nel nome della molecola deve essere specificato anche il segno della rotazione del piano della luce polarizzata, i prefissi (+) e (-) possono essere utilizzati assieme ai prefissi D e L. Ad esempio, il fruttosio, che è levorotatorio, verrà indicato come D-(-)-fruttosio, mentre il glucosio, che è destrorotatorio, sarà indicato come D-(+)-glucosio.
Convenzione di Fischer-Rosanoff e alfa-amminoacidi
Gli amminoacidi, sulla base della posizione del gruppo amminico (–NH2) rispetto a quella del gruppo carbossilico (–COOH) sono classificati come:
α-amminoacidi, se il gruppo amminico è legato al carbonio α;
β-amminoacidi, se il gruppo amminico è legato al carbonio β;
γ-amminoacidi, se il gruppo amminico è legato al carbonio γ;
δ-amminoacidi, se il gruppo amminico è legato al carbonio δ.
Considerando gli α-amminoacidi, questi apparterranno rispettivamente alla serie D o alla serie L se la configurazione dei gruppi –NH2, –COOH, e –R, e dell’atomo di idrogeno legati al carbonio α, il centro chirale, è la stessa di quella dei gruppi idrossile, aldeidico (–CHO), e idrossimetilico (–CH2OH) e dell’atomo di idrogeno rispettivamente della D-gliceraldeide o della L-gliceraldeide.[6][8]
Nelle proiezioni di Fischer le molecole sono disposte in modo che il gruppo carbossilico, ossia il carbonio più ossidato, sia in alto, e il gruppo R in basso.
Tra gli α-amminoacidi, gli amminoacidi proteinogenici, con l’eccezione della glicina il cui carbonio α non è chirale, hanno configurazione L, e si parlerà dunque di L-α-amminoacidi.
Nota: in Natura gli L-α-amminoacidi sono molto più abbondanti rispetto a tutti gli altri tipi di amminoacidi, i quali non partecipano alla sintesi delle proteine.
Configurazioni relative e assolute
Quando Rosanoff assegnò arbitrariamente il prefisso D alla (+)-gliceraldeide e il prefisso L alla (-)-gliceraldeide, aveva il 50% di probabilità di avere ragione.[5]
Nei primi anni ’50 del secolo scorso divenne disponibile la tecnologia per stabilire la configurazione assoluta delle molecole chirali, la cristallografia a raggi X, e nel 1951 un chimico olandese, Johannes Martin Bijvoet, stabilì la configurazione assoluta del (+)-tartrato di rubidio e sodio tetraidrato, e, confrontandola con quella della gliceraldeide dimostrò che la supposizione di Rosanoff era corretta.[1] Di conseguenza, le configurazioni dei composti chirali ottenute mettendole in relazione con quelle della gliceraldeide erano configurazioni assolute, ossia le configurazioni relative divennero configurazioni assolute.
Limiti della convenzione di Fischer-Rosanoff
La convenzione di Fischer-Rosanoff da luogo a incertezze quando si ha a che fare con molecole con più di un centro chirale.[3] Se ad esempio si considera il D-(+)-glucosio, il sistema D-L ci da informazioni circa la configurazione del solo carbonio 2, quando nella molecola sono presenti altri tre centri asimmetrici, i carboni 3, 4 e 5. In questi casi il sistema RS, sviluppato nel 1956 da Robert Sidney Cahn, Christopher Ingold, e Vladimir Prelog, analizzando ogni singolo centro chirale permette di descrivere senza incertezze la stereochimica della molecola.[2][8] Nel caso del D-(+)-glucosio si ha la configurazione (2R,3S,4R,5R).
Va anche notato che, utilizzando la convezione di Fischer-Rosanoff, in base al centro chirale scelto come centro di riferimento, una stessa molecola può appartenere sia alla serie D che a quella L.
Bibliografia
^ Bijvoet J.M., Peerdeman A.F., Van Bommel A.J. Determination of the absolute configuration of optically active compounds by means of X-rays. Nature 1951;168(4268):271. doi:10.1038/168271a0
^ Cahn R.S., Ingold C., Prelog V. Specification of molecular chirality. Angew Chem 1966:5(4); 385-415. doi:10.1002/anie.196603851
^ IUPAC. Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8. doi:10.1351/goldbook
^ Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
^ ab Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ Rosanoff. On Fischer’s classification of stereo-isomers. J Am Chem Soc 1906:28(1);114-121. doi:10.1021/ja01967a014
^ abc Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011
Nel 1891 il chimico tedesco Hermann Emil Fischer, premio Nobel per la chimica nel 1902, sviluppò un metodo sistematico per la rappresentazione bidimensionale delle molecole con centri di chiralità, le cosiddette proiezioni di Fischer o formule proiettive di Fischer.
Le proiezioni di Fischer, pur essendo strutture bidimensionali, preservano l’informazione circa la stereochimica della molecola, e, sebbene non siano una rappresentazione di come le molecole potrebbero apparire in soluzione, sono largamente utilizzate dai biochimici per definire la stereochimica degli amminoacidi, dei carboidrati, degli acidi nucleici, dei terpeni, degli steroidi e di molte altre molecole di interesse biologico.
Considerando una molecola con un solo centro chirale, supponiamo un atomo di carbonio, per la costruzione della sua proiezione di Fischer la struttura tetraedrica viene ruotata in modo che due legami siano orientati verso il basso mentre gli altri legami siano orientati verso l’alto. Si traccia quindi una croce, al centro della quale si fa cadere il centro chirale, e si dispone la molecola in modo che i gruppi orientati verso il basso, ossia al di sotto del piano del foglio, siano legati alle estremità della linea verticale, mentre i gruppi orientati verso l’alto, dunque al di sopra del piano del foglio, siano legati alle estremità della linea orizzontale.
In presenza di più centri chirali si applica lo stesso procedimento a ognuno di essi.
E’ anche possibile convertire una proiezione di Fischer in una rappresentazione tridimensionale, ad esempio utilizzando i cunei e tratteggi delle formule prospettiche, dove i due legami orizzontali sono rappresentati da cunei solidi, mentre quelli verticali da linee tratteggiate.
Regole per utilizzare le formule proiettive di Fischer
Poiché le proiezioni di Fischer rappresentano in forma bidimensionale strutture tridimensionali, ci sono alcune regole che vanno rispettate per evitare un cambio di configurazione.
Le proiezioni non devono essere ribaltate dal piano del foglio ma traslate o girate; con il ribaltamento si ottiene l’altro enantiomero.
Se si ruota sul piano la proiezione, per ottenere lo stesso enantiomero dovremo ruotare la figura di 180°, a prescindere dalla direzione di rotazione, in quanto i gruppi verticali devono giacere sotto il piano del foglio mentre quelli orizzontali sopra. Di contro, se le proiezioni sono ruotate di 90° o 270° non verrebbe rispettata la convenzione e un enantiomero è convertito nell’altro.
Un numero dispari di scambi di due gruppi porta all’altro enantiomero.
IUPAC. Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8. doi:10.1351/goldbook
Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011
Nel 1956, Robert Sidney Cahn, Christopher Ingold, e Vladimir Prelog misero a punto un sistema di nomenclatura che, basandosi su poche e semplici regole, permette di assegnare la configurazione assoluta a ogni centro di chiralità presente in una molecola.[1][5]
Questo sistema di nomenclatura, chiamato sistema RS o convenzione di Cahn-Ingold-Prelog, quando accoppiato con il sistema di nomenclatura IUPAC,[3] permette di assegnare in maniera accurata e priva di ambiguità il nome alle molecole chirali, anche quando è presente più di un centro asimmetrico.
Le molecole chirali sono, nella maggior parte dei casi, in grado di far ruotare il piano della luce polarizzata quando questa attraversa una soluzione che le contenga. A questo riguardo va sottolineato che il segno del potere rotatorio non da alcuna informazione circa la configurazione RS dei centri chirali della molecola.
La convenzione di Fischer-Rosanoff è un altro sistema di nomenclatura per le molecole chirali.[6] Tuttavia, rispetto al sistema RS, non analizza ogni singolo centro chirale ma assegna un nome all’intera molecola, e spesso presenta ambiguità con molecole con più centri chirali.[7]
Il sistema RS assegna un ordine di priorità ai gruppi legati a un centro chirale e, tracciando una circonferenza dal gruppo a priorità maggiore verso quello a priorità minore, assegna la configurazione R o S al centro chirale.[2][4]
Prima regola
Si assegna un ordine di priorità ai gruppi, sulla base del numero atomico dell’atomo direttamente legato al centro chirale.
L’atomo con il numero atomico più alto ha la priorità più alta.
L’atomo con il numero atomico più basso ha la priorità più bassa.
Ad esempio, se un atomo di ossigeno, O, numero atomico 8, di carbonio, C, numero atomico 6, di cloro, Cl, numero atomico 17, e di bromo, Br, numero atomico 35, sono legati a un centro chirale, l’ordine di priorità sarà: Br > Cl> O > C.
Considerando gli isotopi, l’atomo con la massa atomica più alta ha la priorità più alta.
Seconda regola
Quando gruppi differenti sono legati al centro di chiralità attraverso identici atomi, l’ordine di priorità è assegnato in base al numero atomico dell’atomo successivo a quello legato al centro, allontanandoci dal centro chirale finché non si raggiunge il primo punto di differenza.
Se, per esempio, i gruppi –CH3, –CH2CH3 e –CH2OH sono legati al centro di chiralità, ci sono tre atomi identici attaccati direttamente a esso. Analizzando gli atomi successivi, si ha:
per il gruppo metilico –CH3
H, H, H
per il gruppo etilico –CH2CH3
H, H, C
per il gruppo idrossimetilico –CH2OH
H, H, O
Poiché il numero atomico dell’atomo di ossigeno è maggiore di quello dell’atomo di carbonio, che a sua volta è maggiore di quello dell’atomo di idrogeno, l’ordine di priorità sarà –CH2OH > –CH2CH3 > –CH3.
Per alcuni gruppi l’ordine di priorità è il seguente:
Si noti che i gruppi legati a un centro chirale devono avere un differente grado priorità altrimenti il centro non può essere chirale.
Stabilito l’ordine di priorità, si orienta la molecola nello spazio in modo che il gruppo a priorità più bassa sia diretto in direzione opposta a quella dell’osservatore, quindi dietro il centro chirale. A questo punto si congiungono i gruppi rimasti con una circonferenza, in modo che la successione sia secondo priorità decrescente: dal gruppo a priorità più alta verso quello a priorità più bassa.
Se tracciando questa circonferenza si segue una direzione oraria, la configurazione del centro chirale è R, dal latino rectus che significa “destra”.
Se tracciando la circonferenza si segue una direzione antioraria, la configurazione del centro chirale è S, dal latino sinister che significa “sinistra”.
Terza regola
Questa può essere considerata come la terza regola del sistema RS, grazie alla quale è possibile assegnare la configurazione a un centro chirale anche quando sono presenti di doppi o tripli legami nei gruppi legati al centro stesso.
Ai fini dell’attribuzione delle priorità, gli atomi impegnati nei legami multipli sono considerati duplicati, nel caso di un doppio legame, e triplicati, nel caso di un triplo legame. Nel caso di doppio legame C=Y, un atomo Y sarà da legare sull’atomo di carbonio, e un atomo di carbonio sull’atomo Y.
Nel caso di un triplo legame C≡Y, due atomi Y verranno legati sull’atomo di carbonio, e due atomi di carbonio sull’atomo Y.
Sistema RS in presenza di più centri chirali
Quando in una molecola sono presenti due o più centri chirali, si procede analizzando in modo indipendente ciascun centro, utilizzando le regole viste in precedenza.
Consideriamo il 2,3-butandiolo. La molecola ha due centri chirali, il carbonio 2 e il carbonio 3, e tre stereoisomeri: due enantiomeri e un composto meso. Qual è la configurazione RS dei centri chirali dell’enantiomero in figura?
Si consideri il carbonio 2. L’ordine di priorità dei gruppi legati è: –OH > –CH2OHCH3 > –CH3 > –H. Si ruoti la molecola in modo che l’idrogeno, il gruppo con la priorità più bassa, sia diretto in direzione opposta all’osservatore. Disegnando una circonferenza partendo dal gruppo –OH, il gruppo con la priorità più alta, verso il gruppo –CH3, il gruppo a priorità più bassa, ci si muove in senso antiorario, per cui la configurazione del carbonio 2 è R. Applicando la stessa procedura al carbonio 3, si scopre che la sua configurazione è R. Quindi l’enantiomero in figura è il (2R,3R)-2,3-butandiolo.
Aminoacidi e gliceraldeide
Nella convenzione di Fischer-Rosanoff tutti gli aminoacidi proteinogenici sono L-aminoacidi. Nel sistema RS, con l’eccezione della glicina che è achirale, e della cisteina che, per la presenza del gruppo tiolico è la (R)-cisteina, tutti gli altri aminoacidi proteinogenici hanno configurazione S.
La treonina e l’isoleucina posseggono due centri chirali, il carbonio alfa e un atomo di carbonio sulla catena laterale, e tre stereoisomeri: due enantiomeri e un composto meso. Le forme dei due amminoacidi isolate dalle proteine sono la (2S,3R)-treonina e la (2S,3S)-isoleucina, secondo la convenzione di Fischer-Rosanoff la L-treonina e la L-isoleucina.
Nel sistema RS, la L-gliceraldeide ha il centro chirale con configurazione S, per cui è indicata come (S)-gliceraldeide; ovviamente la D-gliceraldeide sarà la (R)-gliceraldeide.
Bibliografia
^ Cahn R.S., Ingold C., Prelog V. Specification of molecular chirality. Angew Chem 1966:5(4); 385-415. doi:10.1002/anie.196603851
^ IUPAC. Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8. doi:10.1351/goldbook
^ Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
^ Prelog V. and Helmchen G. Basic principles of the CIP‐system and proposals for a revision. Angew Chem 1982:21(8);567-583. doi:10.1002/anie.198205671
^ Rosanoff M.A. On Fischer’s classification of stereo-isomers. J Am Chem Soc 1906:28(1);114-121. doi:10.1021/ja01967a014
^ Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011
La chiralità è la proprietà geometrica di un insieme di punti o atomi nello spazio, o di oggetti solidi, di non essere sovrapponibili alla loro immagine speculare. Questi strutture, definite chirali, hanno la caratteristica peculiare di essere prive di elementi di simmetria del secondo ordine, ossia un piano di simmetria, un centro di inversione, o un asse di roto-riflessione. La definizione di chiralità, dal greco cheir che significa “mano”, si deve a Lord Kelvin che la enunciò nel corso delle “Baltimore Lectures”, una serie di lezioni tenute alla Johns Hopkins University di Baltimora a partire dal primo ottobre del 1884, e pubblicate nel 1904.
L’ambiente che ci circonda è ricco di oggetti chirali: le nostre mani ne sono l’esempio per eccellenza, ma ce ne sono moltissimi altri, dal guscio di una chiocciola sino a una galassia a spirale. In chimica, e in special modo in chimica organica, la chiralità è una proprietà di importanza primaria, in quanto molecole come i carboidrati, molti amminoacidi, nonchè moltissimi farmaci, sono chirali. Le molecole chirali possono esistere in due forme, l’una immagine speculare dell’altra e non sovrapponibili, ossia, non esiste una combinazione di rotazioni o traslazioni sul piano del foglio che consenta la loro sovrapposizione. Queste molecole sono dette enantiomeri, dal greco enántios che significa “contrario” e meros che significa “parte”.
La causa più comune di chiralità in una molecola è la presenza di un centro di chiralità o centro chirale, anche detto centro asimmetrico, cioè un atomo che leghi un insieme di ligandi disposti nello spazio in modo che la molecola risultante possa esistere come due enantiomeri. Gli enantiomeri a loro volta sono un tipo di stereoisomeri, i quali possono essere definiti come isomeri che hanno lo stesso numero e tipo di atomi e legami, ma che differiscono nell’orientamento spaziale degli atomi.
Due enantiomeri di una molecola chirale, non essendo sovrapponibili, sono composti differenti. In che cosa differiscono? Ciascuna coppia di enantiomeri possiede identiche proprietà fisiche e chimiche verso tutto ciò che non è chirale come il punto di fusione, il punto di ebollizione, l’indice di rifrazione, lo spettro infrarosso, la solubilità in uno stesso solvente, o la velocità di reazione con i reagenti achirali.
Le differenze emergono quando la coppia enantiomerica viene fatta interagire con fenomeni chimici e fisici che abbiano natura chirale.
Dal punto di vista chimico, due enantiomeri possono essere distinti quando si trovano a interagire con strutture chirali, come il sito di legame di un recettore chirale o il sito attivo di un enzima chirale.
Dal punto di vista fisico due enantiomeri differiscono nelle interazioni con la luce polarizzata, che ha proprietà chirali, sono cioè dotati di attività ottica.
Chiralità e attività ottica
L’attività ottica di materiali come il quarzo e, più importante, di composti organici come zuccheri o l’acido tartarico, fu scoperta nel 1815 dallo scienziato francese Jean-Baptiste Biot. Le molecole chirali possono essere classificate sulla base della direzione in cui viene ruotato il piano della luce polarizzata, rispetto al punto di vista dell’osservatore, quando attraversa una soluzione che le contenga.
Se una soluzione di un enantiomero ruota il piano della luce polarizzata in senso orario, la molecola viene definita destrogira o destrorotatoria, dal latino dexter che significa “destro”, e suo nome è preceduto dai prefissi (+), o d-, da dextro-.
Se una soluzione di un enantiomero ruota il piano della luce polarizzata in senso antiorario, la molecola è definita levogira o levorotatoria, dal latino laevus che significa “sinistro”, e il suo nome è preceduto dai prefissi (-), o l-, da laevo-.
Ovviamente, considerando una coppia di enantiomeri, uno sarà destrogiro e l’altro non potrà che essere levogiro. Al momento non è ancora possibile predire in modo affidabile ne la grandezza, la direzione, o il segno della rotazione del piano della luce polarizzata indotta da un enantiomero. D’altro canto, neppure l’attività ottica di una molecola da alcun tipo di informazione riguardo alla disposizione spaziale dei gruppi chimici legati al centro di chiralità.
Nota: un sistema contenente molecole che abbiamo lo stesso senso di chiralità è chiamato enantiomericamente puro o enantiopuro.
Pasteur e la scoperta degli enantiomeri
Nel 1848, trentatre anni dopo il lavoro di Biot, studi sull’attività ottica delle molecole portarono Louis Pasteur, che di Biot era stato studente, a notare che, a seguito della ricristallizzazione di una soluzione acquosa concentrata di sodio ammonio tartrato, di per se otticamente inattiva, precipitavano due tipi di cristalli che erano l’uno l’immagine speculare dell’altro e non sovrapponibili.
Dopo averli separati con delle pinzette, Pasteur si accorse che le soluzioni ottenute sciogliendo quantità equimolari dei due cristalli erano otticamente attive e, cosa forse più interessante, l’angolo di rotazione del piano della luce polarizzata era lo stesso ma di segno opposto. Poiché queste differenze nell’attività ottica erano dovute alle molecole di sodio ammonio tartrato disciolte, Pasteur ipotizzò che le molecole stesse dovessero essere l’una l’immagine speculare dell’altra e non sovrapponibili, al pari dei loro cristalli, quelli che oggi chiamiamo enantiomeri.
E fu Pasteur che per primo coniò il termine asimmetria per descrivere questa proprietà, definita in seguito chiralità da Lord Kelvin.
Miscele racemiche
Una soluzione che contenga quantità equimolari di ciascun membro di una coppia di enantiomeri è detta miscela racemica o racemo. Queste soluzioni sono otticamente inattive in quanto l’effetto rotatorio di un enantiomero è esattamente compensato da quello dell’altro enantiomero. A differenza di quanto accade nei processi biochimici, la sintesi chimica di molecole chirali che non preveda il ricorso a reagenti chirali, o che non sia seguita da metodiche di separazione degli enantiomeri, porta inevitabilmente alla produzione di una miscela racemica.
Il settore che più a risentito di questo fenomeno è quello della chimica farmaceutica. Come detto in precedenza, due enantiomeri sono molecole differenti. Molti farmaci chirali sono sintetizzati come miscele racemiche, ma molto spesso l’attività farmacologica desiderata è presente solamente in uno dei due enantiomeri, detto eutomero. L’altro enantiomero, detto distomero, è inattivo o meno attivo. Un esempio è il farmaco antiinfiammatorio ibuprofene, un derivato arilpropionico: solamente l’enantiomero S , così definito sulla base delle regola del sistema di nomenclatura detto sistema RS, è dotato di attività farmacologica.
I derivati arilpropionici vengono venduti come miscele racemiche perché a livello epatico una racemasi converte il distomero in eutomero.
Tuttavia è anche possibile che il distomero produca effetti dannosi e debba essere eliminato dalla miscela racemica. Un tragico esempio è la talidomide, un sedativo e anti-nausea commercializzato come miscela racemica dagli anni ‘50 fino al 1961, e utilizzato anche in gravidanza.
Il distomero, l’enantiomero S, poteva causare gravi difetti alla nascita, in particolare la focomelia. Questo è forse l’esempio più eclatante dell’importanza delle proprietà chirali delle molecole, che ha poi spinto gli organismi di salute pubblica a promuovere, da parte dell’industria farmaceutica, la sintesi di farmaci, talidomide compresa, contenenti un singolo enantiomero.
Centri di chiralità
Un qualsiasi atomo tetraedrico che leghi quattro ligandi differenti può essere un centro chirale. L’esempio classico è l’atomo di carbonio, ma anche altri atomi appartenenti al gruppo IVA della tavola periodica, come i semimetalli silicio e il germanio, formano composti a struttura tetraedrica e possono essere centri di chiralità. Anche l’atomo di fosforo negli organofosfati ha una geometria tetraedrica, quindi, quando lega quattro diversi sostituenti, è un centro chirale.
L’atomo di azoto di un’ammina terziaria, un’ammina dove tre gruppi organici differenti sono legati all’azoto, è un centro chirale. In questi composti l’atomo di azoto è disposto al centro di un tetraedro e i suoi quattro orbitali ibridi sp3 sono diretti verso i vertici. Tre di questi sono occupati dai tre sostituenti, mentre verso il quarto è diretta la coppia di elettroni solitaria.
A temperatura ambiente, l’azoto inverte rapidamente la sua configurazione. Il fenomeno è noto come inversione dell’azoto, ossia, una rapida oscillazione dell’atomo e dei suoi ligandi, nel corso della quale l’azoto passa attraverso uno stato di transizione planare in cui ha ibridazione sp2. Come conseguenza, se l’azoto è il solo centro chirale della molecola, non c’è attività ottica in quanto si viene a formare una miscela racemica. L’inversione di configurazione viene evitata solamente in alcuni casi in cui l’azoto fa parte di una struttura ciclica che la impedisce. Dunque, la presenza di un centro chirale può non essere sufficiente per permettere la separazione dei rispettivi enantiomeri.
Nota: nel 1874, Jacobus Henricus van ‘t Hoff e Joseph Achille Le Bel basandosi anche sui risultati ottenuti da Pasteur, per primi ipotizzarono la “teoria dell’atomo di carbonio tetraedrico”. Per questo lavoro van ’t Hoff ricevette il primo premio Nobel per la chimica nel 1901.
Molecole chirali senza centri chirali
La chiralità può essere presente anche in assenza di un centro chirale, ed essere conseguenza della mancanza di libera rotazione attorno a un legame doppio o singolo, come nel caso dei:
derivati allenici, composti organici che presentano due doppi legami cumulati, cioè due doppi legami localizzati sullo stesso atomo di carbonio;
derivati bifenilici.
In questo caso la chiralità è conseguente alla presenza di un asse chirale.
Composti meso
I composti meso o forme meso sono stereoisomeri che possiedono due o più centri chirali ma sono sovrapponibili alla loro immagine speculare, dunque sono achirali, e come tali otticamente inattivi. Inoltre posseggono un piano di simmetria interno che taglia la molecola in due metà, ognuna immagine speculare dell’altra. I composti meso possono quindi essere classificati come diastereoisomeri, ossia stereoisomeri diversi dagli enantiomeri.
Per una molecola con n centri di chiralità il numero massimo di possibili stereoisomeri è 2n.
Si consideri il 2,3-butandiolo. La molecola presenta due centri di chiralità, i carboni 2 e 3, quindi si potrebbero avere 22 = 4 stereoisomeri, le cui strutture sono riportate in figura, secondo le proiezioni di Fischer, e indicate come A, B, C, D.
Le strutture A e B sono l’una l’immagine speculare dell’altra e non sono sovrapponibili, dunque sono una coppia di enantiomeri.
Le strutture C e D sono l’una l’immagine speculare dell’altra, ma sono sovrapponibili. Se infatti si ruota la struttura C o D di 180°, le due strutture si possono sovrapporre. Quindi non sono enantiomeri: sono la stessa molecola scritta con orientamento differente. Inoltre hanno un piano di simmetria interno che le suddivide in due metà, l’una immagine speculare dell’altra. Dunque, la struttura C (o D) è un composto meso perché ha centri chiarali, è sovrapponibile alla sua immagine speculare e presenta un piano di simmetria interno che divide la molecola in due metà speculari.
Bibliografia
Capozziello S. and Lattanzi A. Geometrical approach to central molecular chirality: a chirality selection rule. Chirality 2003;15:227-230. doi:10.1002/chir.10191
IUPAC. Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8. doi:10.1351/goldbook
Il fenomeno per cui due o più composti chimici differenti presentano la stessa formula molecolare è chiamato isomeria, dal greco isos che significa uguale e meros che significa parte, concetto e termine introdotti dallo scienziato svedese Jacob Berzelius nel 1830.[3][4]
L’isomeria è conseguenza del fatto che gli atomi presenti in una stessa formula molecolare possono unirsi in diversi modi a dare composti, detti isomeri, che posseggono proprietà fisiche e chimiche differenti.
L’isomeria può essere di due tipi: isomeria di struttura e stereoisomeria, a loro volta suddividibili in ulteriori sottotipi.[1]
Nella isomeria di struttura, anche detta strutturale o di costituzione, gli isomeri differiscono tra di loro in quanto gli atomi costituenti sono legati in modi e sequenze differenti.
Esistono diversi sottotipi di isomeria di struttura: l’isomeria di posizione, di gruppo funzionale e di catena.
Isomeri di posizione
Nella isomeria di posizione o posizionale gli isomeri hanno gli stessi gruppi funzionali ma disposti in posizioni diverse sulla stessa catena carboniosa.
Un esempio è il composto di formula molecolare C6H4Br2, di cui esistono tre isomeri: l’1,2-dibromobenzene, l’1,3-dibromobenzene e l’1,4-dibromobenzene. I tre isomeri differiscono per la posizione degli atomi di bromo sulla struttura ciclica.
Un altro esempio è il composto di formula molecolare C3H8O, di cui esistono due isomeri: 1-propanolo o alcol n-propilico, e il 2-propanolo o alcol isopropilico. Questi isomeri differiscono per la posizione del gruppo ossidrilico lungo la catena carboniosa.
Isomeri di gruppo funzionale
Nella isomeria di gruppo funzionale, anche detta isomeria funzionale, gli atomi sono organizzati in modo da dare luogo alla formazione di gruppi funzionali diversi.
Un esempio è il composto di formula molecolare C2H6O, di cui esistono due isomeri: il dimetiletere o etere dimetilico e l’etanolo o etil alcool, che presentano gruppi funzionali diversi, un gruppo etere, –O–, e un gruppo ossidrilico, –OH.
Isomeri di catena
Nella isomeria di catena gli isomeri differiscono nella disposizione delle catene carboniose, che possono essere ramificate o lineari.
Un esempio è il composto di formula molecolare C5H12, di cui esistono tre isomeri: l’n-pentano, il 2-metilbutano o isopentano e il 2,2-dimetilpropano o neopentano.Esempi di isomeria di catena si hanno anche tra lipidi. Ad esempio, nel gruppo degli acidi grassi a catena corta risultano isomeri di catena l’acido butirrico e l’acido isobutirrico, che hanno formula molecolare C4H8O2, e l’acido valerico, l’acido isovalerico e l’acido 2-metilbutirrico, che hanno formula molecolare C5H10O2.
Stereoisomeria
Nella stereoisomeria gli isomeri hanno lo stesso numero e tipo di atomi e legami ma differiscono nella orientazione degli atomi nello spazio.[3][4] Questi isomeri sono detti stereoisomeri, dal greco stereos che significa “solido”.
La stereoisomeria può essere di due tipi: isomeria conformazionale e isomeria configurazionale. Quest’ultima è suddividibile in due ulteriori sottotipi, l’isomeria ottica e l’isomeria geometrica.
Isomeria conformazionale
Nella isomeria conformazionale gli stereoisomeri possono essere interconvertiti tra a seguito di rotazioni attorno a uno o più legami singoli, i legami σ. Queste rotazioni producono disposizioni differenti degli atomi nello spazio che non sono sovrapponibili. Inoltre, il numero delle possibili conformazioni che una molecola può assumere è in teoria infinito, variando dalla struttura a più bassa energia, la più stabile, a quella a più alta energia, la meno stabile. Ciascun isomero è definito conformero.
Se ad esempio si considera l’etano, di formula molecolare C2H4, guardando la molecola da una estremità lungo la direzione del legame carbonio-carbonio, gli atomi di idrogeno di un gruppo metilico possono trovarsi rispetto agli atomi di idrogeno dell’altro gruppo metilico in una delle seguenti conformazioni.
In conformazione eclissata, nella quale gli atomi di idrogeno di un gruppo metilico sono nascosti da quelli dell’altro gruppo metilico, quindi l’angolo tra i legami carbonio-idrogeno tra i carboni anteriori e posteriori, detto angolo diedro, può essere 0, 120, 240 o 360 gradi. Questa è la conformazione a più alta energia, dunque la meno stabile.
In conformazione sfalsata, nella quale gli atomi di idrogeno di un gruppo metilico sono completamente sfalsati rispetto a quelli dell’altro gruppo metilico, quindi l’angolo diedro può essere 60, 180 o 300 gradi. Questa è la conformazione a più bassa energia, quindi la più stabile.
In conformazioni sgembe, che corrispondono a una qualsiasi delle conformazioni intermedie tra le due precedenti.
La maggiore o minore stabilità dei conformeri dipende dal grado di sovrapposizione delle coppie di elettroni dei legami carbonio-idrogeno dei due gruppi metilici:
nella conformazione sfalsata la distanza è la massima possibile;
nella conformazione eclissata le coppie elettroniche sono alla distanza minima possibile.
La barriera di energia potenziale tra le due conformazioni opposte è piccola, circa 2,8 kcal/mole (11,7 kJ/mole). A temperatura ambiente l’energia cinetica posseduta dalle molecole è di 15-20 kcal/mole (62,7-83,6 kJ/mole), più che sufficiente a permettere la libera rotazione attorno al legame carbonio-carbonio. Di conseguenza non è possibile isolare i singoli conformeri dell’etano.
Nota: considerando il doppio legame carbonio-carbonio, la barriera di energia potenziale che si oppone alla libera rotazione attorno al doppio legame è di circa 63 kcal/mole (264 kJ/mole), e corrisponde all’energia necessaria per rompere il legame π (Vedi isomeria geometrica). Questa quantità di energia è circa tre volte l’energia cinetica posseduta dalle molecole a temperatura ambiente, alla quale quindi la libera rotazione è impedita. Solamente a temperature superiori ai 300° le molecole acquistano un’energia termica sufficiente a rompere il legame π, permettendo così la libera rotazione intorno al legame σ rimanente, e quindi l’interconversione tra gli isomeri cis e trans.
Isomeria configurazionale
Nella isomeria configurazionale l’interconversione tra gli isomeri non avviene a seguito di rotazioni attorno a legami singoli ma comporta la rottura di legami e la formazione di nuovi legami, quindi a temperatura ambiente non avviene spontaneamente.
Esistono due sottotipi di isomeria configurazionale: l’isomeria ottica e l’isomeria geometrica.
Isomeri ottici
L’isomeria ottica è caratteristica delle molecole che hanno uno o più centri di chiralità o centri chirali, ossia un atomo tetraedrico che leghi quattro ligandi differenti.[2] Il centro chirale può essere un atomo di carbonio, fosforo, zolfo o azoto.
Nota: la parola chiralità deriva dal greco cheiros che significa “mano”.
Gli isomeri ottici mancano di un centro di simmetria o di un piano di simmetria, sono immagini speculari gli uni degli altri, e non sono sovrapponibili. Questi stereoisomeri sono detti enantiomeri, dal greco enántios che significa “contrario”.
A differenza degli altri isomeri, due enantiomeri hanno identiche proprietà fisiche e chimiche con due sole eccezioni.
La direzione di rotazione del piano della luce polarizzata, da cui il nome di isomeria ottica.
Se una soluzione di un enantiomero ruota il piano della luce polarizzata in senso orario, l’enantiomero viene indicato con il simbolo (+). Di contro, una soluzione dell’altro enantiomero ruota il piano della luce polarizzata in senso antiorario dello stesso angolo, e l’enantiomero è indicato con il simbolo (-).
Sebbene spesso indistinguibili dalla maggior parte delle tecniche, due enantiomeri possono essere distinti in un ambiente chirale, come il sito attivo di enzimi chirali.
Si noti che per una molecola con n centri chirali esiste un numero massimo di stereoisomeri pari a 2n.
Isomeri geometrici
L’isomeria geometrica, anche detta isomeria cis-trans, è caratteristica delle molecole dove non è possibile la libera rotazione tra due atomi per la presenza di strutture rigide come:
i composti con doppi legami carbonio-carbonio, carbonio-azoto o azoto-azoto, dove la rigidità è dovuta al doppio legame;
i composti ciclici, dove la rigidità è dovuta alla presenza dell’anello.
Un esempio di isomeria geometrica dovuta a un doppio legame carbonio-carbonio si ha con lo stilbene, composto di formula molecolare C14H12, di cui esistono due isomeri. In uno, definito isomero cis, i gruppi uguali sono dalla stessa parte rispetto al piano individuato dal doppio legame, mentre nell’altro, detto isomero trans, i gruppi uguali sono da parti opposte.
Nota: i termini trans e cis derivano dal latino trans che significa “al di la”, e cis che significa “di qua”.
Tra i composti ciclici, l’isomeria cis-trans non complicata dalla presenza di centri chirali si osserva nei sistemi con numero pari di atomi di carbonio e sostituiti in posizioni opposte, ossia para-sostituiti. Un esempio è l’1,4-dimetilcicloesano, un cicloalcano, idrocarburi ciclici di formula generale CnH2n,, di cui esistono due stereoisomeri, il cis-1,4-dimetilcicloesano e il trans-1,4-dimetilcicloesano.
Questo tipo di stereoisomeria non può esistere nel caso in cui uno dei due atomi non liberi di ruotare leghi due gruppi identici. Perché? Per passare dallo stereoisomero cis a quello trans è necessario scambiare tra di loro i due gruppi legati a uno dei due atomi impegnati nel doppio legame. Se i due gruppi sono uguali lo scambio porta alla formazione della stessa molecola.
Nota: gli isomeri geometrici sono un caso particolare di diastereomeri o diastereoisomeri, che, a loro volta, sono stereoisomeri che non sono l’uno l’immagine speculare dell’altro. Gli altri diastereomeri sono i composti meso e gli isomeri ottici non enantiomerici.
Bibliografia
^ Graham Solomons T. W., Fryhle C.B., Snyder S.A. Solomons’ organic chemistry. 12th Edition. John Wiley & Sons Incorporated, 2017
In una soluzione, le molecole di solvente tendono a spostarsi dalla regione dove la loro concentrazione è maggiore verso quella a concentrazione minore. Se si considerano due soluzioni separate da una membrana semipermeabile, ossia una membrana che consenta il passaggio solo a certi ioni o molecole, in questo caso le molecole di solvente, si verrà a creare attraverso la membrana un flusso netto di molecole di solvente dalla soluzione a concentrazione maggiore di solvente verso quella a concentrazione minore. Questo porta allo sviluppo di una pressione detta pressione osmotica, indicata con Π, che può essere definita come la forza che deve essere applicata per impedire lo spostamento delle molecole di solvente attraverso una membrana semipermeabile.
Assieme all’innalzamento del punto di ebollizione, all’abbassamento del punto di congelamento e alla tensione di vapore, la pressione osmotica è una delle quattro proprietà colligative delle soluzioni, proprietà che dipendono solo dal numero di particelle di soluto presenti in soluzione, ioni, molecole o strutture sopramolecolari che siano, e non dalla natura delle particelle stesse o dalla loro massa.
Per soluzioni con n soluti, l’equazione che descrive la pressione osmotica è la somma dei contributi di ciascun soluto:
Π = RT(i1c1 + i2c2 + … + incn)
L’equazione è conosciuta come equazione di van ‘t Hoff, dove:
T è la temperatura assoluta, che è espressa in Kelvin;
Il fattore di van ‘t Hoff è una misura del grado di dissociazione del soluto in soluzione, ed è descritto dall’equazione:
i = 1 + α(n-1)
dove:
α è il grado di dissociazione delle molecole di soluto, pari al rapporto tra le moli delle molecole di soluto che hanno subito dissociazione e il numero delle moli iniziali, e può assumere valori compresi tra 0, per sostanze che non si ionizzano o dissociano, e 1, per sostanze che si ionizzano o dissociano completamente in soluzione;
n è il numero di ioni ottenuti dalla dissociazione completa della molecola di soluto.
Se si considerano specie non ionizzabili, come il glucosio, il glicogeno o l’amido, n = 1 e i = 1, mentre, per specie che in soluzione diluite si dissociano completamente, come acidi e basi forti o sali, il fattore di van ‘t Hoff è un numero intero maggiore di uno, essendo α = 1 e n pari ad almeno 2. Se ad esempio si considera il cloruro di sodio, NaCl, il cloruro di potassio, KCl, o il cloruro di calcio, CaCl2, si ha:
Quindi nei primi due casi i = 2, mentre con il cloruro di calcio è pari a 3.
Infine, per sostanze che non si ionizzano completamente, come acidi e basi deboli, i non è un numero intero.
Il termine ic, il prodotto del fattore di van ‘t Hoff e la concentrazione molare del soluto, è l’osmolarità della soluzione, ossia la concentrazione delle particelle di soluto osmoticamente attive per litro di soluzione.
Pressione osmotica, osmosi e membrane cellulari
L’osmosi può essere definita come il movimento o flusso netto di un solvente attraverso una membrana semipermeabile, sotto la spinta delle differenze di pressione osmotica tra i due lati della membrana, al fine di cercare di uguagliare la concentrazione del soluto ai due lati della membrana stessa.
Nei sistemi biologici l’acqua è il solvente e le membrane plasmatiche sono le membrane semipermeabili.
Le membrane plasmatiche consentono il passaggio alle molecole d’acqua, grazie alla presenza di canali proteici, le acquaporine, nonché a piccole molecole non polari che possono diffondere rapidamente attraverso esse, mentre sono stanzialmente impermeabili a ioni e macromolecole.
La presenza all’interno della cellula di macromolecole come acidi nucleici, proteine, glicogeno e aggregati sopramolecolari, ad esempio i complessi multienzimatici, ma anche ioni in concentrazione maggiore rispetto a quella dell’ambiente extracellulare, fa si che la pressione osmotica guidi l’acqua dall’esterno all’interno della cellula. Se questo flusso netto di acqua verso l’interno della cellula non fosse controbilanciato si avrebbe in breve una distensione della membrana plasmatica tale da comportarne la rottura, ossia la cellula scoppierebbe a causa di un eccesso di acqua al suo interno, si verificherebbe cioè una lisi osmotica. In condizioni fisiologiche questo non avviene in quanto nel corso dell’evoluzione si sono sviluppati diversi meccanismi che si oppongono, e in alcuni casi addirittura sfruttano, queste forze osmotiche.
Due di questi sono le pompe ioniche energia dipendenti e, nei batteri, nei fungi e nelle cellule vegetali, la parete cellulare.
Pompe ioniche energia dipendenti
Le pompe ioniche riducono, con spesa di ATP, le concentrazioni intracellulari di determinati ioni rispetto alle loro concentrazioni nell’ambiente extracellulare, creando quindi una ineguale distribuzione degli ioni stessi sui due lati della membrana plasmatica, ossia creando un gradiente ionico. In questo modo la cellula controbilancia le forze osmotiche dovute agli ioni e macromolecole intrappolate al suo interno. Un esempio di pompa ionica energia dipendente è la Na+/K+ ATPasi, che riduce la concentrazione intracellulare di Na+ rispetto all’esterno della cellula.
Parete cellulare
Le cellule vegetali sono circondate da una matrice extracellulare, la parete cellulare, che, essendo non espandibile e posizionata in prossimità della membrana plasmatica, permette alla cellula di resistere alle forze osmotiche che potrebbero causarne il rigonfiamento e infine la lisi.
In che modo?
Nelle cellule vegetali mature i vacuoli sono gli organelli di dimensioni maggiori, arrivando a occupare circa l’80% del volume cellulare. Al loro interno vengono accumulate grandi quantità di soluti, per la maggior parte acidi organici e inorganici, i quali osmoticamente richiamano acqua, il che determina il rigonfiamento del vacuolo stesso. A sua volta questo fa si che il tonoplasto, la membrana che circonda l’organello, spinga la membrana plasmatica contro la parete cellulare, la quale, opponendosi meccanicamente a queste forze, permette di evitare la lisi osmotica. Questa pressione osmotica è chiamata pressione di turgore e può raggiungere le 20 atmosfere, 2 MPa, un valore circa 10 volte maggiore rispetto alla pressione dei pneumatici. La pressione di turgore è responsabile della rigidità delle parti non legnose delle piante, è coinvolta nella crescita della pianta, nonchè:
nell’avvizzimento della verdura, a seguito di una sua riduzione;
nei movimenti delle piante, quali:
i movimenti circadiani delle foglie;
i movimenti delle foglie della Dionaea muscipula, una pianta carnivora, o delle foglie delle Mimosa pudica.
Anche nei batteri e funghi la membrana plasmatica è circondata da una parete cellulare sufficientemente rigida e non espandibile da impedire la lisi osmotica della cellula.
Soluzioni isotoniche, ipotoniche e ipertoniche
Dal confronto tra la pressione osmotica di sue soluzioni separate da una membrana semipermeabile è possibile definire tre tipi di soluzioni, di seguito brevemente descritte.
Due soluzioni che abbiano la stessa pressione osmotica sono definite isotoniche.
Se due soluzioni hanno differenti pressione osmotica, quella a pressione maggiore viene definita ipertonica rispetto all’altra.
Se due soluzioni hanno differenti pressioni osmotiche, quella a pressione minore viene definita ipotonica rispetto all’altra.
Nei sistemi biologici la soluzione di riferimento è rappresentata dal citosol; quindi, ponendo una cellula in una soluzione:
isotonica, non si avrà trasferimento netto di acqua tra l’interno e l’esterno della cellula stessa;
ipertonica, sia avrà un trasferimento netto di acqua dalla cellula verso l’esterno, la cellula perde acqua e si raggrinzisce;
ipotonica, si verifica un trasferimento netto di acqua al suo interno, la cellula si rigonfia e può arrivare a scoppiare, ossia si può verificare una lisi osmotica.
In aggiunta alle pompe ioniche e alla parete cellulare, nel corso dell’evoluzione gli organismi pluricellulari hanno sviluppato un’altra soluzione per opporsi alle forze osmotiche: circondare le cellule con soluzioni isotoniche o prossime all’isotonicità che impediscano o comunque limitino un influsso o un efflusso netto di acqua. Un esempio è il plasma, ossia il sangue privato della componente cellulare, che, grazie alla presenza di sali e proteine, nell’uomo principalmente l’albumina, ha un’osmolarità simile a quella presente nel citosol.
Pressione osmotica, amido e glicogeno
Gli organismi immagazzinano glucosio non in forma libera ma come polimeri, glicogeno gli animali, i funghi e i batteri, amido le piante, in questo modo evitando che la pressione osmotica esercitata dalle riserve di carboidrati diventi troppo grande. Infatti, dato che la pressione osmotica, al pari delle altre proprietà colligative, dipende solo dal numero delle molecole di soluto, immagazzinare milioni di molecole di glucosio in forma di un numero notevolmente inferiore di polisaccaridi permette di evitarne un aumento abnorme. Di seguito alcuni esempi.
Se si considera un polisaccaride, come il glicogeno o amido, formato da 1000 unità di glucosio e del peso di un grammo, questi ha un effetto sulla pressione osmotica inferiore a quello di un milligrammo di glucosio libero.
Considerando un epatocita, se il glucosio immagazzinato in forma di glicogeno fosse presente come glucosio libero, la sua concentrazione sarebbe di circa 0,4 M, contro una concentrazione del glicogeno di circa 0,04 μM. Questo causerebbe un flusso netto di acqua verso l’interno della cellula tale da portare a lisi osmotica.
Inoltre, se anche si riuscisse a evitare la lisi osmotica, si avrebbero problemi riguardo al trasporto del glucosio all’interno della cellula. Nell’uomo, in condizioni fisiologiche, il sistema preposto alla regolazione della glicemia agisce in modo da mantenere i livelli glucosio nel sangue compresi tra 3,33 e 5,56 mmol/L o 60-100 mg/dL. Se il glucosio fosse immagazzinato in forma libera la sua concentrazione intracellulare sarebbe da 120 a 72 volte maggiore di quella sanguigna, e il suo trasporto nell’epatocita comporterebbe un grande dispendio energetico.
Diarrea osmotica
In presenza di malattie che causano l’accumulo di soluti non assorbiti e osmoticamente attivi nella porzione distale dell’intestino tenue e nel colon, si verifica una condizione nota come diarrea osmotica.
Tra le cause fisiopatologiche si annoverano, ad esempio, infezioni batteriche, malattie del pancreas, la celiachia, un’enteropatia autoimmune indotta dall’assunzione di glutine in soggetti geneticamente predisposti, o un deficit congenito di una delle disaccaridasi dell’orletto a spazzola degli enterociti, come nel caso dell’intolleranza al lattosio. In queste condizioni la digestione dei carboidrati può essere incompleta a causa di un deficit a carico della alfa-amilasi e/o di una o più disaccaridasi. Inoltre, i soluti osmoticamente attivi non assorbiti passano nel colon dove possono essere fermentati dai batteri del microbiota intestinale, che fa parte del microbiota umano, con conseguente eccessiva produzione di gas, come idrogeno, anidride carbonica e metano, e acidi grassi catena corta, in particolare l’acido butirrico, l’acido propionico e l’acido acetico. Ciò provoca una condizione nota come diarrea osmotico-fermentativa.
La diarrea osmotica può anche essere consente all’uso di lassativi osmotici come polietilenglicole o PEG e il solfato di magnesio.
L’accumulo di soluti osmoticamente attivi derivanti dalla digestione incompleta al pari dei lassativi osmotici portano a un aumento della pressione osmotica intraluminale e inibiscono il normale assorbimento di acqua ed elettroliti, provocando una riduzione della consistenza delle feci e un aumento della motilità intestinale.
Pressione osmotica, galattosemia e cataratta
Il galattosio, con il fruttosio e il glucosio, è uno dei tre monosaccaridi che possono essere assorbiti a livello intestinale.
Metabolizzato per la maggior parte nel fegato, e in misura minore da altri organi e tessuti, previa conversione in UDP-galattosio e UDP-glucosio attraverso la via di Leloir, il galattosio può essere utilizzato a scopi anabolici o catabolici. Mutazioni a carico di uno dei geni che codificano per gli enzimi della via di Leloir ne provocano l’accumulo e causano la galattosemia, una malattia metabolica congenita la cui unica cura è una dieta a ridotto contenuto di galattosio.
L’accumulo del monosaccaride determina l’attivazione di vie metaboliche alternative alla via di Leloir che portano alla sua conversione in galattitolo e galattonato.
Tra i sintomi della galattosemia si ritrova la comparsa precoce di cataratta e tra i fattori scatenanti sembra esserci la sintesi di galattitolo nella lente dell’occhio. Il galattitolo è un poliolo scarsamente metabolizzato e, data la sua scarsa lipofilicità, non diffonde attraverso la membrana plasmatica, accumulandosi nella cellula. Essendo osmoticamente attivo determina un aumento della pressione osmotica intracellulare e di conseguenza un richiamo d’acqua. L’effetto osmotico sembra essere uno dei meccanismi attraverso cui il galattitolo concorre allo sviluppo della cataratta in caso di galattosemia.
Bibliografia
Beauzamy L., Nakayama N., and Boudaoud A. Flowers under pressure: ins and outs of turgor regulation in development. Ann Bot 2014;114(7):1517-1533. doi:10.1093/aob/mcu187
Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002
Coelho A.I., Berry G.T., Rubio-Gozalbo M.E. Galactose metabolism and health. Curr Opin Clin Nutr Metab Care 2015;18(4):422-427. doi:10.1097/MCO.0000000000000189
Coelho A.I., Rubio-Gozalbo M.E., Vicente J.B., Rivera I. Sweet and sour: an update on classic galactosemia. J Inherit Metab Dis 2017;40(3):325-342. doi:10.1007/s10545-017-0029-3
Conte F., van Buuringen N., Voermans N.C., Lefeber D.J. Galactose in human metabolism, glycosylation and congenital metabolic diseases: time for a closer look. Biochim Biophys Acta Gen Subj 2021;1865(8):129898. doi:10.1016/j.bbagen.2021.129898
Gli enzimi multifunzionali sono proteine in cui due o più siti attivi che catalizzano reazioni consecutive di una via metabolica sono presenti in una catena polipeptidica. Sembra probabile che derivino da eventi di fusione genica rappresentando, al pari dei complessi multienzimatici, una soluzione adottata dall’evoluzione per massimizzare l’efficienza catalitica, assicurando vantaggi che non si avrebbero nel caso in cui le singole attività enzimatiche fossero presenti su proteine distinte e libere.
Quali vantaggi offrono gli enzimi multifunzionali?
Gli organismi viventi combattono con l’inevitabile processo di decadimento che, se non contrastato, conduce a un crescente aumento del disordine, sino alla morte.
A livello molecolare il mantenimento della vita è reso possibile dalla grande efficienza raggiunta dagli enzimi nell’accelerare le reazioni chimiche e nell’evitare reazioni collaterali. Un’idea della velocità con cui procede il metabolismo cellulare è fornita dalla velocità del turn over dell’ATP in una cellula di mammifero: ogni 1-2 minuti l’intero pool del trifosfato è idrolizzato e risintetizzato. Tradotto in numeri questo corrisponde al turn over di circa 107 molecole di ATP al secondo, e, per il corpo umano, a circa 1 grammo di ATP al minuto. Alcuni enzimi hanno addirittura raggiunto la perfezione catalitica, ossia sono talmente efficienti che quasi ogni collisione con il proprio substrato porta alla catalisi.
E uno dei fattori limitanti la velocità di una reazione enzimatica è la proprio frequenza con cui enzimi e substrati collidono. Il modo più semplice per aumentare la frequenza delle collisioni sarebbe quello di aumentare la concentrazione di substrati ed enzimi. Tuttavia, dato l’elevatissimo numero di differenti reazioni che avvengono all’interno della cellula, questa strada non è praticabile. Esiste cioè un limite alle concentrazioni che substrati ed enzimi possono raggiungere, concentrazioni che sono dell’ordine delle micromoli per i substrati, e anche più basse per gli enzimi. Fanno eccezione gli enzimi della glicolisi nelle cellule muscolari e negli eritrociti, presenti in concentrazioni dell’ordine delle 0.1 mM e anche maggiori.
Incanalamento dei substrati
Una delle strade percorse dall’evoluzione per aumentare la velocità con cui procedono le reazioni enzimatiche è stata quella di selezionare strutture molecolari, quali gli enzimi multifunzionali e i complessi multienzimatici, che permettano, attraverso l’ottimizzazione dell’organizzazione spaziale degli enzimi di una via metabolica, di minimizzare la distanza che il prodotto della reazione A deve percorrere per raggiungere il sito attivo che catalizza la reazione successiva B, e così via, ottenendo cioè l’incanalamento dei substrati della via stessa. Per alcuni enzimi multifunzionali e complessi multienzimatici l’incanalamento è ottenuto grazie alla presenza di veri e propri tunnel intramolecolari. L’incanalamento dei substrati migliora l’efficienza catalitica, e quindi la velocità di reazione, in vari modi, di seguito brevemente descritti.
Minimizza la diffusione nel mezzo circostante del reagente, quindi la sua diluizione, permettendo così di raggiungere concentrazioni locali elevate, anche quando la sua concentrazione nella cellula è bassa, aumentando quindi la frequenza delle collisioni enzima-substrato.
Riduce il tempo di transito dei substrati da un sito attivo al successivo.
Minimizza la probabilità che si verifichino reazioni collaterali.
Minimizza la probabilità che intermedi chimicamente labili siano degradati.
Gli enzimi multifunzionali offrono vantaggi anche dal punto di vista della regolazione della loro sintesi, essendo possibile coordinare la sintesi di tutte le attività enzimatiche interessate grazie al fatto che la struttura è codificata da un singolo gene.
Infine, analogamente ai complessi multienzimatici, è possibile regolare in modo coordinato le attività enzimatiche presenti. Se a questo si aggiunge che spesso l’enzima che catalizza la tappa di comando della sequenza è quello che catalizza la prima reazione, è possibile sia evitare la sintesi di molecole non necessarie, che sarebbero invece prodotte se la tappa di comando fosse a valle della prima reazione, come pure uno spreco di energia e la sottrazione di metaboliti ad altre vie metaboliche.
Esempi di enzimi multifunzionali
Al pari dei complessi multienzimatici, anche gli enzimi multifunzionali sono molto comuni e coinvolti in vie metaboliche sia anaboliche che cataboliche.
Di seguito alcuni esempi.
Acetil-CoA carbossilasi
L’acetil-CoA carbossilasi o ACC (EC 6.4.1.2), una carbossilasi biotina-dipendente, è formata da due enzimi, la biotina carbossilasi (EC 6.3.4.14) e una transcarbossilasi, più la proteina trasportatrice della biotina o BCCP, acronimo dell’inglese biotin carboxyl-carrier protein. ACC catalizza la sintesi del malonil-CoA via carbossilazione dell’acetil-CoA. La reazione, che rappresenta la tappa di comando della sintesi degli acidi grassi, procede in due tappe.
Nella prima la biotina carbossilasi catalizza il trasferimento di una molecola di anidride carbonica (CO2) dallo ione bicarbonato a un atomo di azoto dell’anello della biotina, che funge da trasportatore temporaneo di CO2. La reazione comporta il consumo di una molecola di ATP.
Nella seconda tappa la transcarbossilasi catalizza il trasferimento del gruppo carbossilico dalla carbossi-biotina all’acetil-CoA a dare malonil-CoA.
Il malonil-CoA verrà di seguito utilizzato come donatore di unità bicarboniose dalla acido grasso sintasi (EC 2.3.1.85) durante l’allungamento degli acidi grassi.
Nei mammiferi e negli uccelli l’acetil-CoA carbossilasi è un enzima multifunzionale essendo le due attività enzimatiche, e BCCP, presenti su un’unica catena polipeptidica. Nei batteri invece ACC è un complesso multienzimatico formato dall’aggregazione di tre catene polipeptidiche, ossia i due enzimi più BCCP.
Nelle piante superiori sono presenti entrambe le forme.
Acido grasso sintasi di tipo I
L’acido grasso sintasi o FAS, acronimo dell’inglese fatty acid synthase, catalizza la sintesi dell’acido palmitico utilizzando il malonil-CoA, che è il prodotto della reazione catalizzata dalla acetil-CoA carbossilasi, come donatore di unità bicarboniose.
Esistono due tipi di acido grasso sintasi.
Negli animali e nei funghi è un enzima multifunzionale, ed è detto di tipo I. Negli animali è un omodimero, e in ogni catena polipeptidica sono presenti le sette le attività enzimatiche e la proteina trasportatrice di acili o ACP, acronimo dell’inglese acyl carrier protein. Nei lieviti e nei funghi FAS è formata da due subunità multifunzionali, dette α and β, disposte a formare una struttura eterododecamerica α6β6.
Nella maggior parte dei procarioti e nelle piante l’acido grasso sintasi, detta di tipo II, non è un enzima multifunzionale ma un complesso multienzimatico essendo composto da enzimi distinti più ACP.
PRA isomerasi:IGP sintasi
La sintesi dell’aminoacido triptofano a partire dal corismato coinvolge diversi passaggi, di seguito brevemente descritti.
Nel primo, la glutammina dona un atomo di azoto all’anello indolico del corismato, che è convertito in antranilato, e la glutammina in glutammato; la reazione è catalizzata dalla antranilato sintasi (EC 4.1.3.27).
L’antranilato è fosforibosilato a spese del 5-fosforibosil-1-pirofosfato o PRPP a dare N-(5’-fosforibosil)-antranilato o PRA, nella reazione catalizzata dalla antranilato fosforibosiltransferasi (EC 2.4.2.18).
Nel passaggio successivo, catalizzato dalla PRA isomerasi (EC 5.3.1.24), PRA è isomerizzato a dare enol-1-o-carbossifenilamino-1-desossiribulosio fosfato o CdRP. PRA e CdRP sono un esempio di isomeria di struttura.
CdRP è convertito in indolo-3-glicerolo fosfato o IGP nella reazione catalizzata dalla indolo-3-glicerofosfato sintasi o IGP sintasi (EC 4.1.1.48).
Infine, la triptofano sintasi (EC 4.2.1.20) catalizza gli ultimi due passaggi della via: la conversione di IGP in indolo, una idrolisi, e la reazione dell’indolo con una serina a dare il triptofano.
In E. coli, PRA isomerasi e IGP sintasi sono presenti su un’unica catena polipeptidica, che è quindi un enzima bifunzionale. In altri microorganismi, come Bacillus subtilis, Salmonella typhimurium e Pseudomonas putida le due attività enzimatiche sono presenti su catene polipeptidiche distinte.
La triptofano sintasi è invece un esempio di complesso multienzimatico, e uno degli esempi meglio caratterizzati di canalizzazione del flusso dei metaboliti.
Glutammina-PRPP amido transferasi
La glutammina-PRPP ammidotransferasi (EC 2.4.2.14) catalizza la prima della 10 tappe che portano alla sintesi de novo delle purine, ossia la formazione della 5-fosforibosilammina a seguito del trasferimento dell’azoto ammidico della glutammina, che funge quindi da fonte di azoto, al PRPP.
La reazione procede in due tappe, catalizzate nei due siti attivi dell’enzima, un sito N-terminale e uno C-terminale.
Nella prima tappa, il sito attivo N-terminale catalizza l’idrolisi dell’azoto ammidico della glutammina a dare ammoniaca e glutammato.
Nella seconda tappa, catalizzata dal sito attivo C-terminale, che ha attività fosforibosiltransferasica, l’ammoniaca precedentemente rilasciata viene legata al C-1 del PRPP a dare la 5-fosforibosilammina. In questa reazione si verifica l’inversione della configurazione del C-1 del ribosio, da α a β, stabilendo la forma anomerica del nucleotide in via di sintesi.
Ci sono tre punti di controllo che cooperano nella regolazione della sintesi de novo dei nucleotidi purinici, e la reazione catalizzata dalla glutammina-PRPP ammidotransferasi, che è anche la prima reazione esclusiva della via, è il primo.
Al pari del complesso della carbamil fosfato sintetasi batterica, anche i siti attivi di questo enzima multifunzionale sono connessi attraverso un canale intramolecolare. Tuttavia questo canale è più corto, essendo lungo circa 20 Å, ed è delimitato da residui amminoacidici non polari, quindi è altamente idrofobico. Mancando gruppi in grado di stabilire legami idrogeno, il tunnel permette la diffusione dell’ammoniaca verso il secondo sito attivo.
CAD
La sintesi de novo dei nucleotidi pirimidinici avviene attraverso una serie di reazioni enzimatiche che, a differenza di quanto accade nella sintesi de novo dei nucleotidi purinici, comporta dapprima la formazione dell’anello pirimidinico e quindi il suo legame al ribosio-5-fosfato. Le prime tre tappe della via sono catalizzate in sequenza dalla carbamil fosfato sintetasi (EC 6.3.4.16), aspartato transcarbamilasi (EC 2.1.3.2) e diidroorotasi (EC 3.5.2.3), e sono comuni a tutte le specie.
Nella prima tappa, la carbamil fosfato sintetasi, che presenta due attività enzimatiche, ossia una attività amidotransferasica glutammina dipendente e un’attività sintasica, catalizza la sintesi del carbamil fosfato a partire da glutammina, ione bicarbonato e ATP.
Nella seconda tappa, che è quella di comando della via metabolica ed è catalizzata dalla aspartato transcarbamilasi, il carbamil fosfato reagisce con l’aspartato a dare l’N-carbamil aspartato.
Infine la diidroorotasi, catalizzando la rimozione di una molecola d’acqua dall’N-carbamil aspartato, porta alla chiusura dell’anello pirimidinico a formare L-diidroorotato.
Negli eucarioti, in particolare nei mammiferi, in Drosofila e Dictyostelium, un genere di amebe, le tre attività sono presenti su una singola catena polipeptidica, codificata da un singolo gene derivante da una fusione avvenuta almeno 100 milioni di anni fa. L’enzima multifunzionale, abbreviato come CAD, è un omomultimero composto da tre subunità o più.
Nei procarioti invece i tre enzimi sono indipendenti, e la carbamil fosfato sintetasi è un esempio di complesso multienzimatico.
Nei lieviti l’attività diidroorotasica è presente su una proteina separata.
Studi sull’attività enzimatica hanno rivelato l’esistenza di un incanalamento dei substrati, più efficace nella proteina del lievito, riguardo ai primi due passaggi, rispetto a quella dei mammiferi.
Bibliografia
Alberts B., Johnson A., Lewis J., Morgan D., Raff M., Roberts K., Walter P. Molecular Biology of the Cell. 6th Edition. Garland Science, Taylor & Francis Group, 2015
Eriksen T.A., Kadziola A., Bentsen A-K., Harlow K.W. & Larsen S. Structural basis for the function of Bacillus subtilis phosphoribosyl-pyrophosphate synthetase. Nat Struct Biol 2000:7;303-308. doi:10.1038/74069
Hyde C.C., Ahmed S.A., Padlan E.A., Miles E.W., and Davies D.R. Three-dimensional structure of the tryptophan synthase multienzyme complex from Salmonella typhimurium. J Biol Chem 1988:263(33);17857-17871. doi:10.1016/S0021-9258(19)77913-7
Hyde C.C., Miles E.W. The tryptophan synthase multienzyme complex: exploring structure-function relationships with X-ray crystallography and mutagenesis. Nat Biotechnol 1990:8;27-32. doi:10.1038/nbt0190-27
Michal G., Schomburg D. Biochemical pathways. An atlas of biochemistry and molecular biology. 2nd Edition. John Wiley J. & Sons, Inc. 2012
Muchmore C.R.A., Krahn J.M, Smith J.L., Kim J.H., Zalkin H. Crystal structure of glutamine phosphoribosylpyrophosphate amidotransferase from Escherichia coli. Protein Sci 1998:7;39-51. doi:10.1002/pro.5560070104
Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
Yon-Kahn J., Hervé G. Molecular and cellular enzymology. Springer, 2009
I complessi multienzimatici sono strutture discrete e stabili formate da enzimi associati in modo non covalente che catalizzano due o più reazioni consecutive di una via metabolica.[7]
Possono essere considerati come un passo in avanti nell’evoluzione dell’efficienza catalitica in quanto assicurano vantaggi che i singoli enzimi, anche quelli che hanno raggiunto la perfezione catalitica, da soli non avrebbero.[1][12]
Quali vantaggi offrono i complessi multienzimatici?
Nel corso dell’evoluzione alcuni enzimi si sono evoluti sino al raggiungimento della perfezione catalitica. Si tratta cioè di enzimi per i quali quasi ogni collisione con il proprio substrato ne determina la conversione in prodotto. Esempi sono:
la fumarasi (EC 4.2.1.2), che catalizza la settima tappa del ciclo dell’acido citrico, ossia l’idratazione/deidratazione reversibile del doppio legame del fumarato a dare malato;
l’acetilcolinesterasi (EC 3.1.1.7), che catalizza l’idrolisi dell’acetilcolina, un neurotrasmettitore, in colina e acido acetico, che a sua volta si dissocia a dare acetato e ioni idrogeno;
la superossido dismutasi (EC 1.15.1.1), che catalizza la conversione, e quindi l’inattivazione, del radicale superossido (O2.-), una specie molto reattiva, in perossido d’idrogeno od acqua ossigenata (H2O2) e acqua;
la catalasi (EC 1.11.1.6), che catalizza la degradazione di H2O2 in acqua e ossigeno.
La velocità con cui avviene una reazione enzimatica è dunque in parte è determinata dalla frequenza con cui i substrati e gli enzimi stessi collidono. Ne deriva che un modo semplice per aumentarla sia quello di aumentare le concentrazioni di enzima e substrato. Tuttavia, visto l’enorme numero di reazioni che si verificano all’interno della cellula le loro concentrazioni non potranno essere elevate. E infatti nella cellule la concentrazione della maggior parte dei metaboliti è dell’ordine delle micromoli (10-6 M), e per la maggior parte degli enzimi anche più bassa.[1]
L’evoluzione ha quindi imboccato strade differenti per aumentare la velocità di reazione. Una delle quali è stata quella di ottimizzare l’organizzazione spaziale degli enzimi con la formazione dei complessi multienzimatici e degli enzimi multifunzionali, ossia strutture che permettono di minimizzare la distanza che il prodotto di una reazione deve percorrere per passare al sito attivo che catalizza la reazione successiva, essendo i siti attivi vicini gli uni agli altri.[2][7]Si verifica cioè l’incanalamento dei substrati, in inglese substrate o metabolic channeling,[8][9][14] incanalamento che può avvenire anche attraverso veri e propri canali intramolecolari che connettono i siti attivi, come nel caso, tra i complessi enzimatici, del complesso della triptofano sintasi (EC 4.2.1.20),[3][4]il cui tunnel fu il primo a essere scoperto, e quello della carbamil fosfato sintetasi batterica (EC 6.3.4.16).[5][13]
L’incanalamento dei substrati è in grado di aumentare la velocità di reazione, ma più in generale l’efficienza catalitica, in più modi, di seguito brevemente descritti.[8][12]</sup>
Viene limitata al minimo la diffusione di substrati e prodotti nel mezzo circostante, quindi la loro diluizione e riduzione della concentrazione, producendo anzi elevate concentrazioni locali anche quando la loro concentrazione cellulare è bassa, il che porta a un aumento della frequenza delle collisioni enzima-substrato.
Viene ridotto il tempo di transito dei substrati da un sito attivo al sito attivo successivo.
Viene ridotta la probabilità che si verifichino reazioni collaterali.
Gli intermedi chimicamente labili sono protetti dalla degradazione da parte del solvente.
Un altro vantaggio metabolico apportato dai complessi multienzimatici, analogamente a quanto accade con gli enzimi multifunzionali, è che consentono di controllare in modo coordinato l’attività catalitica degli enzimi che lo compongono. E se si aggiunge il fatto che spesso l’enzima che catalizza la prima reazione della sequenza è l’enzima regolatorio, è possibile evitare:
la sintesi di intermedi non necessari, altrimenti prodotti se la sequenza di reazioni fosse regolata a valle della prima reazione;
la sottrazione di metaboliti ad altre vie come pure uno spreco di energia.
Esempi
Da quanto detto in precedenza non sorprende che, specialmente nelle cellule eucariote, i complessi multienzimatici, al pari degli enzimi multifunzionali, siamo comuni e coinvolti in differenti vie metaboliche, sia anaboliche che cataboliche, mentre sono pochi gli enzimi liberamente diffusibili. Di seguito alcuni esempi.
Alfa-chetoacido deidrogenasi
Esempi classici di complessi multienzimatici sono i tre complessi appartenenti alla famiglia delle alfa-chetoacido deidrogenasi o 2-ossiacido deidrogenasi, ossia:
il complesso dell’α-chetoacido deidrogenasi a catena ramificata o BCKDH , acronimo dell’inglese branched-chain α-keto acid dehydrogenase complex;
il complesso della α-chetoglutarato deidrogenasi, o 2-oxoglutarato deidrogenasi o OGDH, acronimo dell’inglese 2-oxoglutarate dehydrogenase.[2][6][7][10]
Alfa-Chetoglutarato Deidrogenasi
I tre complessi sono correlati sia dal punto di vista strutturale che funzionale.
Considerando ad esempio il complesso della piruvato deidrogenasi questi è formato da copie multiple di tre enzimi differenti:
piruvato deidrogenasi o E1 (EC 1.2.4.1);
diidrolipoil transacetilasi o E2 (EC 2.3.1.12);
diidrolipoil deidrogenasi o E3 (EC 1.8.1.4).
PDC, sia nei procarioti che negli eucarioti presenta quindi una struttura di base E1-E2-E3, struttura che si ritrova anche negli altri due complessi.[15] In aggiunta, all’interno di una data specie:
la diidrolipoil deidrogenasi è identica;
la piruvato deidrogenasi e la diidrolipoil transacetilasi sono omologhe
E, sebbene questi enzimi siamo specifici per i rispettivi substrati, utilizzano gli stessi cofattori, ossia il coenzima A, il NAD, al tiamina pirofosfato, il FAD e la lipoamide.
Al fine di differenziarle vengono indicate, per il complesso della piruvato deidrogenasi, della α-chetoglutarato deidrogenasi e della α-chetoacido deidrogenasi a catena ramificata, rispettivamente come:
E1p, E1o ed E1b (EC 1.2.4.4);
E3p, E3o, and E3b (EC 1.8.1.4).</li>
Nota: il complesso della piruvato deidrogenasi degli eucarioti è il più grande complesso multienzimatico conosciuto, più grande di un ribosoma e visibile anche al microscopio elettronico.[2]
Il complesso della piruvato deidrogenasi, che rappresenta il ponte di collegamento tra glicolisi e ciclo dell’acido citrico, catalizza la decarbossilazione ossidativa del piruvato, che è la base coniugata dell’acido piruvico e fa parte del gruppo dei chetoacidi, in particolare è un alpha-chetoacido. Nel corso delle reazioni il gruppo carbossilico del piruvato viene rilasciato in forma di anidride carbonica (CO2) e si verifica il trasferimento del gruppo acetilico risultante al coenzima A a dare acetil-coenzima A. Inoltre sono rilasciati due elettroni che sono trasferiti al NAD+.
Anche nel corso delle reazioni catalizzate dal complesso della α-chetoglutarato deidrogenasi e dal complesso della α-chetoacido deidrogenasi a catena ramificata, rispettivamente quarta reazione del ciclo dell’acido citrico, ossia l’ossidazione dell’α-chetoglutarato a succinil-CoA, e l’ossidazione degli α-chetoacidi derivanti dal catabolismo degli amminoacidi a catena ramificata valina, leucina e isoleucina, si verifica:
la liberazione del carbonio carbossilico dell’alfa-chetoacido in forma di CO2;
il trasferimento del gruppo acilico risultante al coenzima A a dare l’acil-CoA corrispondente;
la riduzione del NAD+ a NADH.
Dunque, la notevole somiglianza esistente tra le strutture proteiche, i cofattori richiesti e i meccanismi di reazione riflettono senza dubbio una origine evolutiva comune.
Triptofano sintasi
Il complesso della triptofano sintasi è uno degli esempi di incanalamento dei substrati meglio studiati.[3][12][14] Presente nei batteri e nelle piante ma non negli animali, nei batteri è formato da due subunità α e due β assemblate in unità dimeriche αβ, che sembrano esser l’unità funzionale del complesso, a dare una struttura tetramerica αββα.[4][5][8]
Il complesso catalizza gli ultimi due passaggi della sintesi del triptofano: dall’indolo-3-glicero fosfato, per azione di una liasi (EC 4.1.2.8) presente sulle subunità α, viene liberata una molecola di gliceraldeide-3-fosfato e l’indolo. L’indolo diffonde quindi verso il sito attivo presente sulla subunità β sfruttando un tunnel idrofobico di circa 30 Å che, in ciascuna coppia αβ, connette i due siti attivi. Nel sito attivo della subunità β avviene, in presenza del piridossal-5-fosfato, la condensazione tra l’indolo e una serina a dare il triptofano.
Acetil-CoA carbossilasi
L’acetil-CoA carbossilasi o ACC (EC 6.4.1.2), membro della famiglia delle carbossilasi biotina-dipendenti, catalizza la prima tappa di comando della sintesi degli acidi grassi, ossia la carbossilazione dell’acetil-CoA a malonil-CoA, che è utilizzato come donatore di unità bicarboniose dalla acido grasso sintasi (EC 2.3.1.85) durante il processo di allungamento che porterà alla sintesi dell’acido palmitico.[9][11][12]
Nei batteri ACC è un complesso multienzimatico composto da due enzimi, la biotina carbossilasi (EC 6.3.4.14) e una carbossitransferasi, cui si aggiunge una proteina trasportatrice della biotina o BCCP, acronimo dell’inglese biotin carboxyl-carrier protein.
Nei mammiferi e negli uccelli è invece un enzima multifunzionale, in quanto le due attività enzimatiche sono presenti su una stessa catena polipeptidica, nella quale si ritrova anche BCCP.[2]
Nelle piante superiori sono presenti entrambe le forme.
Carbamil fosfato sintetasi
Altro esempio ben caratterizzato di incanalamento dei substrati è il complesso della carbamil fosfato sintetasi dei batteri,[9][12][14] che catalizza la sintesi del carbamil fosfato, necessario sia per la sintesi delle pirimidine che per quella dell’arginina. Il complesso presenta un tunnel lungo circa 100 Å all’interno del quale si muovono i prodotti delle sue tre attività enzimatiche.[13]
La prima catalizza la cessione dell’azoto ammidico della glutammina in forma di ione ammonio, il quale entra nel tunnel dove nel secondo sito attivo si combina con il bicarbonato, a spese di una molecola di ATP, a dare carbamato, che infine nell’ultimo sito attivo viene fosforilato a carbamil fosfato.
Bibliografia
^ ab Alberts B., Johnson A., Lewis J., Morgan D., Raff M., Roberts K., Walter P. Molecular biology of the cell. 6th Edition. Garland Science, Taylor & Francis Group, 2015
^ ab Hilario E., Caulkins B.G., Huang Y-M. M., You W., Chang C-E. A., Mueller L.J., Dunn M.F., and Fan L. Visualizing the tunnel in tryptophan synthase with crystallography: insights into a selective filter for accommodating indole and rejecting water. Biochim Biophys Acta 2016;1864(3):268-279. doi:10.1016/j.bbapap.2015.12.006
^ ab Hyde C.C., Ahmed S.A., Padlan E.A., Miles E.W., and Davies D.R. Three-dimensional structure of the tryptophan synthase multienzyme complex from Salmonella typhimurium. J Biol Chem 1988;263(33):17857-17871. doi:10.1016/S0021-9258(19)77913-7
^ ab Hyde C.C., Miles E.W. The tryptophan synthase multienzyme complex: exploring structure-function relationships with X-ray crystallography and mutagenesis. Nat Biotechnol 1990:8;27-32. doi:10.1038/nbt0190-27
^ Koolman J., Roehm K-H. Color atlas of Biochemistry. 2nd Edition. Thieme, 2005
^ abc Michal G., Schomburg D. Biochemical pathways. An atlas of biochemistry and molecular biology. 2nd Edition. John Wiley J. & Sons, Inc. 2012
^ abc Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
^ abc Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ ^ Perham R.N., Jones D.D., Chauhan H.J., Howard MJ. Substrate channeling in 2-oxo acid dehydrogenase multienzyme complexes. Biochem Soc Trans 2002;30(2):47-51. doi:10.1042/bst0300047
Il complesso della piruvato deidrogenasi o PDC è uno dei complessi multienzimatici mitocondriali, ed è composto da tre differenti enzimi:
piruvato deidrogenasi o E1 (EC 1.2.4.1);
diidrolipoil transacetilasi o E2 (EC 2.3.1.12);
diidrolipoil deidrogenasi o E3 (EC 1.8.1.4).
Ognuno di questi enzimi è presente in più copie il cui numero, e quindi le dimensioni del complesso stesso, varia da specie a specie, con una massa molecolare che oscilla da 4 a 10 milioni di Dalton.
Fanno parte del complesso multienzimatico anche:
cinque differenti coenzimi;
nelle piante, nei funghi e, tra gli animali, nei volatili e nei mammiferi, due enzimi con attività regolatoria: la piruvato deidrogenasi chinasi (EC 2.7.1.99), enzima Mg2+-dipendente, e la piruvato deidrogenasi fosfatasi) (EC 3.1.3.43), enzima attivato dagli ioni calcio (Ca2+);
negli eucarioti, una proteina con funzione di legame, E3BP.
Il complesso della piruvato deidrogenasi catalizza, attraverso una sequenza di cinque reazioni, la decarbossilazione ossidativa del piruvato, la base coniugata dell’acido piruvico, a dare il gruppo il gruppo acetilico dell’acetil-coenzima A o acetil-CoA, una molecola di CO2, e due elettroni trasportati dal NAD. La stechiometria complessiva della sequenza delle cinque reazioni è:
PDC: Reazione Complessiva
La reazione complessiva è essenzialmente irreversibile, con un ΔG°’ pari a -8,0 kcal/mol (-33,4 kJ/mol), e richiede l’intervento sequenziale di tutti e tre gli enzimi, le cui attività risultano coordinate. Nel corso delle reazioni i prodotti intermedi rimangono legati agli enzimi e al termine della sequenza catalitica il complesso multienzimatico è pronto per il ciclo successivo.
Le Cinque Reazioni Catalizzate da PDC
Nota: il complesso della piruvato deidrogenasi catalizza le stesse reazioni attraverso meccanismi simili in tutti gli organismi in cui è presente.
Coenzimi del complesso della piruvato deidrogenasi
Cinque coenzimi partecipano alle reazioni catalizzate dal complesso della piruvato deidrogenasi: la tiamina pirofosfato (TPP), la flavina adenina dinucleotide (FAD), il coenzima A (CoA), la nicotinammide adenina dinucleotide (NAD), e l’acido lipoico.
La tiamina pirofosfato deriva dalla tiamina o vitamina B1 di cui rappresenta la forma biologicamente attiva. TPP è il coenzima della piruvato deidrogenasi cui è strettamente, ma non covalentemente, legata. Il suo ruolo è quello di trasportatore di gruppi idrossietilici o “aldeidici attivati”.
La flavina adenina dinucleotide deriva dalla riboflavina o vitamina B2, di cui rappresenta una delle forme attive; l’altra è la flavina mononucleotide o FMN.
Forma Ridotta e Ossidata della Flavina Adenina Dinucleotide
Il FAD è il coenzima della diidrolipoil deidrogenasi cui è saldamente legato. Il suo ruolo, analogamente al NAD, è quello di trasportatore di elettroni o ioni idruro (:H– o H+ + 2e–).
Il coenzima A è formato da una β-mercaptoetilamina legata attraverso legame ammidico all’acido pantotenico o vitamina B5. A sua volta la vitamina è unita, attraverso un ponte pirofosfato, a una 3’-fosfoadenosina.
Il CoA partecipa alla reazione catalizzata dalla diidrolipoil transacetilasi. Il suo ruolo è quello di trasportatore di gruppi acilici.
CoA e Acetil-CoA
La porzione del coenzima A corrispondente alla β-mercaptoetilamina termina con un gruppo sulfidrilico (–SH). Si tratta di un tiolo reattivo, cruciale per il ruolo svolto dal coenzima in quanto i gruppi acilici trasportati, legati attraverso un legame tioestere, hanno una elevata energia libera standard di idrolisi. Ciò fornisce loro un elevato potenziale di trasferimento, pari a -31,5 kcal/mol (-7,5 kJ/mol). Degno di nota che questo potenziale di trasferimento è più esoergonico, anche se di solo 0,2 kcal/mol (1 kJ/mol), rispetto a quello per l’idrolisi dell’ATP ad ADP e Pi. Pertanto i tioesteri hanno un elevato potenziale di trasferimento del gruppo acilico e sono in grado di donarlo a numerose molecole accettrici. In altre parole il gruppo acilico legato al coenzima A può essere considerato come una forma attivata pronta per il trasferimento.
E’ anche possibile affermare che la formazione del legame tioestere permette di conservare parte dell’energia metabolica derivante dall’ossidazione del carburante metabolico. E va sottolineato che il coenzima A è anche abbreviato come CoA-SH, proprio per enfatizzare il ruolo svolto dal gruppo tiolico.
Nota: nel legame tioestere è presente un atomo di zolfo nella posizione in cui nel legame estere è presente un atomo di ossigeno.
La nicotinammide adenina dinucleotide può essere prodotta a partire dal triptofano, un aminoacido essenziale, o dalla niacina o vitamina B3 o vitamina PP, da Pellagra-Preventing, la fonte della parte nicotinammidica.
Forma Ridotta e Ossidata della Nicotinammide Adenina Dinucleotide
Il NAD è coinvolto nella reazione catalizzata dalla diidrolipoil deidrogenasi. Il suo ruolo, analogamente al FAD, è quello di trasportatore di elettroni, o ioni idruro.
A differenza degli altri coenzimi del complesso della piruvato deidrogenasi, l’acido lipoico non deriva, direttamente o indirettamente, da vitamine e/o aminoacidi essenziali, ossia da precursori che non possono essere sintetizzati de novo dall’organismo e devono essere assunti con la dieta. L’acido lipoico è il coenzima della diidrolipoil transacetilasi. Il coenzima è legato all’enzima a mezzo di un legame ammidico con l’ε-ammino gruppo di un residuo di lisina a formare un residuo di lipoil-lisina o lipoammide. Il coenzima accoppia il trasferimento di elettroni con quello di gruppi acilici.
Lipoammide o Lipoil-lisina
L’acido lipoico possiede due gruppi tiolici che possono andare incontro a ossidazione intramolecolare reversibile a dare un ponte disolfuro (-S-S-), similmente a quanto può accadere tra due residui di cisteina (Cys) di una proteina. Poiché il ponte disolfuro (nota: un disolfuro ciclico) può andare incontro a reazioni di ossidoriduzione, nel corso delle reazioni catalizzate dal complesso è dapprima ridotto a diidrolipoammide, un ditiolo o la forma ridotta del gruppo prostetico, e quindi riossidato nel disolfuro ciclico.
Molti enzimi necessitano per la loro attività catalitica di piccoli componenti non proteici chiamati cofattori. I cofattori possono essere ioni metallici o piccole molecole organiche o metallo-organiche e sono classificati come coenzimi e gruppi prostetici.
Un gruppo prostetico è un cofattore che si lega in odo permanente alla proteina, a mezzo di un legame non covalente o covalente.
Un coenzima è invece un cofattore che non è legato in modo permanente all’enzima.
Localizzazione del complesso della piruvato deidrogenasi
Negli eucarioti il complesso della piruvato deidrogenasi, al pari degli enzimi del ciclo dell’acido citrico e di quelli per l’ossidazione degli acidi grassi, si trova nel mitocondrio, associato alla superficie della membrana mitocondriale interna che guarda la matrice.
Nei procarioti si trova nel citosol.
Funzioni del complesso della piruvato deidrogenasi
Il complesso della piruvato deidrogenasi ha essenzialmente due funzioni: produrre acetil-CoA e NADH.
Il gruppo acetilico legato al coenzima A, un acetato attivato, a seconda delle condizioni metaboliche della cellula e/o del tipo di cellula, potrà andare incontro a due differenti destini.
Può essere ossidato a due molecole di anidride carbonica attraverso le reazioni del ciclo dell’acido citrico, il che permette la “raccolta” di una parte dell’energia potenziale immagazzinata in forma di ATP o GTP.
Può essere utilizzato per la sintesi di acidi grassi, colesterolo, steroidi, isoprenoidi, corpi chetonici e acetilcolina.
E’ quindi possibile affermare che, a seconda delle condizioni metaboliche e/o del tipo di cellula, il complesso della piruvato deidrogenasi indirizza intermedi carboniosi dal catabolismo degli aminoacidi e del glucosio verso:
il ciclo dell’acido citrico, e quindi la produzione di energia, come ad esempio nel muscolo scheletrico in condizioni aerobiche, e, sempre, nel muscolo cardiaco;
la sintesi dei lipidi e di acetilcolina.
Negli organismi aerobi il NADH potrà essere ossidato a NAD+ a seguito della cessione di uno ione idruro alla catena di trasporto degli elettroni mitocondriale. Quest’ultima a sua volta trasferisce i due elettroni all’ossigeno molecolare (O2), trasferimento che permette la produzione di 2,5 molecole di ATP per coppia di elettroni. Nota: negli organismi anaerobi è presente un accettore di elettroni alternativo all’ossigeno, come ad esempio un nitrato o un solfato.
Dal punto di vista concettuale il complesso della piruvato deidrogenasi rappresenta il ponte di collegamento tra glicolisi e ciclo dell’acido citrico. Tuttavia, vista l’irreversibilità della reazione complessiva catalizzata dal complesso, questo ponte è “a senso unico”. Il piruvato può essere decarbossilato, ossidato e l’unità acetilica rimanente legata al CoA. Non è invece possibile compiere il percorso inverso, ossia convertire acetil-CoA in piruvato.
L’irreversibilità di questa reazione e l’assenza di vie metaboliche alternative spiega perché non è possibile utilizzare l’acetil-CoA, e quindi gli acidi grassi, per la gluconeogenesi.
Altre fonti di acetil-CoA
Il gruppo acetilico dell’acetil-CoA può derivare, oltre che dal piruvato, dall’ossidazione degli acidi grassi e dal catabolismo di molti aminoacidi. Tuttavia, a prescindere dalla sua origine, l’acetil-CoA rappresenta un veicolo di ingresso di nuove unità carboniose nel ciclo dell’acido citrico. E’ anche possibile affermare che il gruppo acetilico dell’acetil-CoA rappresenta la forma in cui la maggior parte del carbonio entra nel ciclo.
Trasporto del piruvato nella matrice mitocondriale
Negli eucarioti, la glicolisi avviene nel citosol mentre tutti i passaggi successivi del metabolismo aerobico, quindi le reazioni catalizzate dal complesso della piruvato deidrogenasi, il ciclo dell’acido citrico, la catena di trasporto degli elettroni e la fosforilazione ossidativa, si svolgono nel mitocondrio.
Similmente a quanto accade per la maggior parte degli altri anioni e metaboliti, il passaggio del piruvato attraverso la membrana mitocondriale esterna è probabilmente mediato da un canale anionico voltaggio-dipendente relativamente non specifico.
Il passaggio attraverso la membrana mitocondriale interna è invece assicurato da uno specifico trasportatore formato da due proteine denominate MPC1 e MPC2 che vanno a formare un complesso etero-oligomerico nella membrana.
Struttura del complesso della piruvato deidrogenasi
Sebbene il complesso della piruvato deidrogenasi sia formato da copie multiple di tre enzimi differenti e catalizzi le medesime reazioni tramite meccanismi simili in tutti gli organismi in cui è presente, ha una struttura quaternaria molto varia.
Il primo complesso di cui fu analizzata la struttura fu quello di E. coli, grazie al lavoro di Lester Reed. Nel complesso multienzimatico, che ha un peso di circa 4600 kD e un diametro di circa 300 Å, 24 unità di diidrolipoil transacetilasi vanno a formare una struttura con simmetria cubica, ossia, gli enzimi, associati in trimeri, sono disposti agli angoli del cubo. Dimeri di piruvato deidrogenasi si associano al nucleo di diidrolipoil transacetilasi, al centro di ognuno dei 12 bordi del cubo, per un totale 24 unità. Infine, dimeri di diidrolipoil deidrogenasi si vanno a disporre al centro di ognuna delle sei facce del cubo, per un totale di 12 unità. Si noti che l’intero complesso è costituito da 60 unità. Anche nella maggior parte degli altri batteri Gram-negativi si osserva una analoga struttura con simmetria cubica.
In alcuni batteri Gram-positivi e negli eucarioti il complesso della piruvato deidrogenasi presenta una struttura dodecaedrica, ossia quella di un poliedro regolare con 20 vertici, 12 facce pentagonali e 30 bordi, con simmetrica icosaedrica, anche detta simmetria I. Se si considera ad esempio il complesso presente nei mitocondri, questi è il più grande complesso multienzimatico conosciuto. Ha un peso di circa 1000 kD e un diametro di circa 500 Å, dunque ben 5 volte le dimensioni di un intero ribosoma. Inoltre è abbastanza grande da poter essere visualizzato con il microscopio elettronico.
Il complesso è costituito da un nucleo dodecaedrico avente un diametro di circa 25 nm e formato, come nei batteri Gram-negativi, dalla diidrolipoil transacetilasi, ma composto da 20 trimeri dell’enzima, per un totale di 60 unità, disposti ai vertici della struttura. Il nucleo è circondato da 30 unità di piruvato deidrogenasi, una centrata su ogni bordo, e 12 unità di diidrolipoil deidrogenasi, una centrata su ogni faccia. L’intero complesso è quindi costituito da 102 unità.
La struttura quaternaria del complesso è ulteriormente complicata dalla presenza di tre subunità addizionali: la piruvato deidrogenasi chinasi, la piruvato deidrogenasi fosfatasi, e la E3BP.
La chinasi e la fosfatasi sono legate al nucleo di diidrolipoil transacetilasi.
E3BP è legata a ognuna della 12 facce pentagonali, e dunque presente in circa dodici copie. La proteina è necessaria per il legame della diidrolipoil deidrogenasi al nucleo formato dalla diidrolipoil transacetilasi, come dimostrato dal fatto che la sua proteolisi parziale riduce la capacità di legame della deidrogenasi. Nella E3-bindign protein è possibile individuare un dominio C-terminale, privo di attività catalitica, e un dominio che contiene una lipoil-lisina, simile a quella della diidrolipoil transacetilasi, in grado anche di accettare un gruppo acetilico. Tuttavia, la rimozione di questo dominio non comporta alcuna riduzione dell’attività catalitica del complesso multienzimatico.
Struttura della piruvato deidrogenasi o E1
La piruvato deidrogenasi degli eucarioti e di alcuni batteri Gram-positivi è composta da due differenti catene polipeptide, indicate come α e β, disposte a formare un eterotetramero α2β2 simmetrico. Di contro in E. coli e in altri batteri Gram-negativi le subunità sono fuse a formare un’unica catena polipeptidica e l’enzima risulta un omodimero.
L’enzima presenta due siti attivi.
Considerando la struttura eterotetramerica della piruvato deidrogenasi di Bacillus stearothermophilus, un batterio Gram-positivo, ogni tiamina pirofosfato si lega tra i domini N-terminali di una subunità α e di una β, al termine di un canale a forma di imbuto profondo circa 21 Å che porta al sito attivo, con l’anello tiazolico, il suo gruppo reattivo, posizionato vicino all’ingresso del canale stesso.
All’ingresso del canale sono inoltre presenti due anse peptidiche conservate, essenziali sia per l’attività catalitica dell’enzima che per la sua regolazione.
L’analisi ai raggi X dell’enzima di B. stearothermophilus, quando lega sia la tiamina pirofosfato che PSBD della diidrolipoil transacetilasi, che si lega al dominio C-terminale delle subunità β, ha evidenziato, oltre a un eterotetramero con un struttura estremamente compatta, che i due siti attivi hanno una struttura differente, in particolare riguardo alla disposizione delle due anse peptidiche conservate. Infatti, in una subunità dell’enzima, in presenza della forma attivata della tiamina pirofosfato:
l’ansa più interna è disposta in modo da bloccare l’ingresso al sito attivo;
l’ansa all’ingresso dell’altro sito attivo ha una conformazione tale da non bloccarne l’ingresso.
Questo spiega dal punto di vista strutturale le differenze osservate nella velocità di legame del substrato esibite dai due siti attivi. Una disposizione e una asimmetria simili sono state osservate anche in tutti gli altri enzimi tiamina pirofosfato dipendenti di cui sia nota la struttura.
Oltre alla tiamina pirofosfato e a uno ione magnesio (Mg2+), localizzati in ciascuno dei due siti attivi dell’enzima, un terzo Mg2+ è posizionato al centro del tetramero, all’interno di un tunnel lungo circa 20 Å, riempito di solvente, che collega i due siti attivi. Il tunnel è in gran parte delimitato da 10 residui amminoacidici conservati, rispettivamente sei residui di glutammato (Glu) e quattro di aspartato (Asp), che provengono da tutte e quattro le subunità, più altri residui acidi attorno all’anello amminopirimidinico della TPP. Va invece sottolineata l’assenza di residui basici che neutralizzino i precedenti. Tunnel simili sono stati osservati in tutti gli enzimi tiamina pirofosfato dipendenti con struttura dimerica o tetramerica di cui sia nota la struttura cristallina. Un esempio è la transchetolasi, enzima della via del pentoso fosfato.
Nota: Bacillus stearothermophilus è un membro del phylum Firmicutes ed è stato da poco rinominato Geobacillus stearothermophilus.
Qual’è la funzione del tunnel acido?
Attraverso esperimenti di mutagenesi condotti sulla piruvato deidrogenasi di B. stearothermophilus è stato dimostrato che il tunnel ha un ruolo nel meccanismo catalitico.
La sostituzione di alcuni dei suddetti residui acidi con amminoacidi neutri non alterava, rispetto alla forma wild-type:
l’efficienza dell’incorporazione dell’enzima modificato nel complesso multienzimatico;
la struttura dei siti attivi;
la struttura quaternaria dell’enzima.
Tuttavia, la velocità di decarbossilazione risultava ridotta di oltre il 70% rispetto all’enzima wild-type, come pure, una volta che l’enzima mutante era inserito nel complesso della piruvato deidrogenasi, l’attività del complesso stesso di oltre l’85%, sempre in confronto alla forma wild-type. Ma a che cosa sono dovute queste riduzioni dell’efficienza catalitica?
Poiché la distanza tra gli amminoacidi sostituiti e i siti attivi dell’enzima è di 7 o più Å, questi amminoacidi sono troppo lontani dai siti attivi per poterne influenzare direttamente l’attività catalitica. E’ stato quindi proposto il meccanismo di seguito descritto.
Considerando l’apoenzima, la TPP si lega velocemente e fortemente al primo sito attivo, viene attivata, e il sito attivo viene chiuso. In questo modo lo zwitterione tiazolico risulta protetto dall’ambiente esterno.
Nel secondo sito attivo invece la tiamina pirofosfato si lega, ma non viene attivata, e quindi il sito attivo rimane in conformazione aperta.
Nel primo sito attivo il piruvato reagisce con il C-2 tiazolico e la tiamina pirofosfato del secondo sito attivo, che è un acido generale, dona un protone al primo sito. Il risultato è una reazione di decarbossilazione nel primo sito attivo e l’attivazione del coenzima nel secondo sito, che quindi viene chiuso.
Si noti che mentre l’attivazione della prima tiamina pirofosfato è conseguente al legame al sito attivo, l’attivazione della seconda, e quindi del secondo sito attivo, è accoppiata alla decarbossilazione del piruvato nel primo sito attivo. O, da un altro punto di vista, mentre un sito attivo richiede un acido generale, l’altro richiede una base generale.
I protoni sono necessari per l’attività catalitica, e il loro trasferimento tra i siti attivi avviene attraverso il tunnel acido. Si verifica cioè il loro trasporto reversibile lungo una catena di gruppi accettori-donatori forniti dai residui di glutammato e aspartato e dalle molecole d’acqua presenti nel tunnel, che nell’insieme agiscono come un vero e proprio filo protonico, in inglese proton wire.
Sembra quindi che, a differenza di quanto accade in molti altri enzimi, in cui la comunicazione tra i siti attivi avviene attraverso modificazioni conformazionali e riarrangiamenti delle subunità che costituiscono l’enzima stesso, nella piruvato deidrogenasi e negli altri enzimi tiamina pirofosfato dipendenti il “proton wire” sia la base molecolare di tale comunicazione.
A questo punto l’oloenzima è formato e i siti attivi si trovano in un equilibrio dinamico, passando vicendevolmente dallo stato dormiente a quello attivato. Questo sembra essere lo stato in cui si trova l’enzima in vivo all’inizio di ogni ciclo catalitico.
Una conseguenza di un tale meccanismo è che, a mano a mano che i cicli catalitici si susseguono, i due siti attivi sono fuori fase tra di loro, ossia mentre uno richiede una base generale, l’altro necessita di un acido generale, e vice versa.
Da notare infine che un tale meccanismo permette anche il cambio di conformazione delle anse che chiudono i siti attivi, in modo da:
coordinare l’uptake dei substrati e il rilascio dei prodotti:
spiegare l’asimmetria esistente tra i due siti attivi.
Nota: un apoenzima è un enzima privo dei suoi cofattori, mentre un oloenzima è un enzima che lega tutti i suoi cofattori. L’apoenzima è la forma cataliticamente inattiva dell’enzima, mentre l’oloenzima ne è la forma cataliticamente attiva.
Struttura della diidrolipoil transacetilasi o E2
Nella struttura della diidrolipoil transacetilasi è possibile individuare tre domini funzionalmente distinti: un dominio lipoilico N-terminale, un dominio centrale di legame o PSBD, e un dominio catalitico C-terminale o acetiltransferasico.
Questi domini sono connessi da sequenze di circa 20-40 residui amminoacidici ricche in alanina e prolina, aminoacidi idrofobici che sono inframezzati da residui dotati di carica. Queste sequenze di collegamento sono altamente flessibili ed estese, il che permette di tenere lontani gli uni dagli altri i tre domini.
Domini di E2
Nota: analoghe sequenze di collegamento flessibili sono presenti anche in E3BP.
Il dominio lipoilico N-terminale, composto da circa 80 residui amminoacidici, è così chiamato in quanto lega l’acido lipoico. Il numero di questi domini dipende dalla specie considerata, variando da uno a tre. Ad esempio è presente con una copia in B. stearothermophilus e nei lieviti, con due nello Streptococcus faecalis e nei mammiferi, e con tre in Azotobacter vinelandii ed E. coli.
Il legame tra l’ɛ-amino gruppo della catena laterale di una lisina e l’acido lipoico porta alla formazione di un braccio flessibile, la lipoil-lisina o lipoammide, che alla sua massima estensione ha una lunghezza di circa 14 Å. Se a questo si aggiunge la sequenza che collega il dominio N-terminale al dominio adiacente, la cui lunghezza è superiore ai 140 Å, si ottiene un braccio flessibile in grado di far oscillare il gruppo lipoilico tra i siti attivi della piruvato deidrogenasi e della diidrolipoil deidrogenasi, nonché di interagire con le unità di diidrolipoil transacetilasi vicine. Si noti che il numero di queste braccia flessibili in E. coli è pari a 3 x 24 = 72, mentre nei mammiferi a 2 x 60 = 120, sulla base del numero dei domini N-terminali e delle unità di diidrolipoil transacetilasi.
Da tutto ciò consegue che una piruvato deidrogenasi può acetilare numerose diidrolipoil transacetilasi, e una diidrolipoil deidrogenasi può riossidare diversi gruppi diidrolipoamidici.
Inoltre, si verifica anche:
un interscambio di gruppi acetilici tra i gruppi lipoilici del nucleo di diidrolipoil transacetilasi;
lo scambio sia di gruppi acetilici che di disolfuri tra le braccia flessibili sopracitate.
PSBD è composto da circa 35 residui amminoacidici disposti a formare una struttura globulare che si lega sia alla piruvato deidrogenasi che alla diidrolipoil deidrogenasi, ossia tiene insieme il complesso multienzimatico.
Il dominio catalitico C-terminale, che, ovviamente, contiene il sito attivo, è composto da circa 250 residui amminoacidici. La disposizione dei residui aminoacidici è tale da formare una struttura simile a una gabbia vuota che contiene canali sufficientemente larghi da permettere ai substrati e prodotti di diffondere fuori e dentro. Ad esempio, la lipoamide e il coenzima A, i due substrati della diidrolipoil transacetilasi, si legano in conformazione estesa alle estremità opposte di un canale che si trova localizzato all’interfaccia tra ogni paio di subunità di ciascun trimero.
Struttura della diidrolipoil deidrogenasi o E3
La struttura della diidrolipoil deidrogenasi è stata desunta dalla studio dell’enzima in numerosi microrganismi.
Presenta una struttura omodimerica, con ciascuna catena polipeptidica, formata da circa 470 residui amminoacidici, ripiegata a dare quattro domini, dall’estremità N-terminale a quella C-terminale:
un dominio legante il FAD;
uno legante il NAD;
un dominio centrale;
un dominio di interfaccia.
Tutti prendono parte alla formazione del sito attivo.
Il FAD si trova quasi completamente all’interno della proteina. Ciò è dovuto al fatto che, a differenza del NADH o dei tioli, è facilmente ossidabile e quindi deve essere protetto dalla soluzione circostante, ossia dall’ossidazione ad opera di O2. E infatti, in assenza del NAD+, la catena laterale fenolica di uno specifico residuo di tirosina (Tyr), ad esempio nel batterio Gram-negativo Pseudomonas putida la Tyr181, va a coprire la tasca dove si va a legare la nicotinamide proteggendo il FADH2 dal contatto con la soluzione.
Quando invece il NAD+ è in posizione nel sito attivo, la catena laterale fenolica della suddetta tirosina si va a disporre tra l’anello nicotinamidico e quello flavinico.
Nel sito attivo della forma ossidata dell’enzima è presente anche un ponte disolfuro redox-attivo che si forma tra due residui di cisteina presenti in un segmento della catena polipeptidica altamente conservato, ad esempio in P. putida tra la Cys43 e la Cys48. Il disolfuro è localizzato dal lato dell’anello flavinico opposto rispetto a quello che guarda l’anello nicotinammidico, e lega due giri consecutivi di un segmento di un’α-elica distorta.
Da notare che in assenza della distorsione dell’ α-elica i Cα delle due cisteine sarebbero troppo distanti per poter permettere la formazione del ponte disolfuro.
La diidrolipoil deidrogenasi possiede quindi due accettori di elettroni: il FAD e il ponte disolfuro redox-attivo.
Nota: i due anelli eterociclici del NAD e del FAD sono paralleli e in contatto attraverso interazioni di van der Waals; anche S48 è in contatto di van der Waals con l’anello flavinico, dal lato opposto rispetto all’anello nicotinamidico.
Reazione della piruvato deidrogenasi o E1
La piruvato deidrogenasi, nella sequenza delle cinque reazioni catalizzate dai componenti del complesso della piruvato deidrogenasi, ne catalizza le prime due, ovvero:
la decarbossilazione del piruvato a dare CO2 e l’intermedio idrossietil-TPP;
l’acetilazione riduttiva del gruppo lipoilico della diidrolipoil transacetilasi.
La prima reazione è essenzialmente identica alla reazione catalizzata dalla piruvato decarbossilasi (EC 4.1.1.1), che opera una decarbossilazione non ossidativa nel corso della fermentazione del glucosio a etanolo. Quello che differisce è il destino del gruppo idrossietilico legato alla tiamina pirofosfato:
nella reazione catalizzata dalla piruvato deidrogenasi viene trasferito all’enzima successivo della sequenza, la diidrolipoil transacetilasi;
nella reazione catalizzata dalla piruvato decarbossilasi è convertito in acetaldeide.
Meccanismo di reazione della piruvato deidrogenasi o E1
Nelle reazioni catalizzate da enzimi TPP-dipendenti, l’anello tiazolico rappresenta la parte attiva, ma solo in forma di carbanione dipolare o ilide, ossia uno ione dipolare o zwitterione, con carica positiva sull’N-3 e carica negativa sul C-2. Di contro, l’anello tiazolico con carica positiva, ossia una carica positiva sull’azoto e nessuna carica sul C-2, può essere definito come la forma “dormiente” o inattiva.
La reazione ha inizio con l’attacco nucleofilo da parte del carbanione sul C-2 al carbonio carbonilico del piruvato, che ha lo stato di ossidazione di un’aldeide. L’attacco porta alla formazione di un legame covalente tra il coenzima e il piruvato.
Segue la scissione del legame tra il C-1 e il C-2 del piruvato. Questo porta al distacco del gruppo carbossilico, ossia del C-1, in forma di CO2, mentre i due restanti atomi di carbonio, il C-2 e il C-3, rimangono legati alla tiamina pirofosfato in forma di gruppo idrossietilico.
La scissione del legame C-1–C-2, e quindi la decarbossilazione del piruvato, è favorita dal fatto che la carica negativa sul C-2, che è instabile, viene stabilizzata grazie alla presenza nell’anello tiazolico dell’azoto imminico carico positivamente, N-3, (C=N+), ossia grazie alla presenza di una struttura elettrofila o elettron deficiente che funge da “pozzo” o “trappola” per gli elettroni, nella quale gli elettroni dei carbanioni possono delocalizzarsi per risonanza.
A questo punto l’intermedio stabilizzato per risonanza può essere protonato a dare idrossietil-TPP.
Nota: questo primo passaggio della reazione catalizzata dalla piruvato deidrogenasi è quello dove il complesso della privato deidrogenasi esercita la sua specificità di substrato. Inoltre è anche il più lento dell’intera sequenza, ossia è quello che limita la velocità della reazione complessiva.
Meccanismo Catalitico della Piruvato Deidrogenasi
Di seguito, l’enzima catalizza l’ossidazione del gruppo idrossietilico a gruppo acetilico e il suo trasferimento sul braccio lipoamidico della diidrolipoil transacetilasi.
La reazione ha inizio con la formazione di un carbanione sul carbonio idrossilico della idrossietil-TPP, a seguito della rimozione del protone legato al carbonio stesso da parte di una base dell’enzima.
Segue l’attacco nucleofilo del carbanione al disolfuro della lipoamide, con formazione di un legame acetil-tioestere ad alta energia con uno dei due tioli. In questa reazione l’ossidazione del gruppo idrossietilico a gruppo acetilico è accompagnata dalla concomitante riduzione del ponte disolfuro della lipoamide. I due elettroni rimossi dal gruppo idrossietilico sono utilizzati per ridurre il ponte disolfuro.
Questa reazione è quindi una acetilazione riduttiva, cui si accoppia la rigenerazione della forma attiva della piruvato deidrogenasi, ossia l’enzima con il C-2 dell’anello tiazolico nella forma deprotonata, la forma ilide o carbanione dipolare.
Si noti che l’energia derivante dall’ossidazione del gruppo idrossietilico a gruppo acetilico permette la formazione del legame tioestere tra il gruppo acetilico e il coenzima A.
Nota: il braccio di lipoil-lisina, presente in conformazione estesa nel canale dove si trova anche la TPP, permette il trasferimento dell’idrossietile dalla idrossietil-TPP al coenzima A. Dunque il braccio di lipoil-lisina si può spostare dal sito attivo della piruvato deidrogenasi a quelli della diidrolipoil transacetilasi e quindi della diidrolipoil deidrogenasi.
Proprietà della tiamina pirofosfato
Nella molecola della tiamina pirofosfato è possibile individuare tre parti distinte, da cui dipendono le sue proprietà chimiche ed enzimologiche: l’anello tiazolico, l’anello 4’-amminopirimidinico, e il gruppo difosfato.
Il difosfato lega il cofattore all’enzima attraverso la formazione di legami elettrostatici tra le cariche negative portate dai suoi gruppi fosforici e quelle positive degli ioni Ca2+ e Mg2+. I due ioni sono a loro volta legati a sequenze altamente conservate, rispettivamente GlyAspGly (GDG) per il Ca2+ e GlyAspGly-X26-AsnAsn (GDG-X26-NN) per l’Mg2+.
L’anello tiazolico ha un ruolo centrale nella catalisi grazie alla capacità di formare un carbanione, ossia un centro nucleofilo sul C-2.
Nota: una volta legatasi all’enzima, la TPP si dispone nel sito attivo in modo che l’anello tiazolico sia posizionato vicino all’ingresso del canale che porta al sito attivo.
L’anello amminopirimidinico ha una duplice funzione.
Ancora il coenzima tenendolo in posizione.
Ha uno specifico ruolo catalitico, partecipando a una catalisi acido/base. Questo è evidenziato da studi condotti utilizzando analoghi della tiamina pirofosfato in cui, a turno, erano stati sostituiti i tre atomi di azoto dell’anello. Gli studi hanno dimostrato che l’atomo N-1’ e il gruppo amminico legato a N-4’ sono necessari per l’attività del coenzima, mentre l’atomo N-3’ lo è meno.
Come si forma il carbanione dipolare della tiamina pirofosfato?
E’ possibile individuare tre forme tautomeriche in cui si presenta l’anello amminopirimidinico della tiamina pirofosfato legata allo specifico enzima ancora non impegnato nella reazione:
la forma canonica, ossia la 4’-amminopirimidina;
la forma protonata su N-1’, ossia lo ione 4’-amminopirimidinio;
l’1’,4’-imminopirimidina.
E sembra che la 1’,4’-imminopirimidina sia il tautomero che, prima dell’arrivo del substrato nel sito attivo, subisce la deprotonazione.
Il C-2 dell’anello tiazolico è assai più acido rispetto alla maggior parte degli altri gruppi =C-H presenti in altre molecole. La maggior acidità, ossia il fatto che il protone legato al C-2 sia facilmente dissociabile, è dovuta alla presenza dell’atomo di azoto quaternario con carica positiva dell’anello tiazolico che è in grado di stabilizzare elettrostaticamente il carbanione risultante.
Nel processo di deprotonazione sembra avere un ruolo essenziale il gruppo amminico dell’anello amminopirimidinico. Il gruppo funge infatti da base ed è posizionato opportunamente per accettare il protone.
Tuttavia, nella forma canonica, la 4’-amminopirimidina, uno dei suoi protoni collide stericamente con il protone legato al C-2. In aggiunta, anche il suo pK è troppo basso per operare la deprotonazione in modo efficiente. E’ stato quindi proposto un meccanismo in cui la catena laterale di un residuo conservato di glutammato, ad esempio βGlu59 in B. stearothermophilus, o Glu51 nella piruvato decarbossilasi di Saccharomyces uvarum, donando un protone all’amminopirimidina la converte nella sua forma immino tautomerica, la 1’,4’-imminopirimidina, che, accettando il protone dal C-2, torna alla forma canonica 4’-amminopirimidinica e permette la formazione del carbanione.
Nota: la formazione del carbanione sul C-2 è quindi conseguenza di un trasferimento protonico intramolecolare.
Deprotonazione della tiamina pirofosfato e chiusura del sito attivo
La perdita del protone dal C-2 dell’anello tiazolico porta, da una situazione con carica positiva sull’anello, alla formazione di uno ione dipolare o zwitterione.
Questo cambio nello stato di carica innesca una modificazione conformazionale in una delle due anse peptidiche conservate presenti all’ingresso del canale che immette al sito attivo, nello specifico la più interna, modificazione che, a sua volta, porta alla chiusura dello canale rispetto all’ambiente acquoso circostante. In questa conformazione chiusa il carbanione tiazolico è protetto dall’attacco di reagenti vari, come elettrofili.
Riassumendo, la deprotonazione della tiamina pirofosfato determina la chiusura del sito attivo e la protezione del carbanione dipolare neoformato; in altri termini, gli enzimi TPP-dipendenti sarebbero attivi solo nella conformazione chiusa.
Se invece si considera l’altro sito attivo, la sua tiamina pirofosfato non è in forma di ilide, il è canale aperto, e il sito stesso risulta inattivo.
Reazione della diidrolipoil transacetilasi o E2
La diidrolipoil transacetilasi catalizza la terza reazione della sequenza delle cinque catalizzate dai componenti del complesso della piruvato deidrogenasi. La reazione comporta il trasferimento del gruppo acetilico dall’acetil-diidrolipoammide al coenzima A, a dare acetil-CoA e la forma completamente ridotta della lipoammide, la diidrolipoammide, il ditiolo.
Va notato che il gruppo acetilico, inizialmente legato tramite legame estere a uno dei gruppi –SH della lipoammide è di seguito legato al gruppo –SH del coenzima A, di nuovo attraverso legame estere, da cui il termine di transesterificazione.
Meccanismo di reazione della diidrolipoil transacetilasi o E2
Nel corso della reazione, il gruppo sulfidrilico del coenzima A porta un attacco nucleofilo al carbonio carbonilico del gruppo acetilico dell’acetil diidrolipoammide-diidrolipoil transacetilasi a formare un intermedio tetraedrico transiente che si “decompone” a dare acetil-CoA e diidrolipoammide-diidrolipoil transacetilasi.
Meccanismo Catalitico della Diidrolipoil Transacetilasi
Come già detto, nel meccanismo di reazione gioca un ruolo centrale la mobilità della lipoammide.
Reazione della diidrolipoil deidrogenasi o E3
La diidrolipoil deidrogenasi catalizza la quarta e la quinta reazione della sequenza delle cinque catalizzate dai componenti del complesso della piruvato deidrogenasi.
L’enzima catalizza trasferimenti di elettroni necessari per rigenerare il ponte disolfuro della lipoammide della diidrolipoil transacetilasi, ossia per rigenerare la forma ossidata del gruppo prostetico e completare così il ciclo catalitico della transacetilasi.
La reazione segue un meccanismo a ping-pong, procedendo attraverso due semireazioni consecutive nelle quali i due substrati, la diidrolipoammide e il NAD+, reagiscono uno in assenza dell’altro. Inoltre durante la prima semireazione, il rilascio del primo prodotto e la formazione di un complesso enzima-intermedio si verificano prima che il secondo substrato si leghi, mentre l’enzima va incontro a un cambio strutturale. Nella seconda semireazione si verificano il rilascio del secondo prodotto e il ritorno dell’enzima allo stato iniziale, di nuovo attraverso una modificazione strutturale.
Considerando la cinetica del meccanismo a ping-pong della diidrolipoil deidrogenasi:
nella prima semireazione si verifica l’ossidazione della diidrolipoamide a lipoammide;
nella seconda semireazione si verifica la riduzione del NAD+ a NADH.
Meccanismo di reazione della diidrolipoil deidrogenasi o E3
Di seguito viene descritto il meccanismo di reazione della diidrolipoil deidrogenasi di P. putida.
Nella prima semireazione l’enzima in forma ossidata (E), ossia con il ponte disolfuro tra la Cys43 e la Cys48, lega la diidrolipoammide (LH2) a formare il complesso enzima-diidrolipoammide (E●LH2). A questo punto un atomo di zolfo della diidrolipoammide porta un attacco nucleofilo allo zolfo della Cys43, con formazione di un ponte disolfuro lipoammide-Cys43, (E–S–S–L), mentre lo zolfo della Cys48 viene rilasciato in forma di ione tiolato (S–48).
Il protone che è rimasto sul secondo gruppo tiolico della lipoammide è quindi estratto ad opera dell’istidina (Hys) 451. L’aminoacido agisce come catalizzatore acido-base generale, il che porta alla formazione di un secondo ione tiolato, stavolta sulla lipoamide (E–S–S–L●S–), il quale, attraverso un attacco nucleofilo, va a sostituire lo zolfo della Cys43, S43, aiutato in questo attacco dalla catalisi acida generale ad opera della Hys451 che dona un protone a S43. L’azione catalitica della Hys451 è essenziale, come dimostrato da studi di mutagenesi in cui la sua sostituzione con un residuo di glutammina fa si che l’enzima mantenga solamente circa lo 0,4% dell’attività catalitica rispetto alla forma wild-type.
L’anione tiolato S–48 entra quindi in contatto, mediante interazioni non covalenti, con l’anello flavinico in vicinanza della sua posizione 4a, ossia una coppia di elettroni di S–48, che funge da donatore di elettroni, è parzialmente trasferita all’anello flavinico ossidato, che a sua volta funge da accettore di elettroni. La struttura derivante è chiamata complesso a trasferimento di carica.
Nel frattempo la catena laterale fenolica della Tyr181 continua a bloccare l’accesso all’anello flavinico, proteggendolo così dall’ossidazione da parte di O2.
Ossidazione della Diidrolipoammide da Parte di E3
Quello che si verifica è quindi una reazione di interscambio di ponti disolfuro. I prodotti sono la forma ossidata della lipoammide, il primo prodotto, che è rilasciato, e la forma ridotta della diidrolipoil deidrogenasi.
La seconda semireazione comporta la riduzione del NAD+ a NADH + H+ mediante il trasferimento di elettroni dal disolfuro reattivo dell’enzima via FAD.
Ossidazione di E3 Ridotta
Il tutto ha inizio con l’ingresso nel sito attivo del NAD+ e il suo legame a formare il complesso EH2●NAD+. Si noti che l’ingresso del coenzima determina lo spostamento a lato della catena laterale fenolica della Tyr181 ad opera dell’anello nicotinammidico.
A seguito del collasso del complesso a trasferimento di carica si forma un legame covalente tra il C-4a dell’anello flavinico e S48. A ciò si accompagna l’estrazione di un protone da parte dell’N-5 flavinico, con formazione del corrispondente anione tiolato S–43.
S–43 porta un attacco nucleofilo a S48. L’attacco determina la formazione del ponte disolfuro redox-attivo tra la Cys43 e la Cys48, cui segue la rottura del legame covalente tra S48 e il C-4a dell’anello flavinico a dare l’anione FADH–, con carica negativa su N-1. Si noti che la diidrolipoil deidrogenasi è nuovamente nella forma ossidata (E).
Il FADH– ha un’esistenza transitoria in quanto si verifica il trasferimento praticamente immediato del protone legato a N-5, in forma di ione idruro, al C-4 dell’anello nicotinamidico, che è giustapposto all’N-5 flavinico. Questo porta alla formazione di FAD e del secondo prodotto della reazione, il NADH, che lascia l’enzima.
In definitiva gli elettroni che sono stati rimossi dal gruppo idrossietilico derivato dal piruvato passano, via FAD, al NAD+. In questo modo si è completato il ciclo catalitico della diidrolipoil deidrogenasi, essendo l’enzima e il suo coenzima nella loro forma ossidata.
A questo punto anche il ciclo catalitico dell’intero complesso della piruvato deidrogenasi si è completato, e il complesso stesso è pronto per un altro ciclo di reazioni.
Nota: a differenza dell’anello tiazolico della tiamina pirofosfato, il FAD non ha la funzione di “trappola” o “pozzo” per gli elettroni quanto piuttosto di condotta per gli elettroni tra il disolfuro redox-attivo, nella sua forma ridotta, e il NAD+.
Il meccanismo catalitico della diidrolipoil deidrogenasi è stato in gran parte determinato in analogia con quello della glutatione reduttasi (EC 1.8.1.7), al 33% identica ma strutturalmente molto meglio caratterizzata. Va tuttavia notato che, sebbene i due enzimi catalizzino reazioni simili, queste normalmente corrono in direzione opposta:
la diidrolipoil deidrogenasi utilizza il NAD+ per ossidare due gruppi –SH a dare un disolfuro (–S–S–);
la glutatione reduttasi utilizza il NADPH per ridurre un –S–S– a due gruppi tiolici.
Nonostante ciò, i siti attivi dei due enzimi sono strettamente sovrapponibili.
Regolazione dell’attività del complesso della piruvato deidrogenasi
Nei mammiferi, la regolazione dell’attività del complesso della piruvato deidrogenasi è essenziale, sia nello stato di digiuno che in quello alimentato. Infatti, il complesso multienzimatico svolge un ruolo centrale nel metabolismo in quanto, catalizzando la decarbossilazione ossidativa irreversibile del piruvato, rappresenta la porta di ingresso dello scheletro carbonioso dei carboidrati e di circa il 50% dello scheletro carbonioso degli amminoacidi glucogenici, che nell’insieme costituiscono circa il 60% dell’apporto calorico giornaliero, verso:
il ciclo dell’acido citrico, e quindi alla completa ossidazione a CO2;
la sintesi degli lipidi, nello stato alimentato, e dell’acetiolcolina.
L’importanza della regolazione della conversione del piruvato in acetil-CoA è sottolineata anche dal fatto che i mammiferi, sebbene siano in grado di produrre glucosio dal piruvato, non sono in grado di farlo dall’acetil-CoA. Ciò è conseguenza sia della irreversibilità della reazione catalizzata dalla piruvato deidrogenasi che dell’assenza di vie metaboliche alternative in grado di farlo.
L’inibizione dell’attività del complesso permetterà quindi di risparmiare glucosio e gli aminoacidi che possono essere convertiti in piruvato, come l’alanina, quando sono disponibili altri carburanti, ad esempio l’acetil-CoA derivante dall’ossidazione degli acidi grassi.
Per questi motivi l’attività del complesso è finemente regolata attraverso:
inibizione a feed-back, anche nota come inibizione da prodotto finale;
nucleotidi;
modificazioni covalenti, ossia fosforilazioni e defosforilazioni di specifiche proteine bersaglio.
Regolazione attraverso inibizione da prodotto finale e stato energetico della cellula
L’attività della forma defosforilata del complesso della piruvato deidrogenasi è regolata attraverso inibizione a feed-back o inibizione da prodotto finale.
Acetil-CoA e NADH inibiscono allostericamente gli enzimi che ne catalizzano la sintesi, rispettivamente diidrolipoil transacetilasi e la diidrolipoil deidrogenasi.
Inoltre, il CoA e l’acetil-CoA, così come il NAD+ e il NADH, competono per i siti di legame sui rispettivi enzimi, i quali catalizzano reazioni reversibili.
Questo significa che, in presenza di elevati valori dei rapporti [NADH]/[NAD+] e [Acetil-CoA]/[CoA], le reazioni di transacetilazione e deidrogenazione vanno nella direzione opposta rispetto a quella della formazione dell’acetil-CoA. Di conseguenza la diidrolipoil transacetilasi non può accettare il gruppo idrossietilico dalla TPP in quanto è mantenuta nella forma acetilata. Questo fa si che la tiamina pirofosfato rimanga legata alla piruvato deidrogenasi nella sua forma idrossietilica. Questo a sua volta riduce la velocità di decarbossilazione del piruvato. Quindi, elevati valori dei rapporti [NADH]/[NAD+] e [Acetil-CoA]/[CoA] influenzano indirettamente l’attività della piruvato deidrogenasi.
PDC: Inibizione a Feed-back
Acetil-CoA e NADH sono prodotti anche durante l’ossidazione degli acidi grassi. Anche questa via metabolica che, al pari delle reazioni catalizzate dal complesso della piruvato deidrogenasi, si verifica nel mitocondrio.
Ciò significa che la cellula, attraverso la regolazione dell’attività del complesso multienzimatico, è in grado di preservare le riserve di carboidrati quando sono disponibili acidi grassi per la produzione di energia. Questo per esempio è quanto accadde nel digiuno, quando il fegato, il muscolo scheletrico e molti altri organi e tessuti si affidano principalmente all’ossidazione degli acidi grassi per la produzione di energia.
Di contro, l’attività del complesso è aumentata nello stato alimentato. In questa condizione molti differenti tipi di cellule e tessuti utilizzano in prevalenza il glucosio come fonte di energia.
Più in generale, quando la produzione di NADH e/o acetil-CoA supera la capacità della cellula di utilizzarli per la produzione di ATP, l’attività del complesso della piruvato deidrogenasi è inibita.
Analogo discorso vale nella condizione in cui non ci sia necessità di produrre ulteriore ATP. Infatti l’attività catalitica del complesso multienzimatico è sensibile anche allo stato energetico della cellula. Attraverso meccanismi allosterici:
livelli di ATP elevati inibiscono l’attività della componente piruvato deidrogenasi del complesso multienzimatico;
elevati livelli di ADP, che segnalano che la cellula potrebbe entrare in una fase di carenza di energia, lo attivano, indirizzando quindi lo scheletro carbonioso dei carboidrati e di alcuni aminoacidi verso la produzione di energia.
Nota: nel muscolo scheletrico l’attività del complesso della piruvato deidrogenasi aumenta con l’aumento dell’attività aerobica. Questo si traduce in una maggiore dipendenza del muscolo dal glucosio come fonte di energia.
Regolazione attraverso fosforilazione/defosforilazione
A differenza di quanto accade nei procarioti, nei mammiferi l’attività del complesso della piruvato deidrogenasi è regolata anche attraverso modificazioni covalenti. Nello specifico si tratta di fosforilazioni e defosforilazioni di tre specifici residui di serina presenti sulla subunità α della piruvato deidrogenasi, l’enzima che catalizza il primo passaggio, irreversibile, dell’intera sequenza di reazioni.
Nota: poiché la piruvato deidrogenasi dei mammiferi è un eterotetramero, ci sono sei potenziali siti di fosforilazione.
PDC: Regolazione Mediante Modificazione Covalente
La fosforilazione, che inattiva la piruvato deidrogenasi, e quindi blocca l’intera sequenza di reazioni, è catalizzata dalla piruvato deidrogenasi chinasi. Due delle suddette serine si trovano su una delle due anse presenti all’ingresso del canale per il substrato che porta al rispettivo sito attivo, quella più vicina all’estremità C-terminale. La fosforilazione di uno solo di questi residui inattiva la piruvato deidrogenasi, dimostrando così la l’accoppiamento fuori fase tra i due siti attivi.
Nello stato defosforilato invece il complesso risulta attivo. La defosforilazione è catalizzata da una specifica protein fosfatasi, la piruvato deidrogenasi fosfatasi.
Le attività della piruvato deidrogenasi chinasi e della piruvato deidrogenasi fosfatasi sono a loro volta soggette a regolazione allosterica da parte di numero effettori.
Regolazione della piruvato deidrogenasi chinasi
L’attività della piruvato deidrogenasi chinasi dipende dal valore dei rapporti [NADH]/[NAD+], [acetil-CoA]/[CoA], e [ATP]/[ADP], come anche dalla concentrazione del piruvato, nella matrice mitocondriale.
Elevati valori dei rapporti [NADH]/[NAD+] e [acetil-CoA]/[CoA], come nel corso dell’ ossidazione degli acidi grassi e dei corpi chetonici, attivano la chinasi. La piruvato deidrogenasi viene quindi fosforilata, e il complesso multienzimatico risulta inibito. Questo permette ai tessuti, come ad esempio il muscolo cardiaco, di risparmiare glucosio quando si stanno utilizzando acidi grassi e/o corpi chetonici per la produzione di energia, in quanto la sintesi di acetil-CoA dal piruvato, e quindi dai carboidrati (e da alcuni aminoacidi) e bloccata. Quando invece le concentrazioni di NAD+ e coenzima A sono elevate, l’attività catalitica della chinasi è inibita e il complesso risulta attivo. Quindi, acetil-CoA e NADH, due dei tre prodotti finali delle reazione catalizzate dal complesso della piruvato deidrogenasi, controllano allostericamente la propria sintesi regolando direttamente e indirettamente, tramite la regolazione dell’attività della piruvato deidrogenasi chinasi, l’attività del complesso multienzimatico.
Elevati valori del rapporto [ATP]/[ADP] attivano la chinasi, e quindi inibiscono il complesso multienzimatico. Nota: a differenza di molte altre chinasi, come quelle che intervengono nel controllo del metabolismo del glicogeno, la piruvato deidrogenasi chinasi non è regolata dai livelli di cAMP, bensì da molecole che segnalano variazioni nel livello della carica energetica della cellula e nella disponibilità di intermedi biosintetici, ossia rispettivamente ATP e NADH e acetil-CoA.
Il piruvato è un effettore allosterico negativo della piruvato deidrogenasi chinasi. Quando i suoi livelli sono elevati, il suo legame alla chinasi la inattiva, la piruvato deidrogenasi non viene fosforilata, e il complesso della piruvato deidrogenasi rimane attivo.
La piruvato deidrogenasi chinasi è attivata anche a seguito dell’interazione con la diidrolipoil transacetilasi nella sua forma acetilata, ossia quando è presente la acetil-diidrolipoamide.
Altri attivatori della piruvato deidrogenasi chinasi sono gli ioni potassio e magnesio.
Regolazione della piruvato deidrogenasi fosfatasi
L’attività della piruvato deidrogenasi fosfatasi dipende dal valore dei rapporti [NADH]/[NAD+] e [acetil-CoA]/[CoA], come anche dalla [Ca2+], nella matrice mitocondriale.
Bassi valori dei rapporti [NADH]/[NAD+] e [acetil-CoA]/[CoA] attivano la fosfatasi, la piruvato deidrogenasi viene defosforilata, e il complesso multienzimatico risulta attivo.
Al contrario, in presenza di elevati valori dei suddetti rapporti l’attività della fosfatasi si riduce, quella della chinasi aumenta, e il complesso multienzimatico viene inibito.
Lo ione calcio attiva la piruvato deidrogenasi fosfatasi.
Ca2+ è un importante secondo messaggero che segnala la necessità da parte della cellula di ulteriore energia. Quindi, quando è presente in elevate concentrazioni, come nelle cellule muscolari cardiache a seguito della stimolazione da parte dell’adrenalina, o nella cellule muscolari scheletriche nel corso della contrazione muscolare, la fosfatasi è attiva, il complesso è defosforilato, e quindi attivato.
Anche l’insulina interviene nel controllo dell’attività catalitica del complesso della piruvato deidrogenasi attraverso l’attivazione della piruvato deidrogenasi fosfatasi. L’ormone, in risposta all’aumento della glicemia, stimola sia la sintesi del glicogeno che dell’acetil-CoA, precursore nella sintesi degli lipidi.
Anche il digiuno e la successiva rialimentazione influiscono sull’attività del complesso multienzimatico.
In tessuti come il muscolo scheletrico, il muscolo cardiaco o il rene, il digiuno riduce in modo significativo l’attività del complesso, mentre la rialimentazione inverte la situazione.
Nel cervello invece non si osservano queste variazioni poiché l’attività del complesso della piruvato deidrogenasi è essenziale per la produzione di ATP.
Bibliografia
Frank R.A.W., Titman C.M., Pratap J.V., Luisi B.F., and Perham R.N. A molecular switch and proton wire synchronize the active sites in thiamine enzymes. Science 2004;306(5697):872-876. doi:10.1126/science.1101030
Gray L.R., Tompkins S.C., Taylor E.R. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-2604. doi:10.1007/s00018-013-1539-2
McCommis K.S. and Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J 2015;466(3):443-454. doi:10.1042/BJ20141171
Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
Nemeria N.S., Chakraborty S., Balakrishnan A., and Frank Jordan. Reaction mechanisms of thiamin diphosphate enzymes: defining states of ionization and tautomerization of the cofactor at individual steps. FEBS J 2009;276:2432-2446. doi:10.1111/j.1742-4658.2009.06964.x
Patel M.S. and Korotchkina L.G. Regulation of the pyruvate dehydrogenase complex. Biochem Soc T 2006;34(2):217-222. doi:10.1042/bst0340217
Patel M.S., Nemeria N.S., Furey W., and Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem 2014;289(24):16615-16623. doi:10.1074/jbc.R114.563148
Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009
Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011
Wang J., Nemeria N.S., Chandrasekhar K., Kumaran S., Arjunan P., Reynolds S., Calero G., Brukh R., Kakalis L., Furey W., and Jordan F. Structure and function of the catalytic domain of the dihydrolipoyl acetyltransferase component in Escherichia coli pyruvate dehydrogenase complex. J Biol Chem 2014;289(22):15215-15230. doi:10.1074/jbc.M113.544080
Zhou Z.H., McCarthy D.B., O’Connor C.M., Reed L.J., and J.K. Stoops. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA 2001;98(26):14802-14807. doi:10.1073/pnas.011597698
La via del pentoso fosfato, anche detta via del fosfogluconato, è una via metabolica, comune a tutti gli organismi viventi, per il metabolismo ossidativo del glucosio, alternativa alla glicolisi da cui si ramifica a livello del glucosio-6-fosfato, e le cui funzioni principali sono la produzione, in rapporti variabili, di NADPH, un coenzima ridotto, e ribosio-5-fosfato, uno zucchero fosforilato a cinque atomi di carbonio, ossia un pentoso fosfato, da cui il nome della via.[14]
La via del fosfogluconato, ramificandosi dalla glicolisi, è anche detta shunt dell’esoso monofosfato, dove shunt può essere tradotto come derivazione o smistamento.
Dal punto di vista concettuale, nella via del pentoso fosfato è possibile individuare due fasi: la fase ossidativa, nel corso della quale viene prodotto il NADPH, e la fase non ossidativa, nella quale si formano il ribosio-5-fosfato e altri zuccheri fosforilati.[16]
Via del Pentoso Fosfato
E’ stato stimato che più del 10% del glucosio metabolizzato è trasportato attraverso questa via metabolica che, degno di nota, pur ossidando il monosaccaride non comporta alcuna produzione diretta, ma neppure consumo, di ATP.[7][13]
Le prime evidenze dell’esistenza della via del fosfogluconato emersero negli anni trenta del secolo scorso dagli studi di Otto Warburg, premio Nobel per la Fisiologia o la Medicina nel 1931, che scoprì il NADP durante studi sull’ossidazione del glucosio-6-fosfato a 6-fosfogluconato.[22]
Ulteriori indicazioni emersero dall’osservazione che nei tessuti il glucosio continuava a essere metabolizzato anche in presenza di inibitori della glicolisi, quali gli ioni fluoruro e iodoacetato, inibitori rispettivamente della enolasi (EC 4.2.1.11) e della gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12).
Tuttavia la delucidazione completa della via avvenne solo negli anni cinquanta del secolo scorso grazie al lavoro di diversi ricercatori e principalmente dei gruppi di Efraim Racker, Fritz Lipmann, vincitore del premio Nobel per la Fisiologia o la Medicina nel 1953 anche grazie alla scoperta del coenzima A, Bernard Horecker e Frank Dickens.[8]
Funzione principale
La principale funzione della via del pentoso fosfato è la produzione di NADPH e ribosio-5-fosfato.[22]
Il NADPH è necessario per le biosintesi riduttive, quali la sintesi degli acidi grassi, del colesterolo, degli ormoni steroidei e degli aminoacidi non essenziali prolina e tirosina, rispettivamente dal glutammato e dalla fenilalanina, nonché per la riduzione del glutatione ossidato.[14] Nelle suddette reazioni il coenzima ridotto funge da donatore di elettroni, o meglio da donatore di uno ione idruro (:H–), equivalente a un protone e due elettroni.
Nicotinammide Adenina Dinucleotide Fosfato
Nota: nei vertebrati circa la metà del NADPH necessario per i passaggi riduttivi della sintesi degli acidi grassi deriva dalla via del pentoso fosfato, mentre la restante parte dalla reazione catalizzata dall’enzima malico (EC 1.1.1.40), di cui di seguito è indicata la reazione.[13]
Malato + NADP+ ⇄ Piruvato + NADPH + H++ HCO3–
Il ribosio-5-fosfato viene utilizzato nella sintesi dei nucleotidi e degli acidi nucleici, DNA e RNA, dell’ATP, di coenzimi come il coenzima A, il NAD, il NADP e il FAD, e degli aminoacidi essenziali triptofano e istidina.[5] Lo zucchero fosforilato non viene utilizzato direttamente come tale ma previa conversione, attivazione, a 5-fosforibosil-1-pirofosfato o PRPP, acronimo dell’inglese 5-phosphoribosyl 1-pyrophosphate, nella reazione catalizzata dalla ribosio fosfato pirofosfochinasi o PRPP sintetasi (EC 2.7.6.1).[13]
Ribosio-5-fosfato + ATP → 5-Fosforibosil-1-pirofosfato + AMP
Funzioni aggiuntive
In aggiunta alla produzione di NADPH e ribosio-5-fosfato, la via del pentoso fosfato ha altre funzioni, sia anaboliche che cataboliche.[18]
In molti batteri e nei lieviti è coinvolta nel catabolismo degli zuccheri a cinque atomi di carbonio ribosio, xilosio e arabinosio.
Anche nell’uomo questa è coinvolta nel catabolismo dei suddetti pentosi e dei meno comuni carboidrati a tre, quattro e sette atomi di carbonio assunti con la dieta, nonché per il catabolismo dei:
pentosi derivanti dal catabolismo dei carboidrati strutturali;
ribosio-5-fosfato derivante dal catabolismo dei nucleotidi.
Negli organismi fotosintetici contribuisce all’incorporazione dell’anidride carbonica (CO2) nella molecola del glucosio nel corso del ciclo di Calvin.[18]
Porta alla sintesi oltre che di ribosio-5-fosfato, anche di altri intermedi utilizzati in vari processi biosintetici come:
eritrosio-4-fosfato, utilizzato per la sintesi dei tre aminoacidi aromatici triptofano, tirosina e fenilalanina;
ribulosio-5-fosfato, utilizzato per la sintesi della riboflavina;
sedoeptulosio-7-fosfato che, nei batteri Gram-negativi, è utilizzato per la sintesi delle unità di eptosio dello strato lipopolisaccaridico della membrana esterna.[19]
Dove avviene la via del pentoso fosfato?
Nelle cellule animali la via del pentoso fosfato, al pari della glicolisi, della sintesi degli acidi grassi, e della maggior parte delle reazioni della gluconeogenesi, avviene nel citosol, analogamente a quanto accade nei batteri.[5] E, considerando glicolisi, gluconeogenesi e via del pentoso fosfato è possibile affermare che queste tre vie sono interconnesse tramite diversi enzimi e/o intermedi condivisi.
Nelle cellule vegetali la via del fosfogluconato avviene nei plastidi, e i suoi intermedi possono raggiungere il citosol attraverso i pori di membrana di questi organelli.[13]
Nell’uomo, i livelli di espressione degli enzimi della via variano da tessuto a tessuto. Livelli relativamente elevati si ritrovano nel fegato, nella corteccia surrenale, nei testicoli, nelle ovaie, nella tiroide, nelle ghiandole mammarie durante l’allattamento e nei globuli rossi.[14] In tutte queste sedi è richiesto un apporto continuo di NADPH per sostenere le biosintesi riduttive e/o contrastare gli effetti dei radicali liberi dell’ossigeno su strutture cellulari sensibili come il DNA, i lipidi di membrana e le proteine.[1]
Elevati livelli degli enzimi di questa via metabolica sono presenti anche in cellule in rapida divisione quali quelle dell’embrione nelle prime fasi di sviluppo, gli enterociti, le cellule della pelle e del midollo osseo e, in condizioni patologiche, le cellule tumorali, tutte cellule che richiedono elevate quantità di ribosio-5-fosfato per la sintesi dei nucleotidi.[16]
Di contro nel muscolo scheletrico sono presenti livelli estremamente bassi degli enzimi della via del pentoso fosfato, via che in questa sede è praticamente assente e dove il glucosio-6-fosfato è utilizzato principalmente per la produzione di energia attraverso la glicolisi e il ciclo dell’acido citrico.[5]
Fase ossidativa
La fase ossidativa della via del pentoso fosfatosi compone di tre tappe, due ossidazioni irreversibili, rispettivamente la prima e la terza reazione, e una idrolisi.
In questa fase una molecola di glucosio-6-fosfato viene convertita in una di ribulosio-5-fosfato, uno zucchero fosforilato a 5 atomi di carbonio e substrato di partenza per le reazioni della fase non ossidativa, con la concomitante produzione di due molecole di NADPH e la liberazione del C-1 del glucosio in forma di CO2.[7] L’equazione complessiva è:
Ossidazione del glucosio-6-fosfato a 6-fosfoglucono-delta-lattone
Nella prima tappa della fase ossidativa della via del pentoso fosfato, il glucosio-6-fosfato viene ossidato a 6-fosfoglucono-δ-lattone, un estere intramolecolare, reazione catalizzata dalla glucosio-6-fosfato deidrogenasi.
Nella reazione il NADP+ funge da agente riducente accettando uno ione idruro dal carbonio 1 del glucosio-6-fosfato, e riducendosi quindi a NADPH.
Si noti che questa è la prima molecola di NADPH prodotta nella via del pentoso fosfato.
La reazione catalizzata dalla glucosio-6-fosfato deidrogenasi o G6PD (EC 1.1.1.49) è una reazione “esclusiva” della via del pentoso fosfato.[1] Analogamente a quanto accade nella maggior parte delle altre vie metaboliche, anche in questo caso, la prima reazione esclusiva della via è essenzialmente irreversibile, con un ΔG nel fegato pari a -4,21 kcal/mol (-17,6 kJ/mol), e altamente regolata per via allosterica.[22] L’enzima infatti rappresenta il principale punto di controllo del flusso di metaboliti attraverso la via stessa.
Nell’uomo i livelli più elevati di G6PD sono presenti nei neutrofili e macrofagi, cellule fagocitarie in cui, durante l’infiammazione, il NADPH viene utilizzato per la produzione di radicali superossido (O2–.) dall’ossigeno molecolare nella reazione catalizzata dalla NADPH ossidasi (EC 1.6.3.1 ).[2][16]
2 O2 + NADPH → 2 O2–. + NADP+ + H+
Di seguito i radicali superossido potranno essere utilizzati per la sintesi, a scopi difensivi, ossia l’uccisione dei microrganismi fagocitati, di altri ROS come anche di radicali liberi dell’azoto o RNS, acronimo dell’inglese reactive nitrogen species, quali:
il perossido di idrogeno o acqua ossigenata (H2O2), nella reazione catalizzata dalla superossido dismutasi o SOD (EC 1.15.1.1)
2 O2–. + 2 H+ → H2O2 + O2
il perossinitrito (O=N–O–O–), a seguito della reazione con il monossido di azoto (•NO)
O2–. + •NO → O=N–O–O–
il radicale idroperossido (HOO•)
O2–. + H+ → HOO•
Meccanismo catalitico della glucosio-6-fosfato deidrogenasi
Il meccanismo catalitico dell’enzima è stato ampiamente studiato nel microorganismo Leuconostoc mesenteroides, la cui glucosio-6-fosfato deidrogenasi ha la peculiare caratteristica di poter utilizzare come coenzima il NAD+ e/o il NADP+.[12]
Meccanismo Catalitico della Glucosio-6-fosfato Deidrogenasi
L’enzima non necessita della presenza di ioni metallici per la propria attività; uno dei residui presenti nel sito attivo funge da base generale, ossia è in grado di estrarre uno ione idruro dal gruppo ossidrilico legato al C-1 del glucosio-6-fosfato.[4]
Nell’enzima batterico questo è effettuato dall’atomo Nɛ2 dell’anello imidazolico della catena laterale di un residuo di istidina, azoto che presenta un doppietto elettronico disponibile portare un l’attacco nucleofilo. Questa fa si che il glucosio-6-fosfato, un emiacetale ciclico con il C-1 nello stato di ossidazione aldeidico, sia ossidato a estere ciclico, ossia un lattone. Ciò consente il trasferimento dello ione idruro legato al C-1 del glucosio sul C-4 dell’anello nicotinamidico del NADP+ a dare NADPH.
Poiché l’istidina suddetta è conservata in molte delle glucosio-6-fosfato deidrogenasi sequenziate, è probabile che questo meccanismo catalitico sia generalizzabile a tutte le glucosio-6-fosfato deidrogenasi.
Regolazione della glucosio-6-fosfato deidrogenasi
La glucosio-6-fosfato deidrogenasi rappresenta l’enzima chiave nella regolazione del flusso metabolico attraverso la via del pentoso fosfato, e quindi il principale punto di controllo della velocità di produzione del NADPH.[2]
Nell’uomo l’enzima è presente in due forme: quella monomerica, inattiva, e in un equilibrio tra una forma dimerica e una tetramerica, le conformazioni attive.[1]
Uno dei principali modulatori della sua attività è il rapporto citosolico NADP+/NADPH.[15] Elevati livelli di NADPH inibiscono l’enzima poiché il coenzima ridotto è un potente inibitore competitivo della G6PD, mentre il NADP+ è necessario sia per la sua attività catalitica che per il mantenimento della conformazione attiva. Infatti il legame del coenzima ossidato a un sito specifico, distante dal sito attivo ma vicino all’interfaccia del dimero, è necessario per mantenerne la conformazione dimerica.
Regolazione dell’Attività della G6PD
Nella maggior parte delle condizioni metaboliche il rapporto NADP+/NADPH è basso, meno NADP+ è disponibile per legarsi all’enzima e di conseguenza si riduce l’attività dell’enzima stesso, a prescindere dai livelli della sua espressione genica. In queste condizioni la fase ossidativa è praticamente inattiva.
Se invece si considerano cellule in cui siano particolarmente attive vie metaboliche e/o reazioni che utilizzano NADPH, si verifica la riduzione della sua concentrazione citosolica, un aumento di quella del NADP+ e quindi una stimolazione dell’attività dell’enzima. Ne consegue un’attivazione della fase ossidativa della via dell’esoso monofosfato.
Quindi è anche possibile affermare che il destino del glucosio-6-fosfato, un intermedio comune alla glicolisi e alla via del fosfogluconato, dipende anche dal fabbisogno contingente di NADPH.[15]
Un secondo meccanismo di regolazione dell’attività della glucosio-6-fosfato deidrogenasi chiama in causa l’accumulo di acil-CoA, intermedi nella sintesi degli acidi grassi.[5] Queste molecole, legandosi alla forma dimerica dell’enzima ne causano la dissociazione nei due monomeri costitutivi, e quindi la perdita dell’attività catalitica.
L’insulina up-regola l’espressione del gene per la glucosio-6-fosfato deidrogenasi e per la 6-fosfogluconato deidrogenasi, per cui l’ormone, nello stato ben alimentato, concorre ad aumentare il flusso di carbonio attraverso la via del pentoso fosfato, promuovendo quindi la sintesi di NADPH.[7]
Nota: l’insulina promuove anche la sintesi degli acidi grassi.
Idrolisi del 6-fosfoglucone-delta-lattone a 6-fosfogluconato
Nella seconda tappa della fase ossidativa si verifica l’idrolisi del 6-fosfoglucone-δ-lattone a dare il 6-fosfogluconato, una molecola lineare. Il 6-fosfoglucone-δ-lattone è di per se idroliticamente instabile e va incontro a una idrolisi spontanea che apre l’anello, reazione che avviene a una velocità significativa.[22] Tuttavia nella cellula la reazione è accelerata dall’azione catalitica di una specifica lattonasi (EC 3.1.1.31).[3]
Decarbossilazione ossidativa del 6-fosfogluconato a ribulosio-5-fosfato
Nella terza tappa della fase ossidativa si verifica la decarbossilazione ossidativa del 6-fosfogluconato a dare ribulosio-5-fosfato, un cheto pentoso, una molecola di CO2 e una di NADPH. La reazione è catalizzata dalla 6-fosfogluconato deidrogenasi (EC 1.1.1.44), enzima che richiede la presenza di ioni magnesio, Mg2+.
6-Fosfogluconato + NADP+ → Ribulosio-5-fosfato + NADPH + CO2
Si noti che questa è la seconda molecola di NADPH prodotta nella via del pentoso fosfato.[15]
Meccanismo catalitico della 6-fosfogluconato deidrogenasi
Il meccanismo catalitico, simile a quello dell’enzima del ciclo dell’acido citrico isocitrico deidrogenasi (EC 1.1.1.41), consiste in una catalisi acido/base e procede attraverso tre passaggi in cui sono coinvolti in particolare due residui conservati presenti nel sito attivo, una lisina (Lys) e un glutammato (Glu), nell’uomo rispettivamente la Lys185 e il Glu192.[22] La lisina agisce sia come acido che come base mentre il glutammato come acido.[6][17]
Meccanismo Catalitico della 6-Fosfogluconato Deidrogenasi
Nel primo passaggio, il passaggio ossidativo, il 6-fosfogluconato viene ossidato in un β-cheto acido, il 3-cheto-6-fosfogluconato.
In questo passaggio l’ε-amino gruppo della catena laterale del suddetto residuo di lisina funge da base, da nucleofilo, estraendo un protone dal gruppo ossidrilico legato sul C-3, cui segue il trasferimento dello ione idruro legato al C-3 sul C-4 dell’anello nicotinamidico del NADP+. In questo modo si viene a formare il 3-cheto intermedio e una molecola di NADPH che lascia il sito attivo.
Nel secondo passaggio, il passaggio decarbossilativo, il 3-cheto-6-fosfogluconato, che è estremamente suscettibile a decarbossilazione, perde, in forma di CO2, il C-1 del glucosio-6-fosfato a dare il cis-1,2-enediolo del ribulosio-5-fosfato, un intermedio a elevata energia. In questo passaggio la lisina agisce come acido donando un protone all’ossigeno carbonilico legato al C-3.
Infine l’enzima catalizza una conversione stereospecifica enolo-chetone con formazione del ribulosio-5-fosfato. In questo passaggio il residuo di acido glutammico suddetto funge da acido donando un protone al C-1 del cis-1,2-enediolo, mentre l’ε-amino gruppo della lisina accetta un protone dal gruppo ossidrilico legato sul C-2. Il risultato è la formazione del ribulosio-5-fosfato.
Fase non ossidativa
Nella fase non ossidativa della via del pentoso fosfato viene prodotta una varietà di carboidrati fosforilati il cui destino dipende dalle necessità relative di NADPH, ribosio-5-fosfato e ATP da parte della cellula.[7]
Questa fase si compone di cinque tappe, tutte facilmente reversibili, nel corso delle quali si verifica una serie di interconversioni di zuccheri fosforilati.
La fase non ossidativa ha inizio con due possibili reazioni a carico del ribulosio-5-fosfato: una isomerizzazione e una epimerizzazione che portano alla formazione rispettivamente di ribosio-5-fosfato e xilulosio-5-fosfato.[14]
Le Epimerasi (EC 5.1), un sottogruppo delle Isomerasi (EC 5.), catalizzano l’inversione reversibile della configurazione di un atomo di carbonio asimmetrico, in genere attraverso la rimozione e l’aggiunta di un atomo di idrogeno.
Nelle reazioni di isomerizzazione lo scambio di gruppi chimici si verifica tra atomi di carbonio.
Nota: le reazioni enzimatiche di isomerizzazione ed epimerizzazione svolgono un ruolo importante nel metabolismo dei carboidrati.
Isomerizzazione del ribulosio-5-fosfato a ribosio-5-fosfato
Nella prima tappa della fase non ossidativa della via del pentoso fosfato, una reazione di isomerizzazione, il ribulosio-5-fosfato, un chetoso, viene convertito nell’aldoso corrispondente, il ribosio-5-fosfato. La reazione è catalizzata dalla fosfopentoso isomerasi o ribosio-5-fosfato isomerasi (EC 5.3.1.6).
Ribulosio-5-fosfato ⇄ Ribosio-5-fosfato
Le due molecole sono un esempio di isomeria di gruppo funzionale.
Meccanismo catalitico della ribosio-5-fosfato isomerasi
Il meccanismo catalitico della ribosio-5-fosfato isomerasi, simile a quello dell’enzima glicolitico fosfoglucosio isomerasi (EC 5.3.1.9), procede attraverso la formazione dell’intermedio ad alta energia cis-1,2-enediolo del ribulosio-5-fosfato, formazione conseguente a un meccanismo di trasferimento protonico comune alle isomerizzazione aldoso-chetoso.
Di seguito viene descritto il possibile meccanismo catalitico dell’enzima di E. coli nella direzione della formazione del ribulosio-5-fosfato dal ribosio-5-fosfato, che è anche la direzione seguita nel ciclo di Calvin della fotosintesi.[23]
Meccanismo Catalitico della Fosfopentoso Isomerasi
Il primo passaggio consiste nell’apertura dell’anello furanosico del ribosio-5-fosfato, apertura rara in soluzione acquosa (<0.5%), e indotta dall’interazione con un residuo di acido aspartico (Asp81) che accetta un protone dal gruppo idrossilico legato al C-1, mentre è probabile che sia l’acqua il donatore di un protone.
Una volta aperta la catena un residuo di acido glutammico (Glu103) funge da base generale, da nucleofilo, estraendo il protone legato sul C-2, mentre Asp81 dona un protone. Come risultato si forma il cis-1,2-enediolo.
Di seguito, la base iniziale, Glu103 adesso protonata, funge da acido e dona un protone al C-1 dell’intermedio enediolico, mentre Asp81 funge da base generale accettando un protone dal gruppo ossidrilico legato al C-2. Il risultato è la formazione del ribulosio-5-fosfato.
Il meccanismo di reazione per la sintesi del ribosio-5-fosfato dal ribulosio-5-fosfato è semplicemente l’inverso di quanto appena descritto.
Epimerizzazione del ribulosio-5-fosfato a xilulosio-5-fosfato
L’altro destino metabolico del ribulosio-5-fosfato nella via del pentoso fosfato è quello di essere epimerizzato a xilulosio-5-fosfato, un chetoso come il ribulosio-5-fosfato, nella reazione catalizzata dalla fosfopentoso epimerasi (EC 5.1.3.1).
Ribulosio-5-fosfato ⇄ Xilulosio-5-fosfato
Nota: lo xilulosio-5-fosfato, oltre a essere un intermedio della via del pentoso fosfato ha anche azione regolatoria: stimola la glicolisi e inibisce la gluconeogenesi, intervenendo nel controllo della concentrazione del fruttosio-2,6-bisfosfato nel fegato.[10]
Meccanismo catalitico della fosfopentoso epimerasi
Anche questa reazione, come quelle catalizzate dalla 6-fosfogluconato deidrogenasi e dalla ribosio-5-fosfato isomerasi, procede attraverso la formazione di un intermedio enediolico, ma con il doppio legame non tra i carboni 1 e 2 ma tra quelli 2 e 3.
Quello che accade nel corso della reazione è che un residuo aminoacidico presente nel sito attivo dell’enzima funge da base generale, da nucleofilo, ed estrae un protone dal C-3, il che porta alla formazione dell’intermedio cis-2,3-enediolico. Di seguito un residuo aminoacidico acido dona un protone al C-3, ma sul lato opposto, e dunque determinando l’inversione della configurazione sul C-3 a formare lo xilulosio-5-fosfato.[5][9]
Meccanismo Catalitico della Fosfopentoso Epimerasi
Arrivati a questo punto la via dell’esoso monofosfato ha portato alla formazione, per ogni molecola di glucosio-6-fosfato metabolizzata, di:
un pool di tre pentoso-5-fosfati, ossia ribulosio-5-fosfato, ribosio-5-fosfato e xilulosio-5-fosfato, che coesistono all’equilibrio;
2 molecole di NADPH.
Nelle tre tappe successive, dalla sesta all’ottava, la transchetolasi (EC 2.2.1.1) e la transaldolasi (EC 2.2.1.2), enzimi esclusivi della via del pentoso fosfato, catalizzano una serie di riarrangiamenti degli scheletri carboniosi che porteranno alla formazione di unità carboniose a tre, quattro, sei e sette atomi che potranno essere utilizzate per vari scopi metabolici, a seconda delle necessità della cellula.[2]
In termini di analisi dei flussi di metaboliti tra le differenti vie metaboliche, l’azione concertata della transchetolasi e della transaldolasi permette l’interazione della via del pentoso fosfato, in particolare la sua fase non ossidativa, con la glicolisi, e la gluconeogenesi, nonché con le vie che portano alla formazione di numerose vitamine, coenzimi e precursori degli acidi nucleici.
Transchetolasi
La transchetolasi è l’enzima che catalizza la tappa limitante della fase non ossidativa della via del pentoso fosfato, e il primo enzima che agisce a valle delle reazioni catalizzate dalla ribosio-5-fosfato isomerasi e della fosfopentoso epimerasi.[11]
Scoperta indipendentemente nel 1953 da Horecker e Racker, e così chiamata da quest’ultimo, catalizza, nella sesta e ottava tappa, il trasferimento di unità a due atomi di carbonio da un donatore chetoso, ossia lo xilulosio-5-fosfato, il sedoeptulosio-7-fosfato o il fruttosio-6-fosfato, a un aldoso accettore, ribosio-5-fosfato, gliceraldeide-3-fosfato o eritrosio-4-fosfato.[8]
Reazioni della Transchetolasi
Considerando come esempi le reazioni “in avanti”, nella sesta tappa il chetoso donatore è lo xilulosio-5-fosfato mentre l’aldoso accettore è il ribosio-5-fosfato, a formare gliceraldeide-3-fosfato, il frammento a tre atomi di carbonio rimanente dello xilulosio-5-fosfato, e sedoeptulosio-7-fosfato, uno zucchero fosforilato a sette atomi di carbonio che sarà utilizzato nel passaggio successivo, il settimo.
Nell’ottava tappa il chetoso donatore è lo xilulosio-5-fosfato mentre l’aldoso accettore è l’eritrosio-4-fosfato, a dare una seconda molecola di gliceraldeide-3-fosfato e una di fruttosio-6-fosfato.
Da notare che tre dei quattro prodotti delle reazioni catalizzate dalla transchetolasi, due molecole di gliceraldeide-3-fosfato e una di fruttosio-6-fosfato, sono anche intermedi della glicolisi.[11]
La transchetolasi può utilizzare come substrati, oltre allo xilulosio-5-fosfato, al sedoeptulosio-7-fosfato e al fruttosio-6-fosfato anche altri 2-chetozuccheri, nonché diversi altri aldoso fosfati.
Transchetolasi e tiamina pirofosfato
La transchetolasi appartiene al gruppo degli enzimi che richiedono la tiamina pirofosfato o TPP, acronimo dell’inglese thiamine pyrophosphate, come cofattore.
La tiamina pirofosfato è la forma biologicamente attiva della tiamina o vitamina B1, ed è saldamente legata all’enzima.
Tiamina Pirofosfato
Altri enzimi che richiedono la TPP come cofattore sono:
la piruvato decarbossilasi (EC 4.1.1.1), che interviene nella fermentazione alcolica;
la componente alfa-chetoacido deidrogenasi o subunità E1 (EC 1.2.4.4) del complesso della alfa-chetoacido deidrogenasi dei chetoacidi a catena ramificata;
la componente 2-chetoglutarato deidrogenasi o subunità E1 (EC 1.2.4.2) del complesso della alfa-chetoglutarato deidrogenasi, enzima del ciclo dell’acido citrico.
La funzione della tiamina pirofosfato è quella di contribuire al trasferimento di intermedi aldeidici attivati stabilizzando carbanioni a due atomi di carbonio che si forma durante la reazione.
Meccanismo catalitico della transchetolasi
L’atomo di carbonio compreso tra l’atomo di zolfo e quello di azoto dell’anello tiazolico della tiamina pirofosfato, ossia l’atomo C-2, è molto più acido rispetto alla maggior parte dei gruppi =C- presenti in altre molecole a causa dell’adiacente atomo di azoto caricato positivamente che stabilizza elettrostaticamente il carbanione derivante dalla dissociazione del protone. Questo fa si che l’atomo di idrogeno a esso legato sia facilmente dissociabile a formare un carbanione, ossia un atomo di carbonio con carica negativa. L’estrazione del protone è catalizzata dalla transchetolasi.[5][14][22]
I carbanioni reagiscono con il carbonio carbonilico del substrato chetonico, nella tappa 6 lo xilulosio-5-fosfato o il sedoeptulosio-7-fosfato nella reazione in direzione opposta, oppure, considerando la tappa 8, di nuovo lo xilulosio-5-fosfato o il fruttosio-6-fosfato nella reazione in direzione opposta.
Prendendo come esempio la reazione “in avanti” della tappa 6, il composto di addizione tra la tiamina pirofosfato e lo xilulosio-5-fosfato va incontro a frammentazione a seguito della scissione del legame C2-C3 dello xilulosio-5-fosfato, a dare gliceraldeide-3-fosfato, che è rilasciata, e un’unità a due atomi di carbonio, un gruppo idrossietilico con carica negativa, che rimane legato al C-2 dell’anello tiazolico.
Meccanismo Catalitico della Transchetolasi
La carica negativa presente sull’intermedio idrossietilico, ossia l’intermedio carbanionico, viene stabilizzata dall’anello tiazolico della tiamina pirofosfato grazie alla presenza dell’atomo di azoto carico positivamente che funge da “trappola” o “pozzo” per gli elettroni. Quindi l’anello tiazolico fornisce una struttura elettron deficiente o elettrofila nella quale gli elettroni del carbanione possono delocalizzarsi per risonanza.
Nell’ultimo passaggio si verifica la condensazione tra il gruppo idrossietilico e il ribosio-5-fosfato, l’aldeide accettore, attraverso l’attacco del carbanione sul carbonio aldeidico del ribosio-5-fosfato, a formare un addotto covalente legato alla tiamina pirofosfato.
La successiva scissione dell’addotto porta alla liberazione di sedoeptulosio-7-fosfato, rigenerando il carbanione della tiamina pirofosfato.
Transaldolasi
La transaldolasi, scoperta nel 1953 da Horecker e Smyrniotis nel lievito Saccharomyces cerevisiae, interviene nella settima tappa della via del pentoso fosfato, dove catalizza il trasferimento di una unità a tre atomi di carbonio da un substrato donatore, il sedoeptulosio-7-fosfato, a un substrato accettore, la gliceraldeide-3-fosfato, a dare fruttosio-6-fosfato ed eritrosio-4-fosfato.[18]
Da notare che, in analogia con quanto visto per le reazioni catalizzate dalla transchetolasi, anche in questo caso il donatore dell’unità carboniosa trasferita è un chetoso mentre l’accettore è un aldoso.
Nella reazione in direzione opposta il chetoso donatore sarà il fruttosio-6-fosfato mentre l’aldoso accettore l’eritrosio-4-fosfato.
Meccanismo catalitico della transaldolasi
A differenza della transchetolasi, la transaldolasi non necessita per la propria attività della presenza di cofattori.
La reazione si compone di una parte equivalente a una scissione aldolica e di una equivalente a una condensazione aldolica. Di seguito verrà analizzato il meccanismo di catalitico della transaldolasi B di E. coli, considerando la reazione nella direzione della formazione dell’eritrosio-4-fosfato e del fruttosio-6-fosfato.
Nel primo passaggio l’ε-amino gruppo della catena laterale di un residuo di lisina (Lys132) presente nel sito attivo, a seguito del trasferimento di un protone a un residuo di acido glutammico (Glu96), trasferimento mediato da una molecola d’acqua, porta un attacco nucleofilo al C-2 del sedoeptulosio-7-fosfato, il carbonio carbonilico. Il risultato è la formazione di una carbinolammina con il sedoeptulosio-7-fosfato.[5][14][18][22]
Meccanismo Catalitico della Transaldolasi
Segue la perdita di una molecola d’acqua dalla carbinolammina con formazione di una immina, o base di Schiff, legata all’enzima; anche in questo passaggio interviene il trasferimento di un protone da Glu96 alla molecola d’acqua “catalitica”.
Nota: l’intermedio covalente enzima-substrato che si è formato è simile a quello che quello che si forma nella reazione catalizzata dalla aldolasi (EC 4.1.2.13) nella quarta tappa della glicolisi.
Nel passaggio successivo, il gruppo carbossilico di un residuo di acido aspartico (Asp17) estrae un protone dal gruppo idrossilico legato sul C-4, con conseguente rottura del legame covalente tra C-3 e C-4, una scissione aldolica che porta alla liberazione dell’aldoso eritrosio-4-fosfato, il primo prodotto della reazione, mentre rimane legato all’enzima un carbanione a tre atomi di carbonio che è stabilizzato per risonanza, analogamente a quanto visto per la transchetolasi. Infatti, al pari dell’atomo di azoto dell’anello tiazolico della tiamina pirofosfato, anche l’atomo di azoto con carica positiva della base di Schiff agisce da trappola per gli elettroni stabilizzando la carica negativa del carbanione.
Una volta che il secondo substrato, la gliceraldeide-3-fosato, si trova in posizione nel sito attivo, il carbanione porta un attacco nucleofilo al carbonio carbonilico della gliceraldeide-3-fosfato a formare, mediante una condensazione aldolica, un nuovo legame C-C e un chetoso legato all’enzima.
Infine l’idrolisi della base di Schiff porta al rilascio del fruttosio-6-fosfato, un chetoso e il secondo prodotto della reazione, e permette l’inizio di un nuovo ciclo di reazione.
A questo punto, come visto in precedenza, nell’ottava tappa della via del pentoso fosfato, interviene nuovamente la transchetolasi che catalizza la formazione di fruttosio-6-fosfato e glicealdeide-3-fosfato a partire da eritrosio-4-fosfato e xilulosio-5-fosfato.
Fabbisogno di ribosio-5-fosfato, NADPH e ATP
Dal punto di vista molecolare il destino del glucosio-6-fosfato dipende in larga parte dalle attività relative degli enzimi che lo metabolizzano nella via glicolitica e nella via del pentoso fosfato, in particolare dalla fosfofruttochinasi-1 o PFK-1, acronimo dell’inglese phosphofructokinase-1 (EC 2.7.1.11) e la glucosio-6-fosfato deidrogenasi, enzimi la cui attività è altamente regolata.
La PFK-1 è inibita quando aumenta la concentrazione dell’ATP e/o di citrato, ossia quando la carica energetica della cellula è alta, mentre è attivata quando aumenta la concentrazione dell’AMP e/o del fruttosio-2,6-bisfosfato, ossia quando la carica energetica della cellula è bassa. Ne consegue che, quando la carica energetica della cellula è alta il flusso di carbonio, e quindi di glucosio-6-fosfato, attraverso la glicolisi si riduce.[20][21]
La glucosio-6-fosfato deidrogenasi è invece inibita dal NADPH e dagli acil-CoA, intermedi nella biosintesi degli acidi grassi.[15] Se quindi la concentrazione citosolica del NADPH aumenta il flusso di glucosio-6-fosfato attraverso la via del pentoso fosfato è inibito. Viceversa, se i livelli di NADPH cadono, l’inibizione viene meno, la via si “riaccende” e vengono sintetizzati NADPH e ribosio-5-fosfato.
Pertanto, in base alle necessità della cellula di NADPH, ribosio-5-fosfato e ATP, quello che accadrà è una “combinazione” di alcune reazioni della via glicolitica, della gluconeogenesi e della via del pentoso fosfato al fine di sintetizzare i metaboliti richiesti, sfruttando anche il fatto che la fase non ossidativa dello shunt dell’esoso monofosfato è controllata essenzialmente dalla disponibilità dei substrati.
Di seguito sono analizzate le quattro principali possibilità.[2]
Fabbisogno di NADPH molto maggiore di quello di ribosio-5-fosfato e ATP
Quando il fabbisogno di NADPH è molto maggiori di quello di ribosio-5-fosfato e la cellula non ha bisogno di ulteriore produzione di ATP, ossia la carica energetica della cellula è alta, il glucosio-6-fosfato entra nella via del pentoso fosfato e può essere completamente ossidato a CO2. Una condizione metabolica del genere si ritrova ad esempio nel tessuto adiposo nel corso di una intensa sintesi di acidi grassi.
Nella fase ossidativa della via, per ogni molecola di glucosio-6-fosfato ossidata a ribulosio-5-fosfato, sono prodotte due molecole di NADPH. Attraverso una combinazione delle reazione della fase non ossidativa e di alcune reazioni della gluconeogenesi, nello specifico quelle catalizzate dagli enzimi trioso fosfato isomerasi (EC 5.3.1.1), aldolasi (EC 4.1.2.13), fosfoglucosio isomerasi (EC 5.3.1.9), e fruttosio-1,6-bisfosfatasi (EC 3.1.3.11), sei molecole di ribulosio-5-fosfato vengono convertite in cinque di glucosio-6-fosfato. E’ quindi anche possibile affermare che le reazioni della fase non ossidativa permettono alle reazioni della fase ossidativa di procedere.
In questo processo è possibile individuare tre gruppi di reazioni che operano in sequenza
Nel primo gruppo si ritrovano le reazioni catalizzate dagli enzimi della fase ossidativa, e si ha la formazione di due molecole di NADPH e una di ribulosio-5-fosfato.
Al secondo gruppo appartengono le reazioni catalizzate dagli enzimi fosfopentoso epimerasi, ribosio-5-fosfato isomerasi, transchetolasi e transaldolasi, ossia quelli della fase non ossidativa della via, che portano alla conversione del ribulosio-5-fosfato in fruttosio-6-fosfato e gliceraldeide-3-fosfato.
Infine, attraverso le suddette reazioni della gluconeogenesi il fruttosio-6-fosfato e la gliceraldeide-3-fosfato sono “riciclati” a glucosio-6-fosfato, di modo che il ciclo possa ricominciare.
Sommando le ultime due reazioni si evince che sei molecole di ribulosio-5-fosfato sono convertite in cinque di glucosio-6-fosfato.
6 Ribulosio-5-fosfato + H2O → 5 Glucosio-6-fosfato + Pi
Dalla somma delle reazioni del primo, secondo e terzo gruppo si ottiene la reazione complessiva:
Glucosio-6-fosfato + 12 NADP+ + 7 H20 → 6 CO2 + 12 NADPH + 12 H+ + Pi
Dunque, una molecola di glucosio-6-fosfato, attraverso sei cicli della via del pentoso fosfato accoppiati con alcune reazioni della reazioni gluconeogenesi, sarà convertita in 6 molecole di CO2, con la concomitante produzione di 12 molecole di NADPH, ma nessuna produzione netta di ribosio-5-fosfato.
Fabbisogno di NADPH e ATP molto maggiore di quello di ribosio-5-fosfato
Quando il fabbisogno di NADPH è molto maggiore di quello di ribosio-5-fosfato e la carica energetica della cellula è bassa, ossia la cellula necessita di ATP, il ribulosio-5-fosfato prodotto nella fase ossidativa della via del pentoso fosfato viene convertito in fruttosio-6-fosfato e gliceraldeide-3-fosfato attraverso le reazioni della fase non ossidativa. Di seguito i due intermedi, attraverso le reazioni della glicolisi, sono ossidati a piruvato, la base coniugata dell’acido piruvico, con concomitante produzione di ATP.
La reazione complessiva è:
Se la cellula necessita di altro ATP, il piruvato prodotto potrà essere ossidato attraverso il ciclo dell’acido citrico. Se invece non è necessario produrre ulteriore ATP, il piruvato potrà essere utilizzato come precursore in diverse vie biosintetiche.
Da notare che anche in questo caso, come nel precedente, non si verifica alcuna produzione netta di ribosio-5-fosfato.
Fabbisogno di ribosio-5-fosfato molto maggiore di quello di NADPH
Quando il fabbisogno di ribosio-5-fosfato è molto maggiore rispetto a quello di NADPH, come nelle cellule che si dividono rapidamente, nelle quali è quindi in corso una rapida sintesi di nucleotidi precursori del DNA, le reazioni della fase ossidativa della via del pentoso fosfato non hanno luogo, e non vi è sintesi di NADPH. Al contrario, poiché le reazioni della fase non ossidativa sono facilmente reversibili, la riduzione della concentrazione del ribosio-5-fosfato, causata dal suo rapido utilizzo, avrà come effetto quello di stimolarne la sintesi.
Quello che accade è che, attraverso la via glicolitica, molto del glucosio-6-fosfato viene convertito in fruttosio-6-fosfato e gliceraldeide-3-fosfato. A questo punto la transaldolasi e la transchetolasi portano alla sintesi di ribosio-5-fosfato e xilulosio-5-fosfato. Quest’ultimo, attraverso le reazione catalizzate dalla fosfotopentoso epimerasi e di seguito dalla ribosio-5-fosfato isomerasi, verrà convertito in ribosio-5-fosfato.
La reazione complessiva è:
6 Glucosio-6-fosfato + ATP → 6-Ribosio-5-fosfato + ADP + H+
In questo caso quindi quello che si verifica è una combinazione di reazioni della glicolisi e della fase non ossidativa della via del fosfogluconato, con queste ultime nella direzione della sintesi del ribosio-5-fosfato.
Da notare che nessun metabolita torna alla glicolisi.
I fabbisogni di ribosio-5-fosfato e NADPH si equivalgono
Se una molecola di ribosio-5-fosfato e due molecole di NADPH per molecola di glucosio-6-fosfato metabolizzata sono sufficienti a soddisfare le necessità metaboliche della cellula, quello che si verifica sono le prime quattro reazioni della via del pentoso fosfato, ossia l’intera fase ossidativa, più la reazione catalizzata dalla ribosio-5-fosfato isomerasi.
La reazione complessiva è:
Anche in questa condizione metabolica nessun metabolita torna alla glicolisi.
Bibliografia
^ abc Au S.W.N., Gover S., Lam V.M.S., Adams M.J. Human glucose-6-phosphate dehydrogenase: the crystal structure reveals a structural NADP+ molecule and provides insights into enzyme deficiency. Structure 2000;8(3):293-303. doi:10.1016/S0969-2126(00)00104-0
^ abcd Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002
^ Brodie A.F., Lipmann F. Identification of a gluconolactonase. J Biol Chem 1955;212(2):677-85. doi:10.1016/S0021-9258(18)71006-5
^ Cosgrove M.S., Naylor C., Paludan S., Adams M.J., Richard Levy H. On the mechanism of the reaction catalyzed by glucose 6-phosphate dehydrogenase. Biochemistry 1998;37(9);2759-2767. doi:10.1021/bi972069y
^ Hanau S., Montin K., Cervellati C., Magnani M., Dallocchio F. 6-Phosphogluconate dehydrogenase mechanism: evidence for allosteric modulation by substrate. J Biol Chem 2010;285(28):21366-21371. doi:10.1074/jbc.M110.105601
^ ab Horecker B.L. The pentose phosphate pathway. J Biol Chem 2002;277(50):47965-47971. doi:10.1074/jbc.X200007200
^ Jelakovic S., Kopriva S., Süss K-H, Schulz G.E. Structure and catalytic mechanism of the cytosolic D-ribulose-5-phosphate 3-epimerase from rice. J Mol Biol 2003;326:127-135. doi:10.1016/S0022-2836(02)01374-8
^ Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003;100:5107-5112. doi:10.1073/pnas.0730817100
^ ab Kochetov G.A., Solovjeva O.N. Structure and functioning mechanism of transketolase. Biochim Biophys Acta 2014;1844(9):1608-1618. doi:10.1016/j.bbapap.2014.06.003
^ Levy H.R. Glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides. Biochem Soc Trans. 1989 Apr;17(2):313-5. doi:10.1042/bst0170313
^ abcd Michal G., Schomburg D. Biochemical pathways. An atlas of biochemistry and molecular biology. 2nd Edition. John Wiley J. & Sons, Inc. 2012
^ abcdef Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ abcd Patra K.C., Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci 2014;39(8):347-354. doi:10.1016/j.tibs.2014.06.005
^ abc Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009
^ Ruda G.F., Campbell G., Alibu V.P., Barrett M.P., Brenk R., Gilbert I.H. Virtual fragment screening for novel inhibitors of 6-phosphogluconate dehydrogenase. Bioorg Med Chem 2010;18(14):5056-62. doi:10.1016/j.bmc.2010.05.077
^ abcd Samland A.K., Sprenger G.A. Transaldolase: from biochemistry to human disease. Int J Biochem Cell Biol 2009;41(7):1482-1494. doi:10.1016/j.biocel.2009.02.001
^ Sprenger G.A. Genetics of pentose-phosphate pathway enzymes of Escherichia coli K-12. Arch Microbiol 1995;164(5):324-30. doi:10.1007/BF02529978
^ Van Schaftingen E., Hers H-G. Inhibition of fructose-1,6-bisphosphatase by fructose-2,6-bisphosphate. Proc Natl Acad Sci USA 1981;78(5):2861-2863. doi:10.1073/pnas.78.5.2861
^ Van Schaftingen E., Jett M-F., Hue L., Hers H-G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA 1981;78(6):3483-3486. doi:10.1073/pnas.78.6.3483
^ abcdefg Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011
^ Zhang R., Andersson C.E., Savchenko A., Skarina T., Evdokimova E., Beasley S., Arrowsmith C.H., Edwards A.M., Joachimiak A., Mowbray S.L. Structure of Escherichia coli ribose-5-phosphate isomerase: a ubiquitous enzyme of the pentose phosphate pathway and the Calvin cycle. Structure 2003;11(1):31-42. doi:10.1016/S0969-2126(02)00933-4
La glicolisi, dal greco glykys, dolce e lysis, sciogliere, può essere definita come la sequenza di reazioni enzimatiche che, nel citosol della cellula, anche in assenza di ossigeno, porta alla conversione di una molecola di glucosio, un composto a sei atomi di carbonio, in due di piruvato, un composto a tre atomi di carbonio, con la concomitante produzione di due molecole di ATP, la moneta di scambio energetico della cellula.[5]
La Via Glicolitica
La glicolisi, che si è evoluta prima che nell’atmosfera si accumulasse una quantità sostanziale di ossigeno, è la via metabolica con il maggior flusso di carbonio nella maggior parte degli esseri viventi. E’ presente in quasi tutti gli organismi.
Grazie al fatto di poter procedere in assenza di ossigeno, ha svolto un ruolo cruciale nei processi metabolici durante i primi due miliardi di anni di evoluzione della vita sulla terra. Probabilmente rappresenta il modo più antico che gli organismi hanno sviluppato per ottenere energia dalle molecole organiche quando la disponibilità del gas è scarsa. E’ inoltre una fonte di precursori per il catabolismo aerobico e per vari processi biosintetici.
Nota: la glicolisi è conosciuta anche come la via di Embden-Meyerhof, dal nome dei due ricercatori che delucidarono l’intero processo nel muscolo.[2][5]
Lo sviluppo della biochimica è andato di pari passo con la comprensione del metabolismo del glucosio e in particolare della glicolisi, la prima via metabolica a essere stata chiarita.[2][5]
Sebbene la delucidazione di questa via fu completata negli anni quaranta del secolo scorso, la scoperta chiave sul metabolismo del glucosio venne fatta in modo casuale nel 1897, a seguito di un problema sorto un anno prima quando un chimico tedesco, M. Hahn, nel tentativo di ottenere e conservare estratti proteici privi di cellule dal lievito, incontrò difficoltà nella loro conservazione. Un suo collega, Hans Buchner, ricordando quanto veniva, e viene tuttora fatto per la conservazione delle marmellate, suggerì di addizionare saccarosio all’estratto.
Eduard Buchner, fratello di Hans, mise in pratica l’idea di Hans, osservando però che dalla soluzione si sviluppavano bollicine. Questo indusse Eduard a concludere che fosse in corso una fermentazione, per l’epoca una scoperta sorprendente. Infatti sino ad allora la fermentazione, secondo quanto affermato da Pasteur nel 1860, era stata inestricabilmente legata alle cellule viventi, mentre ora veniva dimostrato che poteva avvenire anche al di fuori delle cellule stesse. In breve, questi due ricercatori demolirono il dogma vitalistico e diedero il via alla moderna biochimica.
Grazie a queste ricerche Eduard Buchner fu insignito del premio Nobel per la Chimica nel 1907. Buchner fu il primo di una serie di ricercatori che vinsero il premio grazie alle loro scoperte sulla glicolisi. In seguito fu dimostrato, lavorando con estratti muscolari, che molte delle reazioni della fermentazione lattica erano le stesse della fermentazione alcolica, il che evidenziò l’esistenza di una unità nella biochimica.
Come anticipato, la glicolisi fu poi chiarita in modo esaustivo nella prima metà del secolo scorso in gran parte grazie al lavoro di ricercatori quali Gerty and Carl Cori, Carl Neuberg, Arthur Harden, William Young, Jacob Parnas, Otto Warburg, Hans von Euler-Chelpin, Gustav Embden e Otto Meyerhof.
Perché è importante?
La glicolisi è una via metabolica essenziale sia dal punto di vista energetico che come fonte di precursori per diverse altre vie metaboliche. E la velocità del flusso di carbonio attraverso di essa, ossia la velocità con cui il glucosio viene convertito in piruvato, è regolata in modo da soddisfare queste due necessità fondamentali della cellula.[7]
Dal punto di vista energetico, sebbene sia un processo relativamente inefficiente, può avvenire in assenza di ossigeno, la condizione in cui si è sviluppata la vita sulla Terra e in cui tutt’oggi molte cellule, sia eucariote che procariote, si ritrovano. Di seguito alcuni esempi.
I muscoli di molti animali presentano una anaerobiosi dipendente dall’attività, ossia possono funzionare in assenza di ossigeno per brevi periodi. Ad esempio, quando gli animali, ma anche gli atleti, compiono esercizi molto intensi, il loro fabbisogno di ATP aumenta più rapidamente della capacità del corpo di rifornire di ossigeno il muscolo. In questa situazione il muscolo ricava l’energia metabolica necessaria al suo funzionamento in modo anaerobico, appunto attraverso la sola glicolisi.
Un altro esempio è la cornea dell’occhio, un tessuto scarsamente vascolarizzato.
Molti microorganismi, come quelli che vivono in ambienti quali il suolo, le profondità marine ma anche i pori della pelle, si ritrovano in zone in cui l’ossigeno è scarso o assente. Ed esistono microrganismi, definiti anaerobi obbligati, che non possono sopravvivere in presenza di ossigeno, una molecola altamente reattiva. Esempi sono il batterio responsabile della gangrena gassosa, Clostridium perfringens, del tetano, Clostridium tetani, e del botulismo, Clostridium botulinum.
Va inoltre sottolineato che la glicolisi gioca un ruolo centrale anche in tutte quelle cellule e tessuti nei quali il glucosio rappresenta la sola fonte di energia, come:
i globuli rossi, privi di mitocondri;
le cellule spermatiche;
il cervello, che solo in particolari condizioni può usare anche i corpi chetonici per produrre energia;
la midollare del surrene.
Una situazione simile si ritrova anche nel mondo vegetale dove molte piante acquatiche e alcuni tessuti vegetali specializzati nell’accumulo di amido, come i tuberi delle patate, utilizzano il glucosio come principale fonte di energia.
Nota: esistono organismi che sono anaerobi facoltativi, ossia possono sopravvivere in presenza e in assenza di ossigeno, come gli animali appartenenti al genere Mytilus, che quindi mostrano un funzionamento anaerobico dipendente dall’ambiente, o anaerobico habitat-dipendente, una situazione simile a quanto visto in precedenza nel muscolo sottoposto a lavori molto intesi.
Infine non va dimenticato che in condizioni aerobiche, nelle cellule dotate di mitocondri, la glicolisi rappresenta la parte iniziale della via metabolica che porta alla completa ossidazione del glucosio ad anidride carbonica (CO2) e acqua a scopi energetici.
Precursori Derivanti dalla Via Glicolitica
Diversi intermedi glicolitici, come il glucosio-6-fosfato (G-6-P), il fruttosio-6-fosfato (F-6-P,), il diidrossiacetone fosfato (DHAP) o il piruvato possono essere utilizzati come precursori biosintetici in vie metaboliche quali ad esempio quelle che portano alla sintesi del glicogeno, degli acidi grassi, dei trigliceridi, dei nucleotidi, di alcuni amminoacidi, o del 2,3-bisfosfoglicerato (2,3-BPG).
Tappe
Le 10 tappe che compongono la glicolisi possono essere suddivise in due fasi.
La prima, detta fase preparatoria, si compone di 5 reazioni e comporta dapprima la conversione di una molecola di glucosio in una di fruttosio-1,6-bisfosfato (F-1,6-BP), attraverso tre passaggi:
una prima fosforilazione sul C-1;
una isomerizzazione;
una seconda fosforilazione, stavolta sul C-6, con consumo complessivo di 2 ATP.
Segue la scissione, la lisi, del fruttosio-1,6-bisfosfato in due intermedi fosforilati a tre atomi di carbonio, la gliceraldeide-3-fosfato e il diidrossiacetone fosfato, e infine l’isomerizzazione di quest’ultimo in gliceraldeide-3-fosfato. Dunque, nella fase preparatoria una molecola di glucosio viene scissa in due di gliceraldeide-3-fosfato e si consumano due ATP.[2][5]
Nella seconda fase, detta fase di recupero energetico, composta dalle restanti 5 reazioni, le due molecole di gliceraldeide-3-fosfato sono convertite in altrettante molecole di piruvato, con concomitante produzione di 4 molecole di ATP. Quindi nella seconda fase si verifica l’estrazione e conservazione in forma di ATP di parte dell’energia chimica presente nella molecola del glucosio. Inoltre sono estratti anche equivalenti riducenti, immagazzinati nella molecola del NADH. Quello che sarà poi il destino del NADH dipende dal tipo di cellula e dalla presenza o meno di quantità adeguate di ossigeno.[2][5]
Nota: nella glicolisi è metabolizzato sia il glucosio di derivazione ematica, che entra nella cellula attraverso un trasporto mediato da specifici trasportatori proteici di membrana, che il glucosio-6-fosfato derivante dalla glicogenolisi.
Reazione 1
Nella prima tappa della via glicolitica il glucosio viene fosforilato a glucosio-6-fosfato a spese di una molecola di ATP.
Glucosio + ATP → Glucosio-6-fosfato + ADP + H+
Nella maggior parte delle cellule questa reazione è catalizzata dalla esochinasi (EC 2.7.1.1), enzima presente nelle cellule di tutti gli organismi, e negli esseri umani con quattro forme isoenzimatiche.
L’esochinasi e la piruvato chinasi (EC 2.7.1.40), l’altra chinasi della glicolisi, al pari di molte altre chinasi, richiede per il corretto funzionamento la presenza dello ione magnesio (Mg2+) o di un altro ione metallico bivalente come il manganese (Mn2+). Considerando lo ione magnesio, questi si va a legare all’ATP a formare il complesso MgATP2-, che è il vero substrato dell’enzima. E va sottolineato che l’attacco nucleofilo da parte di un gruppo ossidrilico (-OH) del glucosio all’atomo di fosforo terminale dell’ATP è facilitato proprio dall’azione degli ioni magnesio che vanno a interagire con le cariche negative dei gruppi fosforici del nucleoside trifosfato.
La formazione del legame fosfoestereo tra un gruppo fosforico e il gruppo ossidrilico sul C-6 è termodinamicamente sfavorita e richiede energia per procedere, energia che è fornita dall’ATP. Infatti, mentre la fosforilazione del glucosio sul C-6 a opera del solo fosfato inorganico ha un ΔG°’ pari a 3,3 kcal/mol (13,8 kJ/mol), ossia è una reazione endoergonica, l’idrolisi dell’ATP a dare ADP e Pi ha un ha un ΔG°’ pari -7,3 kcal/mol (30,5 kJ/mol), ossia è una reazione esoergonica. La reazione netta avrà un ΔG°’ = -7,3 + 3,3 = -4,0 kcal/mol (-30,5 + 13,8 = -16,7 kJ/mol). Nelle condizioni presenti nella cellula la reazione è ancora più favorevole, con un ΔG pari a -8,0 kcal/mol (-33,5 kJ/mol).
Dunque si tratta di una reazione essenzialmente irreversibile.
Nota: in biochimica le fosforilazioni sono reazioni fondamentali catalizzate da enzimi detti chinasi, una sottoclasse delle transferasi. Le chinasi catalizzano il trasferimento del gruppo fosforico terminale, o gruppo fosforico in posizione γ, di un nucleoside trifosfato a un nucleofilo accettore a formare un legame fosfoestereo. Nello specifico, le esochinasi catalizzano il trasferimento del gruppo fosforico in posizione γ dell’ATP a vari zuccheri a sei atomi di carbonio, o esosi, tra cui, oltre ovviamente al glucosio, anche al fruttosio e al mannosio.
Perché la fosforilazione del glucosio è importante?
La fosforilazione della molecola del glucosio è importante per almeno alcuni motivi.[2]
Il glucosio-6-fosfato grazie alla sua carica negativa e all’assenza sulla membrana plasmatica di trasportatori per gli zuccheri fosforilati non può diffondere attraverso di essa. Quindi, dopo la fosforilazione iniziale non è più necessario spendere energia per mantenere la molecola fosforilata all’interno della cellula, nonostante la grande differenza esistente tra le sue concentrazioni intra- ed extracellulari.
Considerazioni analoghe valgono per gli altri otto intermedi fosforilati che si ritrovano tra il glucosio-6-fosfato e il piruvato.
La rapida fosforilazione del glucosio mantiene la concentrazione intracellulare dell’esoso bassa, favorendo così il suo passaggio all’interno della cellula.
La fosforilazione comporta l’aumento del contenuto in energia della molecola, ossia inizia a destabilizzarla, facilitandone il successivo metabolismo.
Altri destini metabolici del glucosio-6-fosfato
Il glucosio-6-fosfato rappresenta un punto di ramificazione importante del metabolismo del glucosio. Infatti, oltre che proseguire nella via glicolitica, in condizioni anaboliche, può avere destini differenti. Di seguito alcuni esempi.
Può essere utilizzato per la sintesi di: glicogeno, immagazzinato principalmente nel fegato e nel muscolo;
polisaccaridi complessi presenti nella matrice extracellulare; galattosio;
glucosammina e altri zuccheri utilizzati per la glicosilazione delle proteine.
Può essere entrare nella via del pentoso fosfato, via attraverso la quale le cellule producono:
NADPH, necessario in generale per le biosintesi riduttive, come quella degli acidi grassi, del colesterolo, degli ormoni steroidei, e dei desossiribonucleotidi, ma anche indispensabile nella prevenzione del danno ossidativo in cellule come i globuli rossi;
riboso-5-fosfato, utilizzato nella sintesi dei nucleotidi ma anche del NADH, FADH2 e del coenzima A.
Reazione 2
Nella seconda tappa della glicolisi si verifica l’isomerizzazione del glucosio-6-fosfato, un aldoso, a fruttosio-6-fosfato, un chetoso, nella reazione catalizzata dalla fosfoglucosio isomerasi (EC 5.3.1.9).
Glucosio-6-fosfato ⇄ Fruttosio-6-fosfato
Glucosio-6-fosfato e fruttosio-6-fosfato sono un esempio di isomeria di gruppo funzionale
Come l’esochinasi, anche la fosfoglucosio isomerasi necessita della presenza di Mg2+.
Il ΔG°’ della reazione è pari a 0,4 kcal/mol (1,7 kJ/mol), mentre il ΔG è di -0,6 kcal/mol (-2,5 kJ/mol). Questo piccolo valore sta a indicare che la reazione è prossima all’equilibrio ed è facilmente reversibile. La reazione consiste essenzialmente nel passaggio del gruppo carbonilico presente sul C-1 della forma a catena aperta del glucosio-6-fosfato al C-2 della forma a catena aperta del fruttosio-6-fosfato.
La reazione enzimatica può essere suddivisa in almeno tre passaggi. Poiché entrambe gli esosi in soluzione acquosa sono presenti principalmente nella forma ciclica, l’enzima deve dapprima aprirne l’anello, catalizzare l’isomerizzazione, e infine richiudere l’anello stesso, portando alla formazione dell’anello a 5 atomi di carbonio del fruttosio-6-fosfato.
Reazione Catalizzata dalla Fosfoglucosio Isomerasi
Questa reazione di isomerizzazione è un passaggio cruciale per glicolisi in quanto prepara per le due tappe successive.
Perché?
La fosforilazione che si verifica nella reazione successiva, la terza, richiede la presenza di un gruppo alcolico sul C-1, e non di un gruppo carbonilico.
Nella quarta tappa si verifica la scissione del legame tra il C-3 e il C-4, scissione facilitata dalla presenza del gruppo carbonilico sul C-2.
Reazione 3
Nella terza tappa della via glicolitica si verifica una seconda reazione di fosforilazione nella quale il fruttosio-6-fosfato viene fosforilato sul C-1 a dare fruttosio-1,6-bisfosfato, a spese di una molecola di ATP. La reazione è catalizzata dalla fosfofruttochinasi-1 o PFK-1 (EC 2.7.1.11).
Fruttosio-6-fosfato + ATP → Fruttosio-1,6-bisfosfato + ADP + H+
La PFK-1 è così chiamata per distinguerla dall’enzima che catalizza la fosforilazione del fruttosio-6-fosfato a fruttosio-2,6-bisfosfato, ossia la fosfofruttochinasi-2 o PFK-2 (EC 2.7.1.105).
Al pari della reazione catalizzata dalla esochinasi/glucochinasi, anche questa fosforilazione è un passaggio essenzialmente irreversibile della glicolisi. E anche in questo caso l’irreversibilità è assicurata dall’accoppiamento, grazie alla PFK-1, con l’idrolisi dell’ATP. Infatti la fosforilazione del fruttosio-6-fosfato a opera del solo fosfato inorganico è una reazione endoergonica, con un ΔG°’ pari a 3,9 kcal/mol (16,3 kJ/mol), mentre, quando la reazione è accoppiata all’idrolisi dell’ATP, la reazione complessiva diviene esoergonica, con un ΔG°’ = 7,3-3,9 = -3,4 kcal/mol (-30,5 + 16,3 = -14,2 kJ/mol) e un ΔG di -5,3 kcal/mol (-22,2 kJ/mol).
Mentre la esochinasi permette alla cellula di “intrappolare” il glucosio al suo interno, la fosfofruttochinasi-1 evita che lo scheletro carbonioso del monosaccaride sia utilizzato per la sintesi del glicogeno o per la produzione di altri zuccheri, ma venga invece metabolizzato nella via glicolitica. Infatti, a differenza del glucosio-6-fosfato, il fruttosio-1,6-bisfosfato è un metabolita esclusivo della glicolisi/gluconeogenesi. In altri termini, la fosfofruttochinasi-1 catalizza la prima tappa di comando della via glicolitica. Reazioni di questo tipo sono di solito sono catalizzate da enzimi regolati per via allosterica, la cui regolazione impedisce l’accumulo sia dei prodotti intermedi che di quelli finali. La PFK-1 non fa eccezione. La chinasi è infatti soggetta a una fine regolazione allosterica da parte di numerosi metaboliti che segnalano il livello di energia e lo stato ormonale dell’organismo.
Alcuni batteri e protisti, e forse tutte le piante, posseggono una fosfofruttochinasi che utilizza, nella sintesi del fruttosio-1,6-bisfosfato, come donatore del gruppo fosforico non l’ATP ma il pirofosfato, una reazione con un ΔG°’ pari a -12,1 kcal/mol (-2,9 kJ/mol).
Fruttosio-6-fosfato + PPi → Fruttosio-1,6-bisfosfato + Pi
Nota: il prefisso bis– in bisfosfato, come fruttosio-1,6-bisfosfato, sta a indicare la presenza di due gruppi fosforici separati, mentre il prefisso di– in difosfato, come in adenosina difosfato, indica che sono presenti due gruppi fosforici connessi da un legame anidridico a dare un gruppo pirofosforico. Regole simili si applicano anche per la denominazione di molecole che presentano tre gruppi fosforici separati, come l’inositolo 1,4,5-trisfosfato, o connessi da legame anidridico come l’ATP o la guanosina trifosfato o GTP.
Reazione 4
Nella quarta tappa della glicolisi si verifica la scissione reversibile del fruttosio-1,6-bisfosfato in gliceraldeide-3-fosfato, un aldoso, e diidrossiacetone fosfato, un chetoso. La reazione è catalizzata dalla fruttosio-1,6-bisfosfato aldolasi o semplicemente aldolasi (EC 4.1.2.13), che catalizza la scissione del legame tra il C-3 e il C-4.
Tutti gli intermedi glicolitici a valle di questa reazione sono molecole a tre atomi di carbonio anziché a 6 come le precedenti.
Il ΔG°’ della reazione nella direzione della produzione di gliceraldeide-3-fosfato e diidrossiacetone fosfato è positivo, pari a 5,7 kcal/mol (23,8 kJ/mol), mentre la Km è pari a circa 10-4 M, valori che indicherebbero che la reazione non procede da sinistra verso destra. In realtà nelle normali condizioni cellulari, a causa delle basse concentrazioni dei reagenti presenti nella cellula, il ΔG è pari a -0,3 kcal/mol (-1,3 kJ/mol), un valore molto piccolo, per cui la reazione è facilmente reversibile, ossia essenzialmente all’equilibrio.
Nota: l’enzima aldolasi deriva il nome proprio nome dalla natura della reazione inversa, ossia una condensazione aldolica.
Reazione 5
Dei due prodotti della reazione catalizzata dalla aldolasi, la gliceraldeide-3-fosfato si trova sulla via diretta della glicolisi, al contrario del diidrossiacetone fosfato che come tale non può proseguire. Per farlo deve essere convertito, isomerizzato, a gliceraldeide-3-fosfato. Suddetta isomerizzazione avviene nella reazione catalizzata dalla trioso fosfato isomerasi (EC 5.3.1.1).
La trioso fosfato isomerasi, nel convertire il diidrossiacetone fosfato in gliceraldeide-3-fosfato, catalizza il trasferimento di un atomo d’idrogeno dal C-1 a C-2, ossia una ossidoriduzione intramolecolare. In pratica l’enzima fa si che i carboni C-1, C-2 e C-3 della molecola di glucosio di partenza divengano equivalenti rispettivamente ai carboni C-6, C-5 e C-4.
Il risultato netto dell’azione della aldolasi e della trisofosfato isomerasi è dunque la produzione di due molecole di gliceraldeide-3-fosfato.
Il ΔG°’ della reazione è pari a 1,8 kcal/mol (7,5 kJ/mol), mentre il ΔG è pari a 0,6 kcal/mol (2,5 kJ/mol). All’equilibrio, circa il 96% del triosofosfato presente sarebbe rappresentato dal diidrossiacetone fosfato. Tuttavia ciò non accade, e la reazione procede rapidamente verso la formazione della gliceraldeide-3-fosfato grazie alla tappa successiva della glicolisi che rimuove la gliceraldeide-3-fosfato prodotta.
Una delle caratteristiche distintive della trioso fosfato isomerasi è la sua grande capacità catalitica. L’enzima è infatti considerato cineticamente perfetto. Perché? Se si considera la velocità con cui avviene l’isomerizzazione in presenza di un catalizzatore basico come può essere l’acetone, l’enzima è in grado di accelerarla di un fattore 1010. Infatti, analizzando il rapporto Kcat/KM questi è pari a 2×108 M-1s-1, valore prossimo alla limite controllato dalla diffusione. Quindi, il passaggio limitante nella catalisi è l’incontro tra enzima e substrato controllato appunto dalla diffusione.
Considerando dal punto di vista energetico gli ultimi due passaggi della glicolisi, questi sono sfavorevoli, con ΔG°’ pari a 7,5 kcal/mol (31,3 kJ/mol), mentre il ΔG°’complessivo dei primi 5 passaggi è pari a 0,5 kcal/mol (2,1 kJ/mol), con una Keq complessiva di circa 0,43. Ed è l’energia libera derivante dall’idrolisi delle due molecole di ATP che, in condizioni standard, sposta la costante di equilibrio complessiva vicino a uno. Se invece si considera il ΔG questi è ampiamente negativo, -13,6 kcal/mol (-56,8 kJ/mol).
Nota: il diidrossiacetone fosfato può anche essere ridotto a glicerolo-3-fosfato nella reazione catalizzata dalla glicerolo-3-fosfafo deidrogenasi (EC 1.1.1.8).
L’enzima fa da ponte tra il metabolismo glucidico e lipidico in quanto il glicerolo-3-fosfato prodotto è utilizzato nella sintesi di lipidi come i trigliceridi.
Questa via è particolarmente importante nel tessuto adiposo e nell’intestino tenue.
Reazione 6
Nella sesta tappa della via glicolitica, la prima della seconda fase, si verifica l’ossidazione della gliceraldeide-3-fosfato a dare 1,3-bisfosfoglicerato (1,3-BPG) e la concomitante riduzione del NAD+ a NADH. La reazione è catalizzata dalla gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12).
Questa reazione è la prima delle due reazione della glicolisi in cui viene resa disponibile l’energia chimica necessaria per la successiva sintesi dell’ATP; l’altra è quella catalizzata dalla enolasi (EC 4.2.1.11). Perché?
La reazione è la somma di due processi.
Nella prima reazione si verifica l’ossidazione del gruppo aldeidico a gruppo carbossilico, passaggio nel quale il NAD+ funge da agente ossidante. La reazione è fortemente esoergonica, con un ∆G’° di -10,3 kcal/mol (-43 kJ/mol).
Nella seconda reazione si verifica la formazione del legame tra l’ortofosfato e il gruppo carbossilico sul C-1 dell’1,3-bisfosfoglicerato a dare un’anidride mista, detta acil fosfato. Questa reazione è fortemente endoergonica, con ∆G’° di 11,8 23kcal/mol (49,3 kJ/mol).
Affinché possa verificarsi la formazione dell’acil fosfato, le due reazioni precedenti non debbono avvenire in successione ma debbono essere accoppiate. In questo modo l’ossidazione dell’aldeide viene utilizzata per guidare la formazione dell’anidride mista, con un ΔG°’ complessivo leggermente endoergonico, 1,5 kcal/mol (6,3 kJ/mol), mentre il valore del ΔG è 0,6 kcal/mol (2,5 kJ/mol).
Dunque la maggior parte dell’energia libera derivante dall’ossidazione del gruppo aldeidico anziché dissiparsi in calore viene conservata grazie alla formazione dell’acil fosfato.
Nota: la riduzione, reversibile dell’anello nicotinamidico del NAD+ o del NADP+ è conseguente alla perdita di due atomi d’idrogeno da parte di un’altra molecola, in questo caso il gruppo aldeidico della gliceraldeide-3-fosfato, che quindi si ossida, e al successivo trasferimento di uno ione idruro, ossia due elettroni e un protone, all’anello suddetto. L’H+ rimanente è rilasciato nel mezzo acquoso. Di seguito le semireazioni per entrambe i coenzimi.
NAD+ + 2 e– + 2 H+ → NADH + H+
NADP+ + 2 e– + 2 H+ → NADPH + H+
Reazione 7
Nella settima tappa della glicolisi si verifica il trasferimento del gruppo fosforico ad alta energia dall’acil fosfato dell’1,3-bisfosfoglicerato all’ADP con formazione di ATP e 3-fosfoglicerato. La reazione è catalizzata dalla fosfoglicerato chinasi (EC 2.7.2.3).
1,3-Bisfosfoglicerato + ADP + H+ ⇄ 3-Fosfoglicerato + ATP
Il ΔG°’ della reazione è pari a -4,4 kcal/mol (-18,5 kJ/mol), quindi si tratta di una reazione esoergonica. Il ΔG è pari a 0,3 kcal/mol (1,3 kJ/mol).
L’elevato potenziale di trasferimento del gruppo fosforico dell’acil fosfato viene sfruttato per la fosforilazione dell’ADP, reazione definita fosforilazione a livello del substrato. In altri termini, parte dell’energia rilasciata nel corso dell’ossidazione del gruppo aldeidico nel passaggio precedente viene ora conservata attraverso la formazione di ATP dall’ADP e Pi.
La reazione catalizzata dalla fosfoglicerato chinasi è la prima delle due reazione della glicolisi in cui parte dell’energia chimica presente nella molecola del glucosio viene conservata in forma di ATP. E, poiché dall’azione dell’aldolasi e della trioso fosfato isomerasi, reazione 4 e 5, si formano due molecole di gliceraldeide-3-fosfato per molecola di glucosio, in questo passaggio sono prodotte due molecole di ATP che vanno a “pareggiare” le due molecole di ATP consumate nella fase preparatoria.
Nota: la fosfoglicerato chinasi prende il nome dalla reazione che procede nella direzione opposta rispetto a quella appena descritta, ossia la fosforilazione del 3-fosfoglicerato a dare 1,3-bisfosfoglicerato a spese di una molecola di ATP. L’enzima infatti, al pari di tutti gli altri enzimi, è in grado di catalizzare la reazione in entrambe le direzioni. E la direzione che porta alla sintesi dell’1,3-bisfosfoglicerato è seguita durante la fissazione fotosintetica della CO2 e la gluconeogenesi.
La reazione sei e la sette nel loro insieme rappresentano un processo di accoppiamento energetico in cui l’intermedio comune è rappresentato dall’1,3-bisfosfoglicerato. Mentre la reazione che porta alla sintesi dell’1,3-bisfosfoglicerato è endoergonica, con un ΔG°’ = 1,5 kcal/mol (6,3 kJ/mol), la seconda è esoergonica, ΔG°’ = -4,4 kcal/mol (-18,5 kJ/mol). Il ΔG°’ complessivo è pari a (-4,4 + 1,5) = 2,9 kcal/mol (-18,5 + 6,3 = 12,2 kJ/mol), ossia la razione catalizzata dalla fosfoglicerato chinasi è sufficientemente esoergonica da “trascinare” anche la precedente, rendendo la reazione complessiva esoergonica.
Gliceraldeide-3-fosfato + ADP + Pi + NAD+ ⇄ 3-Fosfoglicerato + ATP + NADH + H+
In verità, la reazione della fosfoglicerato chinasi è sufficientemente esoergonica da trascinare anche le reazioni catalizzate dalla aldolasi e dalla trioso fosfato isomerasi.
Poiché l’energia libera standard d’idrolisi del gruppo fosforico sul C-3 del 3-fosfoglicerato è pari a -3 kcal/mol (-12,5 kJ/mol), non è possibile alcuna sintesi di ATP dall’ADP e dal 3-fosfoglicerato stesso. Le due reazioni successive della glicolisi porteranno alla conversione del 3-fosfoglicerato in fosfoenolpiruvato, una molecola con un potenziale di trasferimento del gruppo fosforico sufficientemente alto da permettere la sintesi di ATP.
Reazione 8
Nell’ottava tappa della glicolisi il 3-fosfoglicerato viene convertito in 2-fosfoglicerato, in una reazione reversibile catalizzata dalla fosfoglicerato mutasi (EC 5.4.2.1). La reazione richiede la presenza di Mg2+, ha un ΔG molto basso, pari a circa 0,2 kcal/mol (0,8 kJ/mol) e un ΔG°’ pari a 1,1 kcal/mol (4,4 kJ/mol).
L’enzima è una mutasi, enzimi che catalizzano lo spostamento intramolecolare di un gruppo chimico, in questo caso lo spostamento di un gruppo fosforico dal C-3 al C-2 del 3-fosfoglicerato. A loro volta le mutasi sono una sottoclasse delle isomerasi.
Il meccanismo con cui si svolge questa reazione dipende dal tipo di organismo considerato. Ad esempio, nel lievito o nel muscolo di coniglio la reazione avviene in due passaggi e comporta la formazione di un intermedio fosfo-enzima. Nella prima parte della reazione un gruppo fosforico legato covalentemente a un residuo di istidina presente nel sito attivo viene trasferito all’ossidrile presente sul C-2 del 3-fosfoglicerato a dare il 2,3-bisfosfoglicerato. Nel passaggio successivo l’enzima opera come una fosfatasi convertendo il 2,3-bisfosfoglicerato in 2-fosfoglicerato; tuttavia il gruppo fosforico presente sul C-3 non viene rilasciato ma si lega al residuo d’istidina del sito attivo a rigenerare l’intermedio enzima-His-fosfato. Schematicamente:
Vale la pena notare che il gruppo fosforico del 2-fosfoglicerato prodotto non è lo stesso di quello del substrato 3-fosfoglicerato.
All’incirca una volta ogni 100 passaggi catalitici il 2,3-bisfosfoglicerato si dissocia dal sito attivo dall’enzima, lasciandolo privo dell’istidina fosforilata, e quindi in forma inattiva. L’enzima in forma inattiva può essere riattivato dal 2,3-BPG, che è quindi necessario sia sempre presente in piccole quantità. E il 2,3-bisfosfoglicerato è in effetti presente in piccole quantità nella maggior parte delle cellule. Fanno eccezione i globuli rossi, dove è prodotto attraverso lo shunt di Rapoport-Luebering e agisce anche come inibitore allosterico riducendo l’affinità dell’emoglobina per l’ossigeno, e dove la sua concentrazione è di circa 4-5 mM.
Nota: il 3-fosfoglicerato, oltre che proseguire nella via glicolitica, può essere utilizzato anche per la sintesi della serina, da cui derivano glicina e cisteina. La sintesi della serina ha inizio con la reazione catalizzata dalla fosfoglicerato deidrogenasi (EC 1.1.1.95), in cui si verifica l’ossidazione del 3-fosfoglicerato a 3-fosfoidrossipiruvato e la concomitante riduzione del NAD+ a NADH. Questa reazione costituisce anche il passaggio limitante dell’intera via biosintetica, in quanto la serina inibisce l’attività della deidrogenasi.
Reazione 9
Nella nona tappa della glicolisi si verifica la deidratazione, la rimozione di una molecola di acqua, del 2-fosfoglicerato a dare fosfoenolpiruvato, un enolo. La reazione, reversibile, è catalizzata dalla enolasi.
2-Fosfoglicerato ⇄ Fosfoenolpiruvato + H2O
La reazione richiede la presenza di Mg2+ che va a stabilizzare l’intermedio enolico che si forma nel corso del processo.
Il ΔG°’ della reazione è pari a 1,8 kcal/mol (7,5 kJ/mol), mentre il ΔG è pari a -0,8 kcal/mol (-3,3 kJ/mol).
Come anticipato fosfoenolpiruvato e 1,3-bisfosfoglicerato sono gli unici due intermedi della glicolisi a possedere un potenziale di trasferimento del gruppo fosforico sufficiente a permettere la sintesi dell’ATP. Da dove deriva l’elevata energia libera standard d’idrolisi del gruppo fosforico del fosfoenolpiruvato. Sebbene fosfoenolpiruvato e 2-fosfoglicerato contengano all’incirca la stessa quantità totale di energia, rispetto alla decomposizione a CO2 e H2O e Pi, la deidratazione del 2-fosfoglicerato porta a una diversa ridistribuzione della stessa, tale che l’energia libera standard d’idrolisi dei due gruppi fosforici corrisponde a:
-4,2 kcal/mol (-17,6 kJ/mol) per il 2-fosfoglicerato, un estere fosforico;
-14,8 kcal/mol (-61,9 kJ/mol) per il fosfoenolpiruvato, un enol fosfato.
Quello che accade è che il gruppo fosforico del fosfoenolpiruvato intrappola la molecola nella sua forma enolica, instabile. Quando, nell’ultima reazione della glicolisi, il fosfoenolpiruvato cede il gruppo fosforico all’ADP si formano ATP e la forma enolica del piruvato, instabile, che rapidamente e non enzimaticamente tautomerizza alla più stabile forma chetonica, prevalente a pH 7. Quindi, alla base dell’elevato potenziale di trasferimento del gruppo fosforico del fosfoenolpiruvato c’è la successiva tautomerizzazione enolo-chetone del piruvato.
Reazione 10
Nell’ultimo passaggio della via glicolitica si verifica il trasferimento del gruppo fosforico dal fosfoenolpiruvato all’ADP con produzione di una molecola di ATP e una di piruvato.
Fosfoenolpiruvato + ADP + H+ → Piruvato + ATP
La reazione, la seconda fosforilazione a livello del substrato della glicolisi, è catalizzata dalla piruvato chinasi, enzima tetramerico la cui attività, al pari della PFK-1, è altamente regolata. Infatti l’enzima possiede siti di legame per numerosi effettori allosterici. Inoltre nei vertebrati sono presenti tre forme isoenzimatiche, di cui una prevalente nel fegato, indicata come forma L, e un’altra, detta forma M, prevalente nel muscolo e nel cervello. I diversi isoenzimi hanno molte proprietà in comune, ma differiscono, oltre che nella distribuzione tissutale, anche nella risposta a ormoni quali glucagone, epinefrina e insulina.
L’attività dell’enzima è stimolata dallo ione potassio, K+, e da alcuni altri cationi monovalenti.
La reazione è essenzialmente irreversibile, con un ΔG°’ pari a -7,5 kcal/mol (-31,4 kJ/mol), e un ΔG pari a -4,0 kcal/mol (-16,7 kJ/mol), irreversibilità in gran parte dovuta, come anticipato nel paragrafo precedente, alla rapida tautomerizzazione spontanea del piruvato dalla forma enolica a quella chetonica.
Tautomeri, Enolico e Chetonico, del Piruvato
E, delle 14,8 kcal/mol (-61,9 kJ/mol) derivanti dall’idrolisi dell’enol fosfato del fosfoenolpiruvato, circa la metà viene conservata grazie alla formazione del legame fosfoanidridico tra Pi e ADP, il cui ΔG°’ d’idrolisi è pari a -7,3 kcal/mol (-30,5 kJ/mol). La restante energia, -7,5 kcal/mol (-31,4 kJ/mol), è la forza trainante che spinge la reazione verso la produzione di ATP.
Se, con la reazione catalizzata dalla fosfoglicerato chinasi venivano pareggiate le due molecole di ATP spese nella fase preparatoria della glicolisi, con la reazione catalizzata dalla piruvato chinasi si ha un guadagno netto di due molecole di ATP per molecola di glucosio.
Destino del piruvato e NADH
I prodotti della glicolisi sono tre: ATP, piruvato e NADH.
Il NADH deve essere riossidato a NAD+ per permettere alla glicolisi di procedere.[2]
Il NAD+, un coenzima derivante dalla vitamina niacina, è presente nella cellula in quantità limitata, ≤ 10-5M, valore ben inferiore a quello del glucosio metabolizzato in pochi minuti, e deve essere quindi continuamente rigenerato. E il passaggio finale della via glicolitica è proprio la sua rigenerazione dal NADH attraverso vie metaboliche aerobiche o anaerobiche, ognuna delle quali comporta un ulteriore metabolismo del piruvato. Queste vie permettono quindi il mantenimento del bilancio redox della cellula.
Il piruvato è una molecola assai versatile che può entrare in diverse vie metaboliche, sia anaboliche che cataboliche, a seconda del tipo di cellula, dello stato energetico della cellula e della disponibilità di ossigeno.[5]
Catabolismo del Piruvato
Con l’esclusione di alcune “variazioni” che si incontrano nel mondo dei batteri, sfruttate anche dall’industria alimentare per la produzione di vari alimenti tra cui molti formaggi, sono essenzialmente tre le vie cataboliche che possono essere imboccate dal piruvato:
la riduzione ad acido lattico, attraverso la fermentazione lattica;
la riduzione ad alcol etilico, attraverso la fermentazione alcolica;
l’ossidazione aerobica.
Questo permette alla glicolisi di procedere sia in condizioni anaerobiche che aerobiche.
E’ quindi anche possibile affermare, ampliando la visuale, che il destino catabolico dello scheletro carbonioso del glucosio è influenzato dal tipo di cellula, dal suo stato energetico e dalla disponibilità di ossigeno.
Fermentazione lattica
Negli animali, con poche eccezioni, e in molti microrganismi quando la disponibilità di ossigeno non è sufficiente a soddisfare le richieste energetiche della cellula, o se la cellula è priva di mitocondri, il piruvato prodotto dalla glicolisi viene ridotto a lattato nella reazione catalizzata dall’enzima citosolico lattato deidrogenasi (EC 1.1.1.27).
Piruvato + NADH + H+ ⇄ Lattato + NAD+
Nella reazione il NADH fornisce gli equivalenti riducenti e si ossida a NAD+. L’equilibrio complessivo della reazione favorisce fortemente la formazione del lattato, come testimoniato anche del valore del ΔG°’ pari a -6 kcal/mol (-25,1 kJ/mol).
La conversione del glucosio in acido lattico viene definita fermentazione lattica. L’equazione complessiva del processo è:
Glucosio + 2 Pi + 2 ADP + 2 H+ → 2 Lattato + 2 ATP + 2 H2O
Si noti che la fermentazione, scoperta da Louis Pasteur che la definì “la vie sans l’air”, è un processo che:
estrae energia dalla molecola del glucosio immagazzinandola in forma di ATP;
non consuma ossigeno;
non modifica la concentrazione del NAD+ o del NADH.
Riguardo ai coenzimi si noti che nell’equazione complessiva della fermentazione lattica non compaiono ne NAD+ ne NADH, sebbene entrambe cruciali nel processo. Quindi non si verifica alcuna ossidoriduzione netta. In altri termini, nel passaggio da glucosio C6H12O6, a lattato, C3H6O3, il rapporto H:C non varia.
Dal punto di vista energetico va sottolineato che la fermentazione permette di estrarre una ridotta quantità dell’energia chimica contenuta nella molecola del glucosio.
Nell’uomo molto del lattato prodotto entra nel ciclo di Cori, per essere riutilizzato nella gluconeogenesi. In definitiva si può anche affermare che la produzione di lattato sposta parte del carico metabolico dalla periferia, ad esempio dal muscolo scheletrico di un atleta impegnato in un esercizio molto inteso, come uno sprint, quando la velocità della glicolisi può quasi istantaneamente aumentare anche di 2000 volte, al fegato.
Al contrario del muscolo scheletrico che rilascia in circolo lattato, il muscolo cardiaco è in grado di assumere lattato e utilizzarlo come carburante per produrre ATP. Questo è possibile grazie alle proprietà dell’isoenzima cardiaco della lattato deidrogenasi, indicato come H4, e al suo metabolismo completamente aerobico. Quindi, parte del lattato rilasciato dal muscolo scheletrico sottoposto a un lavoro inteso sarà utilizzato dal muscolo cardiaco come carburante.
Nota: il lattato prodotto dai microorganismi durante la fermentazione lattica è responsabile sia del profumo che del gusto dei crauti, ossia dei cavoli fermentati, come anche del gusto del latte acido.
Fermentazione alcolica
In microorganismi come il lievito di birra e il lievito da panificazione, ma anche in certi tessuti vegetali, e in alcuni invertebrati e protisti, il piruvato, in condizioni ipossiche o anerobiche, può essere ridotto in due passaggi ad alcol etilico o etanolo, con liberazione di CO2.
Il primo passaggio comporta la decarbossilazione non ossidativa del piruvato a dare acetaldeide, una reazione essenzialmente irreversibile. La reazione è catalizzata dalla piruvato decarbossilasi (EC 4.1.1.1), enzima che richiede la presenza di Mg2+ e tiamina pirofosfato, un coenzima derivante dalla vitamina tiamina o vitamina B1. L’enzima è assente nei tessuti dei vertebrati e negli altri organismi che operano la fermentazione lattica.
Nel secondo passaggio si verifica la riduzione dell’acetaldeide a etanolo nella reazione catalizzata dalla alcol deidrogenasi (EC 1.1.1.1), enzima che ha un atomo di zinco legato nel sito attivo. Nella reazione il NADH fornisce gli equivalenti riducenti e si ossida a NAD+. A pH neutro, l’equilibrio della reazione è fortemente spostato verso la formazione dell’alcol etilico.
La conversione del glucosio in etanolo e CO2 viene definita fermentazione alcolica. La reazione complessiva è:
Glucosio + 2 Pi + 2 ADP + 2 H+ → 2 Etanolo + 2 CO2 + 2 ATP + 2 H2O
E, al pari della fermentazione lattica, anche nella fermentazione alcolica non si verifica alcuna ossido-riduzione netta.
La fermentazione alcolica è alla base della produzione della birra e del vino. Da notare che la CO2 prodotta dal lievito di birra è responsabile delle caratteristiche bollicine della birra, dello champagne e dello spumante, mentre quella prodotta dal lievito usato nella panificazione porta alla lievitazione dell’impasto.
Destino del piruvato e NADH in condizioni aerobiche
Nelle cellule dotate di mitocondri e in condizioni aerobiche, la situazione più comune negli organismi multicellulari e in molti unicellulari, l’ossidazione del NADH e il catabolismo del piruvato seguono vie distinte.
Il piruvato viene dapprima convertito in acetil-CoA nelle reazioni catalizzate dal complesso della piruvato deidrogenasi, uno dei complessi multienzimatici mitocondriali. Nella reazione, una decarbossilazione ossidativa, la molecola perde un atomo di carbonio in forma di CO2, e l’unità a due atomi di carbonio rimanente è legata al coenzima A a dare acetil-coenzima A o semplicemente acetil-CoA.
Piruvato + NAD+ + CoA → Acetil-CoA + CO2 + NADH + H+
Il gruppo acetilico dell’acetil-CoA è quindi completamente ossidato a CO2 attraverso le reazioni del ciclo dell’acido citrico, con produzione di ulteriore NADH, e anche di FADH2. Il complesso della piruvato deidrogenasi rappresenta quindi un ponte tra la glicolisi, che si verifica nel citosol, e il ciclo dell’acido citrico, una via metabolica mitocondriale.
Gli elettroni derivati dalle ossidazioni che si verificano durante la glicolisi sono trasportati nel mitocondrio a seguito della riduzione di intermedi metabolici citosolici. In questo modo nel citosol viene rigenerato NAD+ dal NADH, mentre l’intermedio ridotto, una volta nella matrice mitocondriale, è riossidato grazie al trasferimento dei suoi equivalenti riducenti al Complesso I della catena di trasporto degli elettroni mitocondriale. In questa sede gli elettroni passano all’ossigeno a dare H2O, trasferimento che fornisce l’energia necessaria per la sintesi dell’ATP attraverso il processo della fosforilazione ossidativa.
Ovviamente anche gli elettroni trasportati dal NADH prodotto nelle reazioni catalizzate dal complesso della piruvato deidrogenasi e nel ciclo dell’acido citrico, e quelli del FADH2, seguono un analogo destino.
Nota: il FADH2 cede i suoi equivalenti riducenti non al Complesso I ma al Complesso II.
Destino anabolico del piruvato
In condizioni anaboliche lo scheletro carbonioso del piruvato può avere destini diversi dalla completa ossidazione a CO2, o dalla conversione in lattato. Infatti, previa conversione in acetil-CoA, potrà essere utilizzato ad esempio per la sintesi degli acidi grassi, o dell’aminoacido alanina.
Glicolisi e produzione di ATP
Nella glicolisi una molecola di glucosio viene convertita in due di piruvato.
Durante la prima fase, la fase preparatoria, sono consumate due molecole di ATP nelle reazioni catalizzate dalla esochinasi e dalla PFK-1. Nella seconda fase, la fase di recupero energetico, sono prodotte 4 molecole di ATP attraverso fosforilazioni a livello del substrato nelle reazioni catalizzate dalla fosfoglicerato chinasi e dalla piruvato chinasi. Quindi si ha un guadagno netto di due molecole di ATP per molecola di glucosio metabolizzata. In più, nella reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi, per ogni molecola di glucosio sono prodotte due molecole di NADH.[5]
ΔG°’ e ΔG delle Reazioni della Glicolisi
Il ΔG°’ dell’intero processo è pari a -20,3 kcal/mol (-85 kJ/mol), valore che deriva dalla differenza tra il ΔG°’ della conversione del glucosio in due molecole di piruvato, pari a -34,9 kcal/mol (146 kJ/mol), e il ΔG°’ della formazione dell’ATP da ADP e Pi, pari a 2 x 7,3 kcal/mol = 14,6 kcal/mol (2 x 30,5 kJ/mol = 61 kJ/mol). Di seguito le due reazioni.
Glucosio + 2 NAD+ → 2 Piruvato + 2 NADH + 2 H+
2 ADP + 2 Pi → 2 ATP + 2 H2O
La somma delle due reazioni da la reazione complessiva della glicolisi.
Eliminando i termini comuni si ottiene l’equazione complessiva mostrata sopra.
Produzione di ATP in condizioni anaerobiche
In condizioni anaerobiche, a prescindere da quello che sarà il destino metabolico del piruvato, conversione in lattato, etanolo o altre molecole, non sono prodotte altre molecole di ATP a valle della glicolisi.
Quindi in queste condizioni la glicolisi permette di estrarre solamente una frazione dell’energia chimica contenuta nella molecola del glucosio, pari a 679 kcal/mol (2840 kJ/mol) rilasciate a seguito della sua ossidazione a CO2 e H2O. Infatti, la conversione del glucosio in due molecole di piruvato porta al rilascio di 34,9 kcal/mol, il che significa che solamente il 5%, [(34,9/679) x 100], dell’energia chimica contenuta nella molecola del glucosio è rilascia a seguito della sua conversione in piruvato. Quindi il piruvato contiene ancora la maggior parte dell’energia chimica del glucosio.
Analogamente, neppure i 4 elettroni, in forma di ioni idruro, trasportati dalle due molecole di NADH potranno essere utilizzati per la produzione di ATP.
Considerando la fermentazione lattica, il ΔG°’ associato alla conversione di una molecola di glucosio in due di lattosio è pari a -43,9 kcal/mol (-183,6 kJ/mol) e la percentuale dell’energia libera rilasciata e immagazzinata in forma di ATP sarà pari a: (14,6/43,9) x 100 = 33,2%, mentre se si considera la sola glicolisi la percentuale è del 41,8%. Da notare che nelle condizioni intracellulari la quantità di energia libera necessaria per la sintesi dell’ATP da ADP e Pi è molto più elevata di quella in condizioni standard, per cui la percentuale dell’energia libera disponibile immagazzinata è maggiore, pari a circa il 50% del totale.
Produzione di ATP in condizioni aerobiche
In condizioni aerobiche, nelle cellule dotate di mitocondri, la quantità di energia chimica che può essere estratta dalla molecola del glucosio e immagazzinata in forma di ATP è molto maggiore rispetto a quanto accade in assenza di ossigeno. Se si considerano solo le due molecole di NADH prodotte durante la glicolisi, il trasferimento dei loro 4 equivalenti riducenti lungo la catena di trasporto degli elettroni mitocondriale permette la produzione di 2-3 molecole di ATP per coppia di elettroni attraverso la fosforilazione ossidativa. In questo caso si avrà una produzione netta di ATP compresa tra 6 e 8 molecole per ogni molecola di glucosio ossidata a due di piruvato, dalla glicolisi e 4-6 dalla fosforilazione ossidativa.
Nota: la quantità complessiva di ATP prodotta dagli equivalenti riducenti del NADH dipende dal sistema attraverso cui gli equivalenti riducenti del coenzima ridotto entrano nel mitocondrio.
Se invece si analizza l’azione concertata della glicolisi, del complesso della piruvato deidrogenasi, del ciclo dell’acido citrico, della catena di trasporto degli elettroni mitocondriale e della fosforilazione ossidativa, viene estratta e immagazzinata in forma di ATP molta altra dell’energia chimica disponibile presente nel monosaccaride. In questo caso, secondo quanto riportato dal Lehninger sono prodotti 30-32 ATP/molecola di glucosio, sebbene recenti stime suggeriscano una produzione netta pari a 29,85 ATP/molecola di glucosio, o 29,38 ATP/molecola di glucosio se anche l’ATP derivato dal GTP, prodotto dal ciclo dell’acido citrico, viene esportato. Considerando entrambe le stime, la produzione di ATP è circa 15 volte maggiore rispetto a quanto accade in assenza di ossigeno.
Vie di alimentazione
Oltre al glucosio, altri carboidrati, sia semplici che complessi, possono essere catabolizzati attraverso la glicolisi, previa conversione enzimatica in uno degli intermedi della via metabolica stessa.[2][7]
Tra i più importanti si ritrovano:
due polisaccaridi di deposito, il glicogeno e l’amido;
i monosaccaridi galattosio e fruttosio; un terzo monosaccaride è il mannosio, meno comune rispetto ai precedenti.
Vie di Alimentazione della Glicolisi
A livello intestinale, amido e disaccaridi assunti con l’alimentazione incontrano gli enzimi responsabili della digestione dei carboidrati. Segue l’assorbimento dei monosaccaridi liberati e derivanti dall’alimentazione. Una volta nel circolo venoso i monosaccaridi raggiungono, attraverso la vena porta, il fegato, ed è principalmente in questa sede che sono metabolizzati.
Glicogeno e amido
Per quanto riguarda il catabolismo fosforolitico dell’amido e del glicogeno endogeno si rimanda ai rispettivi articoli.
Fruttosio
In condizioni fisiologiche, il fegato rimuove dal circolo ematico molto del fruttosio ingerito, prima che lo stesso possa raggiungere i tessuti extraepatici.
La via metabolica epatica che porta alla conversione del monosaccaride in intermedi della glicolisi si compone di alcuni passaggi.
Nel primo il fruttosio viene fosforilato a fruttosio-1-fosfato nella reazione catalizzata dalla fruttochinasi (EC 2.7.1.4), con consumo di una molecola di ATP.
Fruttosio + ATP → Fruttosio-1-fosfato + ADP + H+
La reazione richiede la presenza di ioni magnesio.
Come per il glucosio, anche per il fruttosio la fosforilazione ha come effetto quello di intrappolare la molecola all’interno della cellula.
Nel secondo passaggio il fruttosio-1-fosfato viene scisso a diidrossiacetone fosfato e gliceraldeide, nella reazione catalizzata dalla fruttosio-1-fosfato aldolasi.
Il diidrossiacetone fosfato è un intermedio della glicolisi e prosegue lungo la via previa conversione in gliceraldeide-3-fosfato. Invece, la gliceraldeide, che non è un intermedio della glicolisi, viene fosforilata a gliceraldeide-3-fosfato nella reazione catalizzata dalla trioso chinasi (EC 2.7.1.28), con consumo di una seconda molecola di ATP.
Gliceraldeide + ATP → Gliceraldeide-3-fosfato + ADP + H+
La reazione richiede la presenza di ioni Mg2+.
Quindi nell’epatocita, una molecola di fruttosio viene convertita in due di gliceraldeide-3-fosfato, con consumo di due molecole di ATP, come per il glucosio.
In sedi extraepatiche come il muscolo scheletrico, il rene o il tessuto adiposo non è presente la fruttochinasi, e il fruttosio entra nella via glicolitica in forma di fruttosio-6-fosfato. Infatti tra i substrati della esochinasi c’è anche il fruttosio, che viene fosforilato in posizione C-6.
Fruttosio + ATP → Fruttosio-6-fosfato + ADP + H+
L’affinità dell’enzima per il fruttosio è però circa 20 volte minore rispetto a quella per il glucosio, per cui nell’epatocita, dove il glucosio è molto più abbondante del fruttosio, o nel muscolo scheletrico in condizioni di scarsa disponibilità di ossigeno, quando cioè il glucosio diviene il carburante preferito, si forma poco fruttosio-6-fosfato. Al contrario, nel tessuto adiposo il fruttosio è più abbondante del glucosio per cui la sua fosforilazione a opera della esochinasi non è inibita competitivamente in modo significativo dal glucosio. In questo tessuto quindi la sintesi del fruttosio-6-fosfato rappresenta il modo principale per incanalare il monosaccaride nella via glicolitica.
Ai fini degli effetti metabolici del fruttosio è importante sottolineare che nel fegato il monosaccaride, essendo fosforilato in posizione C-1, entra nella glicolisi a livello dei trioso fosfati, dunque a valle della reazione catalizzata dalla PFK-1, enzima che gioca un ruolo chiave nella regolazione del flusso del carbonio attraverso la glicolisi stessa. Al contrario, quando il monosaccaride viene fosforilato sul C-6, entra nella via glicolitica a monte della reazione catalizzata dalla PFK-1.
Sorbitolo
Il fruttosio rappresenta il punto d’ingresso nella via glicolitica anche per il sorbitolo. La molecola è presente in molti frutti e vegetali e utilizzata anche come uno dei dolcificanti e stabilizzanti. Nel fegato è convertita in fruttosio nella reazione catalizzata dall’enzima citosolico sorbitolo deidrogenasi (EC 1.1.99.21), enzima che fa parte anche della via dei polioli, per la conversione del glucosio a fruttosio.
Sorbitolo + NAD+ → Fruttosio + NADH + H+
La reazione richiede la presenza di ioni Zn2+.
Galattosio
Il galattosio, che per la maggior parte deriva dalla digestione intestinale del lattosio, raggiunto il fegato viene convertito, attraverso la via di Leloir, in glucosio-1-fosfato. Il destino del glucosio-1-fosfato dipende dallo stato energetico della cellula. In condizioni che favoriscono il deposito di glucosio, la molecola può essere incanalata nella via di sintesi del glicogeno. In condizioni che favoriscono l’utilizzo del glucosio per la produzione di energia, il glucosio-1-fosfato viene isomerizzato in glucosio-6-fosfato nella reazione reversibile catalizzata dalla fosfoglucomutasi (EC 5.4.2.2).
Glucosio-1-fosfato ⇄ Glucosio-6-fosfato
A sua volta il glucosio-6-fosfato prodotto potrà essere incanalato nella via glicolitica per essere utilizzato per la produzione di energia, o essere defosforilato a glucosio nella reazione catalizzata dal complesso proteico della glucosio-6-fosfatasi ed essere quindi rilasciato nel circolo ematico.
Mannosio
Il mannosio è presente in vari polisaccaridi, glicolipidi e glicoproteine della dieta. Una volta liberato da queste fonti, viene assorbito, e raggiunto il fegato è fosforilato in posizione C-6 a dare mannosio-6-fosfato, nella reazione catalizzata dalla esochinasi.
Mannosio + ATP → Mannosio-6-fosfato + ADP + H+
Il mannosio-6-fosfato è quindi isomerizzato a fruttosio-6-fosfato nella reazione catalizzata dalla mannosio-6-fosfato isomerasi (EC 5.3.1.8.).
Mannosio-6-fosfato ⇄ Fruttosio-6-fosfato
Regolazione della glicolisi
Il flusso di carbonio attraverso la glicolisi viene regolato in base alle condizioni metaboliche all’interno e all’esterno della cellula, rispondendo essenzialmente a due bisogni:
la produzione di ATP;
la produzione di precursori per molte vie biosintetiche.
Inoltre, nel fegato, per evitare uno spreco di energia, glicolisi e gluconeogenesi sono finemente regolate in modo che quando una via procede l’altra rallenta. Nel corso dell’evoluzione ciò è stato ottenuto selezionando enzimi differenti per catalizzare le reazioni essenzialmente irreversibili delle due vie, enzimi la cui attività è regolata separatamente. Se infatti queste reazioni procedessero simultaneamente a elevata velocità creerebbero un ciclo futile o ciclo del substrato.[7] Una regolazione così fine non potrebbe essere ottenuta se uno stesso enzima catalizzasse la reazione nelle due direzioni.
Il controllo del flusso di carbonio attraverso la glicolisi coinvolge principalmente le reazioni catalizzate dagli enzimi esochinasi, PFK-1 e piruvato chinasi, la cui attività è regolata mediante:
meccanismi allosterici, che si svolgono in un arco temporale di millisecondi e sono istantaneamente reversibili;
modificazioni covalenti, ossia fosforilazioni e defosforilazioni, che si svolgono in secondi;
modificazioni nella concentrazione degli enzimi coinvolti, conseguenti a modifiche nella loro velocità di sintesi/degradazione, che avvengono in ore.
Nota: per la gluconeogenesi i principali punti di regolazione sono le reazioni catalizzate dagli enzimi piruvato carbossilasi (EC 6.4.1.1) e fruttosio-1,6-bisfosfatasi (EC 3.1.3.11).
Esochinasi
Nell’uomo la esochinasi è presente con quattro forme isoenzimatiche tessuto specifiche, designate da I a IV, e codificate da altrettanti geni differenti.[2][5]
La esochinasi I è l’isoenzima prevalente nel cervello, mentre nel muscolo scheletrico si ritrova sia l’esochinasi I, che costituisce il 70-75% del totale, che la esochinasi II, che rappresenta il restante 25-30%.
L’esochinasi IV, detta anche glucochinasi (EC 2.7.1.2) è presente prevalentemente negli epatociti e nelle cellule β del pancreas, dove è il principale isoenzima. Nel fegato, con la glucosio-6-fosfatasi, catalizza il ciclo del substrato tra glucosio e glucosio-6-fosfato.
La glucochinasi differisce dalle altre isoforme della esochinasi per quanto riguarda la cinetica e le proprietà regolatorie.[1]
Nota: gli isoenzimi o isozimi sono proteine differenti che catalizzano la stessa reazione, e che in genere si differenziano per le proprietà cinetiche e regolatorie, distribuzione subcellulare, o per i cofattori utilizzati. Possono essere presenti contemporaneamente in una stessa specie, tessuto o anche cellula.
Proprietà cinetiche delle esochinasi
Le esochinasi I, II e III hanno proprietà cinetiche simili. La esochinasi I e la II hanno una Km per il glucosio, ossia la concentrazione del glucosio alla quale l’enzima è per metà saturato, rispettivamente pari a 0,03 mM e 0,1 mM. Pertanto questi due isoenzimi lavorano in modo molto efficiente ai normali valori di glicemia, che sono pari a circa 4-5 mM.[7]
Invece la glucochinasi ha una Km per il glucosio assai più alta, pari a circa 10 mM; questo significa che l’attività dell’enzima diviene importante solo quando i valori della glicemia sono alti, come dopo un pasto ricco di carboidrati ad alto indice glicemico.
Regolazione delle esochinasi I-III
Le esochinasi I-III sono inibite allostericamente dal glucosio-6-fosfato, il prodotto della loro reazione. Questo assicura che il glucosio-6-fosfato non si accumuli nella cellula quando non è richiesto ulteriore glucosio per la produzione di energia, la sintesi del glicogeno, la via del pentoso fosfato, o come fonte di intermedi per la sintesi di altre molecole, e al contempo permette di ridurre l’assunzione dal circolo di altro glucosio che rimane quindi disponibile per altri organi e tessuti. Ad esempio, quando la PFK-1 è inibita, si accumula fruttosio-6-fosfato e, grazie alla reazione catalizzata dalla fosfoglucosio isomerasi, glucosio-6-fosfato. Dunque, l’inibizione della PFK-1 porta alla inibizione delle esochinasi I-III.
Nel muscolo scheletrico l’attività della esochinasi I e II è coordinata con quella di GLUT4, un trasportatore del glucosio con una bassa Km per il monosaccaride (5 mM), la cui traslocazione sulla membrana plasmatica è indotta sia dall’insulina che dall’attività fisica. L’azione combinata delle esochinasi e di GLUT4 mantiene un equilibrio tra l’ingresso del glucosio nella cellula e la sua fosforilazione. Considerando che la concentrazione ematica del monosaccaride è compresa tra 4 e 5 mmol/L, il suo ingresso nel miocita per mezzo di GLUT4 può portare la sua concentrazione a valori sufficientemente elevati da saturare o quasi l’enzima, che dunque può lavora vicino o addirittura alla sua Vmax.
Regolazione della glucochinasi epatica
La glucochinasi differisce per almeno tre aspetti dalle esochinasi I-III, ed è particolarmente idonea per il ruolo che il fegato svolge nella regolazione della glicemia.[1] Perché?
Come detto in precedenza, la glucochinasi ha una Km per il glucosio pari a circa 10 mM, molto più elevata rispetto alla Km per il glucosio delle esochinasi I-III, e più alta anche del valore della glicemia a digiuno, pari a circa 4-5 mM.[7] Nel fegato, dove rappresenta l’isoenzima prevalente, il suo ruolo è quello di fornire glucosio-6-fosfato per la sintesi del glicogeno e degli acidi grassi. L’enzima lavora in modo coordinato con il trasportatore del glucosio GLUT2, il principale carrier per il glucosio nell’epatocita, la cui Km per lo zucchero è circa 10 mM.
Quindi GLUT2 è molto attivo quando la glicemia è elevata, equilibrando rapidamente la concentrazione del glucosio nel citosol dell’epatocita con quella presente nel sangue. In queste condizioni la glucochinasi è attiva e catalizza la conversione del glucosio in glucosio-6-fosfato, e, grazie alla elevata Km per il glucosio la sua attività continua ad aumentare anche quando la concentrazione intracellulare dello zucchero raggiunge o supera le 10 mM. Quindi la velocità con cui il monosaccaride entra nella cellula e di seguito viene fosforilato è determinata dal valore della stessa glicemia.
Quando invece la disponibilità del glucosio è scarsa, la sua concentrazione nel citosol dell’epatocita è altrettanto bassa, ben più bassa del valore della Km della glucochinasi. In questa condizione il glucosio prodotto attraverso la gluconeogenesi e/o la glicogenolisi non viene fosforilato e può lasciare la cellula. Una situazione simile si verifica anche nelle cellule β del pancreas, dove il sistema GLUT2/glucochinasi fa si che la concentrazione intracellulare del glucosio-6-fosfato equipari quella del glucosio nel sangue, permettendo alla cellula di rilevare e rispondere a elevate glicemie.
A differenza delle esochinasi I-III, la glucochinasi non è inibita dal glucosio-6-fosfato, per cui continua a catalizzarne la sintesi anche quando questi si accumula.
La glucochinasi viene inibita a seguito del legame reversibile a una specifica proteina regolatrice detta anche GKRP, acronimo dell’inglese glucokinase regulatory protein.[4] Il meccanismo con cui la proteina regolatrice agisce implica l’ancoraggio della glucochinasi all’interno del nucleo, dove rimane separata dagli altri enzimi della glicolisi.
Regolazione della Glucochinasi Epatica
Il legame tra glucochinasi e GKRP è molto più forte in presenza del fruttosio-6-fosfato mentre è indebolito dal glucosio e dal fruttosio-1-fosfato.
In assenza di glucosio la glucochinasi si trova nella sua conformazione “super-aperta” che è dotata di bassa attività. L’aumento nei livelli citosolici di glucosio determina una transizione concentrazione-dipendente dell’enzima verso la sua conformazione chiusa, la conformazione attiva che non è accessibile alla GKRP. Ne consegue che la glucochinasi è attiva e non più inibita.
Da notare che il fruttosio-1-fosfato è presente nell’epatocita solamente quando viene metabolizzato il fruttosio, che quindi, quando disponibile fa venire meno l’inibizione della glucochinasi da parte di GKRP.
Esempio
Quando dopo un pasto ricco di carboidrati la glicemia sale, il glucosio tramite il GLUT2 entra nella cellula epatica, e quindi attraverso i pori nucleari nel suo nucleo dove determina la transizione della glucochinasi verso la sua conformazione chiusa, attiva e non accessibile a GKRP, permettendo così all’enzima di diffondere nel citosol dove potrà fosforilare il glucosio.
Viceversa, quando il glucosio è scarso, come nel digiuno quando la glicemia può scendere sotto il valore di 4 mM, la concentrazione del glucosio nell’epatocita è bassa, e il fruttosio-6-fosfato associandosi a GKRP permette a quest’ultima di legarsi in modo più saldo alla glucochinasi. Da ciò ne risulta una inibizione molto efficace dell’enzima. Questo concorre a evitare che il fegato, in condizioni di bassa glicemia, competa con gli altri organi, in primis il cervello, per il poco glucosio disponibile.
Nella cellula il fruttosio-6-fosfato è presente in equilibrio con il glucosio-6-fosfato grazie alla reversibilità della reazione catalizzata dalla fosfoglucosio isomerasi. Tramite la sua associazione con GKRP, il fruttosio-6-fosfato opera quindi un feedback negativo segnalando alla cellula che non è necessario produrre altro glucosio-6-fosfato, ossia che l’attività della glucochinasi può ridursi per prevenire l’accumulo di intermedi.
Riassumendo si può dire che quando i valori della glicemia sono normali, il glucosio viene fosforilato prevalentemente dalle esochinasi I-III, mentre quando la glicemia è elevata l’esoso è fosforilato dalla glucochinasi.
Regolazione della fosfofruttochinasi-1
La fosfofruttochinasi-1 è il principale punto di controllo del flusso di carbonio attraverso la glicolisi.[7]
L’enzima, oltre ai siti di legame per i substrati, presenta molti altri siti su cui vanno a legarsi effettori allosterici sia inibitori che attivatori. Tra gli effettori allosterici negativi si ha l’ATP, che è anche un substrato dell’enzima e un prodotto finale della glicolisi, il citrato e gli ioni idrogeno.
Tra gli effettori allosterici positivi si ritrovano l’AMP, il Pi e il fruttosio-2,6-bisfosfato.[8][9]
Nota: la PFK-1 presenta due siti di legame distinti per l’ATP, uno nel sito attivo e dotato di grande affinità per la molecola, e l’altro, regolatorio, a bassa affinità.
Che cosa segnalano i diversi effettori allosterici?
ATP, AMP e Pi segnalano lo stato energetico della cellula. L’attività della fosfofruttochinasi-1 aumenta quando la cellula necessita di energia, ossia quando c’è necessità di ATP, mentre si riduce quando lo stato energetico della cellula è alto, ossia quando la cellula è ricca di ATP.
In che modo?
Quando la concentrazione di ATP è elevata, quando cioè il nucleotide viene prodotto a una velocità superiore a quella con cui è consumato, lo stesso legandosi allo specifico sito allosterico va a inibire la fosfofruttochinasi-1, riducendone l’affinità per il fruttosio-6-fosfato. Dal punto di vista cinetico l’aumento della concentrazione dell’ATP determina una cambiamento della rapporto tra la velocità di reazione e la concentrazione del fruttosio-6-fosfato che diviene sigmoide da iperbolico, e quindi si ha un aumento della Km dell’enzima per il fruttosio-6-fosfato stesso.
Tuttavia nella maggior parte delle condizioni cellulari la concentrazione dell’ATP non varia più di tanto. Ad esempio, nel muscolo durante un esercizio vigoroso si possono avere riduzioni della concentrazione di ATP di circa un 10% rispetto alla situazione di riposo mentre la velocità della glicolisi varia molto di più rispetto a quanto ci si aspetterebbe da tale riduzione. Quando la velocità di produzione dell’ATP è inferiore a quella con cui viene consumato, si verifica l’aumento della concentrazione dell’AMP e dell’ADP, e in particolare dell’AMP, grazie all’azione dell’enzima adenilato chinasi (EC 2.7.4.3 ), secondo la reazione di seguito descritta.
ADP + ADP ⇄ ATP + AMP
La costante di equilibrio Keq della reazione è:
Keq = [ATP][AMP]/[ADP]2= 0,44
Nelle normali condizioni cellulari la concentrazione dell’ADP e dell’AMP corrispondono rispettivamente a circa il 10% e a meno dell’1% di quella dell’ATP. Pertanto, considerando che il pool dell’adenilato è costante nel breve periodo, anche una piccola variazione nella concentrazione dell’ATP porterà, grazie all’attività della adenilato chinasi, a una variazione relativa molto maggiore della concentrazione dell’AMP. A sua volta l’AMP agisce rimuovendo l’inibizione dovuta all’ATP.
Quindi, l’attività della fosfofruttochinasi-1 dipende dallo stato energetico della cellula:
quando l’ATP è abbondante l’attività dell’enzima si riduce;
quando i livelli di AMP aumentano l’attività dell’enzima aumenta.
Perché l’ADP non è un effettore allosterico positivo della fosfofruttochinasi-1? Ci sono due ragioni.
Quando la carica energetica della cellula si riduce, l’ADP è utilizzato per riformare l’ATP, nella reazione catalizzata dalla adenilato chinasi. Inoltre, come detto in precedenza, una piccola riduzione nei livelli di ATP porta una più elevata variazione percentuale nei livelli dell’ADP e soprattutto dell’AMP.
Gli ioni idrogeno inibiscono la fosfofruttochinasi-1. Tale inibizione previene, controllando la velocità della glicolisi, l’eccessivo accumulo di lattato e la conseguente caduta del pH ematico.
Il citrato è un inibitore allosterico della fosfofruttochinasi-1 che agisce aumentando l’effetto inibitorio dell’ATP. Il citrato è il prodotto del primo passaggio del ciclo dell’acido citrico, una via metabolica che fornisce intermedi metabolici per le vie biosintetiche e indirizza gli elettroni verso la catena di trasporto degli elettroni mitocondriale per la sintesi dell’ATP via fosforilazione ossidativa. Elevati livelli citosolici di citrato indicano che nel mitocondrio si sta verificando la produzione di un eccesso di precursori e che le richieste energetiche della cellula sono state soddisfatte, ossia, il ciclo dell’acido citrico ha raggiunto la saturazione; pertanto la glicolisi, che rifornisce il ciclo in condizioni aerobiche, può rallentare, risparmiando glucosio.
Quindi, va sottolineato che la PFK-1 accoppia la glicolisi e il ciclo dell’acido citrico.
Nel fegato il punto centrale per la regolazione sia della glicolisi che della gluconeogenesi è rappresentato dal ciclo del substrato tra fruttosio-6-fosfato e fruttosio-1,6-bisfosfato, catalizzato dagli enzimi fosfofruttochinasi-1 e fruttosio-1,6-bisfosfatasi.
Il fegato gioca un ruolo cruciale nel mantenimento della glicemia entro valori normali.
Se la glicemia scende, il glucagone a livello epatico stimola la sintesi, via glicogenolisi e gluconeogenesi, di glucosio, e al contempo segnala all’organo di non consumare l’esoso per soddisfare i propri fabbisogni.
Quando invece la glicemia è elevata, l’insulina induce il fegato a utilizzare il glucosio per produrre energia, sintetizzare glicogeno e trigliceridi. In quest’ottica, la regolazione della glicolisi e della gluconeogenesi è mediata dal fruttosio-2,6-bisfosfato, una molecola che permette all’organo di svolgere un ruolo di primo piano nella regolazione della glicemia segnalando il rapporto tra insulina e glucagone.
A seguito del legame allo specifico sito allosterico sulla fosfofruttochinasi-1, il fruttosio-2,6-bisfosfato aumenta l’affinità dell’enzima per il fruttosio-6-fosfato, il suo substrato, mentre diminuisce quella per gli inibitori allosterici citrato e ATP. Ed è notevole sottolineare che in presenza di concentrazioni fisiologiche dei substrati e degli effettori allosterici sia positivi che negativi l’enzima, in assenza di fruttosio-2,6-bisfosfato, è praticamente inattivo.
Viceversa, il legame del fruttosio-2,6-bisfosfato alla fruttosio-1,6-bisfosfatasi ne comporta l’inibizione, anche in assenza di AMP, un altro inibitore allosterico dell’enzima.
Grazie a queste azioni la molecola aumenta il flusso netto di glucosio attraverso la glicolisi.
Altro metabolita importante per il controllo del flusso del carbonio attraverso la glicolisi e la gluconeogenesi è lo xilulosio-5-fosfato.[3]. Si tratta di un intermedio della via del pentoso fosfato, la cui concentrazione nell’epatocita aumenta a seguito dell’ingestione di un pasto ricco di carboidrati. La molecola, attivando la protein fosfatasi 2A porta infine a un aumento della concentrazione del fruttosio-2,6-bisfosfato e quindi a un incremento del flusso del carbonio attraverso la glicolisi e alla riduzione di quello attraverso la gluconeogenesi.
Regolazione della piruvato chinasi
Un ulteriore punto di regolazione del flusso di carbonio attraverso la glicolisi e la gluconeogenesi è rappresentato dal ciclo del substrato tra il fosfoenolpiruvato e il piruvato. Suddetto ciclo è catalizzato dalla piruvato chinasi, per la glicolisi, e dall’azione combinata della piruvato carbossilasi e della fosfoenolpiruvato carbossichinasi (EC 4.1.1.32) per la gluconeogenesi.[5][7]
Tutte le isoforme della piruvato chinasi sono allostericamente inibite da elevate concentrazioni di ATP, acetil-CoA e acidi grassi a catena lunga, segnali che la cellula si trova in uno stato energetico ottimale. Anche l’alanina, che può essere sintetizzata dal piruvato attraverso una reazione di transaminazione, è un inibitore allosterico dell’enzima. L’accumulo di alanina segnala l’abbondanza di precursori per le vie biosintetiche.
Regolazione della Piruvato Chinasi Epatica
Di contro la piruvato chinasi è allostericamente attivata dal fruttosio-1,6-bisfosfato, il prodotto della prima reazione esclusiva della glicolisi. Questo permette all’enzima di tenere il passo con il flusso di intermedi in arrivo. E va sottolineato il fatto che, in una situazione di concentrazioni fisiologiche di substrato, ossia il fosfoenolpiruvato, e degli inibitori ATP e alanina, la piruvato chinasi sarebbe completamente inibita senza l’effetto stimolatorio del fruttosio-1,6-bisfosfato.
L’isoenzima epatico, ma non quello muscolare, è soggetto anche a regolazione attraverso fosforilazione a opera della:
protein chinasi A, attivata a seguito del legame del glucagone allo specifico recettore o dell’epinefrina ai recettori β-adrenergici;
protein chinasi calcio/calmodulina dipendente, attivata dal legame dell’epinefrina ai recettori α1-adrenergici.
La fosforilazione dell’enzima ne riduce l’attività a seguito di un aumento della sua Km per il fosfoenolpiruvato, e rallenta la glicolisi.
Ad esempio, a seguito di una riduzione della glicemia, la fosforilazione indotta dal glucagone riduce l’attività dell’enzima. Nella forma fosforilata l’enzima è anche meno facilmente stimolato dal fruttosio-1,6-bisfosfato ma più facilmente inibito dall’alanina e dall’ATP.
Di contro, l’enzima in forma defosforilata è più sensibile al fruttosio-1,6-bisfosfato, e meno agli inibitori allosterici ATP e alanina. In questo modo quando la glicemia è bassa, il fegato rallenta l’ossidazione del glucosio a scopi energetici, e lo zucchero rimane quindi disponibile per altri tessuti e organi, come il cervello. Va comunque notato che la piruvato chinasi non subisce la fosforilazione indotta dal glucagone in presenza del fruttosio 1,6-bisfosfato.
Viceversa, un aumento nel rapporto insulina/glucagone porta infine alla defosforilazione dell’enzima e quindi alla sua attivazione. Nella forma defosforilata l’enzima è meno sensibile agli inibitori allosterici alanina e ATP, ma più sensibile al suo attivatore allosterico, il fruttosio-1,6-bisfosfato.
Bibliografia
ab de la Iglesia N., Mukhtar M., Seoane J., Guinovart J.J., & Agius L. The role of the regulatory protein of glucokinase in the glucose sensory mechanism of the hepatocyte. J Biol Chem 2000;275(14):10597-10603. doi: 10.1074/jbc.275.14.10597
^ Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003;100:5107-5112. doi:10.1073/pnas.0730817100
^ Kaminski M.T., Schultz J., Waterstradt R., Tiedge M., Lenzen S., Baltrusch S. Glucose-induced dissociation of glucokinase from its regulatory protein in the nucleus of hepatocytes prior to nuclear export. BBA – Molecular Cell Research 2014;1843(3):554-564. doi:10.1016/j.bbamcr.2013.12.002
^ abcdefghi Nelson D.L., M. M. Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ Oslund R.C., Su X., Haugbro M., Kee J-M., Esposito M., David Y., Wang B., Ge E., Perlman D.H., Kang Y., Muir T.W., & Rabinowitz J.D. Bisphosphoglycerate mutase controls serine pathway flux via 3-phosphoglycerate. Nat Chem Biol 2017;13:1081-1087. doi:10.1038/nchembio.2453
^ abcdefg Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
^ Van Schaftingen E., and Hers H-G. Inhibition of fructose-1,6-bisphosphatase by fructose-2,6-bisphosphate. Proc Natl Acad Sci USA 1981;78(5):2861-2863. doi:10.1073/pnas.78.5.2861
^ Van Schaftingen E., Jett M-F., Hue L., and Hers H-G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA 1981;78(6):3483-3486. doi:10.1073/pnas.78.6.3483
La gluconeogenesi è la via metabolica che permette, anche agli organismi non fotosintetizzanti, di produrre glucosio a partire dal piruvato, la base coniugata dell’acido piruvico, e da altri precursori non glucidici.[1][6]
E’ presente in tutti gli animali, piante, funghi e microorganismi, con essenzialmente le stesse reazioni, che portano, da due molecole di piruvato, alla sintesi di una molecola di glucosio. Dunque è essenzialmente l’inverso della glicolisi, che invece procede dal glucosio fino al piruvato, e con essa condivide molti enzimi.[5]
Gluconeogenesi e Glicolisi
La glicogenolisi è distinta dalla gluconeogenesi, non corrispondendo a una sintesi de novo del monosaccaride, come si può facilmente evidenziare osservando la sua reazione complessiva:
Glicogeno o (glucosio)n → n molecole di glucosio
La discussione successiva verterà sulla gluconeogenesi che avviene negli animali superiori, e in particolare nel fegato dei mammiferi.
La gluconeogenesi è una via metabolica di grande importanza per almeno due motivi.
Assicura il mantenimento di una adeguata concentrazione ematica di glucosio quando le riserve epatiche di glicogeno sono prossime all’esaurimento e lo zucchero non viene assunto con l’alimentazione.[6]
Il mantenimento della glicemia entro il range di normalità, 3,3-5,5 mmol/L (60 e i 99 mg/dL), è essenziale in quanto molte cellule e tessuti dipendono largamente o totalmente dal glucosio per soddisfare le loro richieste di ATP; tra questi i globuli rossi, i neuroni, il muscolo scheletrico quando lavora in anaerobiosi, la midollare del rene, i testicoli, la lente e la cornea dell’occhio, e i tessuti embrionali. Considerando ad esempio il cervello, il suo fabbisogno giornaliero di glucosio è di circa 120 g, una quantità pari a:
oltre il 50% delle riserve corporee totali del monosaccaride, circa 210 g, di cui 190 g immagazzinato come glicogeno muscolare ed epatico, e 20 g in forma libera nei fluidi corporei;
circa il 75% del fabbisogno giornaliero di glucosio dell’intero organismo, quindi sui 160 g.[1]
Nel digiuno breve, come nell’intervallo tra i pasti o durante la notte, la glicemia è mantenuta entro il range di normalità grazie alla glicogenolisi epatica e al rilascio di acidi grassi dal tessuto adiposo e corpi chetonici dal fegato. Acidi grassi e corpi chetonici, utilizzati di preferenza dal muscolo scheletrico, consentono un risparmio di glucosio che sarà disponibile per le cellule e i tessuti che da esso dipendono. Tuttavia, dopo circa 18 ore di digiuno, le riserve di glicogeno sono prossime all’esaurimento, riserve che possono divenire insufficienti anche durante l’attività fisica intensa e prolungata. Ed è a questo punto che, se non sono assunti carboidrati con l’alimentazione, la gluconeogenesi diviene essenziale.
E, a sottolineare ulteriormente l’importanza della sintesi de novo del glucosio il fatto che se i valori della glicemia scendono al di sotto di 2 mmol/L si verifica la perdita di coscienza.
L’eventuale escrezione del piruvato comporterebbe la perdita della possibilità di produrre ATP attraverso la sua ossidazione aerobica, ossia più di 10 molecole di ATP per ogni piruvato ossidato.
Dove avviene?
Negli animali superiori la gluconeogenesi avviene nel fegato, nella corticale del rene e nelle cellule epiteliali dell’intestino tenue, gli enterociti.[5]
Quantitativamente il fegato rappresenta la sede principale, producendo circa il 90% di tutto il glucosio sintetizzato, seguito dalla corticale del rene, con circa il 10%.[1] La maggiore capacità di sintesi del fegato rispetto alla corticale del rene è dovuta solo alle sue maggiori dimensioni; se infatti si considera il rapporto peso/sintesi, la corticale del rene produce più glucosio del fegato.
A livello della corticale del rene, le cellule che portano a termine la gluconeogenesi sono quelle del tubulo prossimale, la porzione del nefrone successiva al glomerulo. Molto del glucosio prodotto nel rene viene utilizzato dalla midollare del rene stesso, mentre l’azione dell’organo sul mantenimento della omeostasi glicemica diviene più importante durante il digiuno prolungato e nell’insufficienza epatica. Va tuttavia sottolineato che il rene, essendo privo di depositi di glicogeno, può contribuire alla regolazione della glicemia solo attraverso la gluconeogenesi e non anche attraverso la glicogenolisi, come invece può fare il fegato.
Parte della via gluconeogenetica può verificarsi anche nel muscolo scheletrico e cardiaco e nel cervello, anche se a velocità estremamente ridotta. Nell’adulto il muscolo ha circa 18 volte la massa del fegato, per cui la sua sintesi di glucosio potrebbe avere una qualche importanza quantitativa.
Tuttavia in questi tessuti la sintesi de novo del glucosio non porta alla sua liberazione in circolo, essendo assente la glucosio-6-fosfatasi (EC 3.1.3.9), l’enzima che catalizza l’ultima tappa della via.[6] Quindi, l’eventuale produzione di glucosio-6-fosfato, compreso quello derivante dalla glicogenolisi, non contribuirà al mantenimento della glicemia ma aiuterà solo a ripristinare le scorte del glicogeno, per la verità nel cervello piccole e limitate per lo più agli astrociti. L’unico contributo diretto al mantenimento della glicemia operato da questi tessuti, e in particolare dal muscolo scheletrico, vista la sua grande massa, deriva dalla piccola quota di glucosio rilasciata dall’enzima deramificante (EC 3.2.1.33) della glicogenolisi.
Per quello che riguarda la localizzazione cellulare, la maggior parte delle reazioni della gluconeogenesi avvengono nel citosol, alcune nel mitocondrio, e la tappa finale all’interno delle cisterne del reticolo endoplasmatico.
Tappe irreversibili
Come detto, glicolisi e gluconeogenesi sono essenzialmente una l’inverso dell’altra. E, delle dieci reazioni che costituiscono la gluconeogenesi, ben 7 sono in comune con la glicolisi. Si tratta di reazioni caratterizzate da un ΔG prossimo allo zero, per cui facilmente reversibili. Ma nelle normali condizioni cellulari, il ΔG complessivo della glicolisi è pari a circa -63 kJ/mole (-15 kcal/mole) e quello della gluconeogenesi a circa -16 kJ/mole (-3,83 kcal/mole), ossia si tratta di processi irreversibili.[5]
Nel caso della glicolisi l’irreversibilità è conseguenza di tre reazioni fortemente esoergoniche, che non potranno essere utilizzate nella gluconeogenesi, e di seguito elencate.
La fosforilazione del glucosio a glucosio-6 fosfato, catalizzata dalla esochinasi (EC 2.7.1.1) o dalla glucochinasi (EC 2.7.1.2).
ΔG = -33,4 kJ/mole (-8 kcal/mole)
ΔG°’ = -16,7 kJ/mole (-4 kcal/mole)
La fosforilazione del fruttosio-6-fosfato a fruttosio-1,6-bisfosfato, catalizzata dalla fosfofruttochinasi-1 o PFK-1 (EC 2.7.1.11).
ΔG = -22,2 kJ/mole (-5,3 kcal/mole)
ΔG°’ = -14,2 kJ/mole (-3,4 kcal/mole)
La conversione del fosfoenolpiruvato o PEP, acronimo dell’inglese phosphoenolpyruvate, in piruvato, catalizzata dalla piruvato chinasi (EC 2.7.1.40).
ΔG = -16,7 kJ/mole (-4,0 kcal/mole)
ΔG°’ = -31,4 kJ/mole (-7,5 kcal/mole)
Nella gluconeogenesi queste tre tappe unidirezionali sono superate grazie a enzimi specifici che catalizzano passaggi irreversibili nella direzione della sintesi del glucosio.[6] In questo modo è assicurata l’irreversibilità dell’intera via metabolica.
Di seguito sono analizzate tali reazioni.
Conversione del piruvato in fosfoenolpiruvato
Il primo passaggio della gluconeogenesi che by-passa una tappa irreversibile della glicolisi, nello specifico quella catalizzata dalla piruvato chinasi, è la conversione del piruvato in fosfoenolpiruvato.[8]
La sintesi del fosfoenolpiruvato è ottenuta attraverso una sequenza di due reazioni catalizzate nell’ordine dagli enzimi:
piruvato carbossilasi (EC 6.4.1.1);
fosfoenolpiruvato carbossichinasi o PEP carbossichinasi (EC 4.1.1.32).
Piruvato → Ossalacetato → Fosfoenolpiruvato
La piruvato carbossilasi catalizza la carbossilazione del piruvato in ossalacetato, con consumo di una molecola di ATP. L’enzima richiede la presenza di ioni manganese o magnesio.
Piruvato + HCO3– + ATP → Ossalacetato + ADP + Pi
L’enzima, scoperto nel 1960 da Merton Utter, è una proteina mitocondriale formata da quattro subunità identiche, ognuna dotata di attività catalitica. Le subunità hanno come coenzima la biotina, legata attraverso legame ammidico al gruppo amminico ε di un residuo di lisina, e la cui funzione è quella di trasportatore di CO2 attivata nel corso della reazione enzimatica. In ogni subunità è presente anche un sito di legame per l’acetil-CoA.
Va notato che la reazione catalizzata dalla piruvato carbossilasi, portando alla produzione di ossalacetato, fornisce intermedi anche al ciclo dell’acido citrico o di Krebs.
La fosfoenolpiruvato carbossichinasi è presente, all’incirca nelle stesse quantità, sia nel mitocondrio che nel citosol dell’epatocita. Le due forme isoenzimatiche sono codificate da distinti geni nucleari.
L’enzima catalizza la decarbossilazione e fosforilazione dell’ossalacetato a dare fosfoenolpiruvato, in una reazione in cui il GTP funge da donatore di un gruppo fosfato, e richiede la presenza sia di ioni manganese che magnesio. La reazione nelle normali condizioni cellulari è reversibile.
Ossalacetato + GTP ⇄ PEP + CO2 + GDP
Nella reazione la CO2 aggiunta nella tappa catalizzata dalla piruvato carbossilasi viene rimossa. La sequenza di carbossilazione e decarbossilazione è un modo per “attivare” il piruvato, poiché la decarbossilazione dell’ossalacetato facilita, rende termodinamicamente possibile la formazione del fosfoenolpiruvato.
Più in generale le sequenze carbossilazione-decarbossilazione sono utilizzate per favorire reazioni che altrimenti sarebbero fortemente endoergoniche, e sono utilizzate anche nel ciclo dell’acido citrico, nella via del pentoso fosfato, detta via dell’esoso monofosfato, e nella sintesi degli acidi grassi.
Prima della nascita i livelli di PEP carbossichinasi sono molto bassi, mentre, poche ore dopo il parto la sua attività aumenta di diverse volte. Questo è il motivo per cui la gluconeogenesi è attiva solo dopo la nascita.
La somma delle reazioni catalizzate dalla piruvato carbossilasi e dalla fosfoenolpiruvato carbossichinasi è:
Piruvato + ATP + GTP + HCO3– → PEP + ADP + GDP + Pi + CO2
Il ΔG°’ della reazione è pari a 0,9 kJ/mole (0,2 kcal/mole), mentre la variazione di energia libera standard associata alla formazione di piruvato dal fosfoenolpiruvato per semplice inversione della reazione catalizzata dalla piruvato chinasi è di + 31,4 kJ/mole (7,5 kcal/mole).
Sebbene il ΔG’° dei due passaggi che portano alla formazione di fosfoenolpiruvato dal piruvato sia leggermente positivo, il ΔG calcolato dalle concentrazioni intracellulari degli intermedi è molto negativo, -25 kJ/mole (-6 kcal/mole), grazie al rapido consumo del fosfoenolpiruvato in altre reazioni, il che mantiene la sua concentrazione molto bassa. Quindi, nelle condizioni esistenti nella cellula la suddetta sintesi del PEP dal piruvato è un processo irreversibile.[5]
Particolarità della sintesi del fosfoenolpiruvato dal piruvato è che la via seguita dipende dal precursore prevalente: piruvato o alanina, oppure il lattato.
Substrato prevalente piruvato o alanina
I passaggi di seguito descritti prevalgono quando i substrati gluconeogenetici prevalenti sono piruvato o alanina.
La piruvato carbossilasi è un enzima mitocondriale, per cui il piruvato dovrà essere trasportato dal citosol nell’organello. Ciò avviene grazie a due trasportatori presenti sulla membrana mitocondriale interna, indicati come MPC1 and MPC2, che, associandosi, formano un eteropolimero che facilita il passaggio della molecola.[4]
Il piruvato può anche essere prodotto direttamente all’interno del mitocondrio dall’alanina per transaminazione, nella reazione catalizzata dalla alanina aminotransferasi o ALT (EC 2.6.1.2), mentre il gruppo amminico sarà infine convertito in urea attraverso il ciclo dell’urea.
Da Piruvato e Alanina a PEP
Poiché gli enzimi che intervengono nelle tappe successive della gluconeogenesi, fino alla formazione del glucosio-6-fosfato, sono citosolici, l’ossalacetato prodotto nei mitocondri dovrà essere trasportato nel citosol. La membrana mitocondriale interna è però priva di trasportatori per l’ossalacetato. Il suo passaggio nel citosol avviene a seguito della sua riduzione a malato, che al contrario può attraversare la membrana mitocondriale interna. La reazione è catalizzata dalla malato deidrogenasi mitocondriale (EC 1.1.1.37), un enzima che interviene anche nel ciclo dell’acido citrico dove però il flusso dei metaboliti procede in direzione opposta. Nella reazione il NADH viene ossidato a NAD+.
Ossalacetato + NADH + H+ ⇄ Malato + NAD+
Sebbene ΔG°’ della reazione sia piuttosto elevato, il ΔG calcolato sulla base della concentrazione intracellulare dell’ossalacetato, molto bassa, è prossimo allo zero per cui la reazione è facilmente reversibile.
Il malato prodotto attraversa la membrana mitocondriale interna grazie a un componente dello shuttle del malato-aspartato, il trasportatore malato-α-chetoglutarato. Una volta nel citosol il malato è riossidato a ossalacetato nella reazione catalizzata dalla malato deidrogenasi citosolica, con produzione di NADH.
Malato + NAD+ → Ossalacetato + NADH + H+
Nota: lo shuttle del malato-aspartato è il più attivo nel trasporto degli equivalenti riducenti del NADH dal citosol all’interno del mitocondrio, ed è presente nei mitocondri del fegato, rene, e cuore.
Grazie a questa reazione equivalenti riducenti mitocondriali, in forma di NADH, sono trasferiti nel citosol.[8] Tale trasferimento è necessario per il proseguimento della gluconeogenesi, in quanto nel citosol il NADH, utilizzato nella reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12), è presente in bassa concentrazione, con un rapporto [NADH]/[NAD+] pari a 8 x 10-4, circa 100000 volte più basso di quello osservato nei mitocondri.[5]
Infine l’ossalacetato viene convertito in fosfoenolpiruvato nella reazione catalizzata dalla PEP carbossichinasi.
Substrato prevalente lattato
Il lattato è un importante precursore gluconeogenico. Esempi di cellule e tessuti che lo producono in grande quantità sono i globuli rossi, che, completamente dipendenti dalla glicolisi anaerobica, lo producono continuamente, e il muscolo scheletrico in forte attività, quando la velocità della glicolisi supera la velocità del ciclo dell’acido citrico e della fosforilazione ossidativa.[5]
Quando il lattato è il precursore gluconeogenico prevalente la sintesi del PEP segue una via differente rispetto a quanto visto in precedenza.
Nel citosol dell’epatocita, dove come detto la concentrazione del NAD+ è elevata, il lattato viene ridotto a piruvato nella reazione catalizzata dall’isoenzima epatico della lattato deidrogenasi (EC 1.1.1.27). Nella reazione il NAD+ viene ridotto a NADH.
Lattato + NAD+ → Piruvato + NADH + H+
La produzione citosolica di NADH rende non necessaria l’esportazione di equivalenti riducenti dal mitocondrio.
Il piruvato passa nella matrice mitocondriale per essere convertito in ossalacetato nella reazione catalizzata dalla piruvato carbossilasi. Nel mitocondrio l’ossalacetato è convertito in fosfoenolpiruvato nella reazione catalizzata dall’isoenzima mitocondriale della piruvato carbossilasi, che, a mezzo di un trasportatore anionico della membrana mitocondriale interna, esce dal mitocondrio per continuare nella via gluconeogenetica.
Nota: la sintesi del glucosio dal lattato può essere considerata anche come una parte del “ramo epatico” del ciclo di Cori.
Conversione del fruttosio-1,6-bisfosfato in fruttosio-6-fosfato
Il secondo passaggio della gluconeogenesi che supera una reazione irreversibile della via glicolitica, quella catalizzata dalla PFK-1, è la defosforilazione del fruttosio-1,6-bisfosfato a fruttosio-6-fosfato.
La reazione, catalizzata dalla fruttosio-1,6-bisfosfatasi o FBPasi-1 (EC 3.1.3.11), enzima citosolico e Mg2+ dipendente, comporta l’idrolisi del fosfato sul C-1, senza alcuna produzione di ATP.
Fruttosio-1,6-bisfosfato + H2O → Fruttosio-6-fosfato + Pi
Il ΔG°’ della reazione è pari a -16,3 kJ/mol (-3,9 kcal/mol), dunque una reazione irreversibile.
Conversione del glucosio-6-fosfato a glucosio
Il terzo passaggio esclusivo della gluconeogenesi permette di superare la reazione della glicolisi catalizzata dalla esochinasi o dalla glucochinasi. Nello specifico si tratta della defosforilazione del glucosio-6-fosfato a glucosio catalizzata dall’unità catalitica della glucosio-6-fosfatasi, un complesso proteico presente nella membrana del reticolo endoplasmatico degli epatociti, enterociti e cellule del tubulo prossimale del rene.[8] La glucosio-6-fosfatasi è composta da una subunità catalitica dotata di attività fosfatasica e un trasportatore bidirezionale specifico per il glucosio-6-fosfato detto glucosio-6-fosfato traslocasi o T1.
La subunità catalitica della glucosio-6-fosfatasi presenta il sito attivo rivolto verso la superficie luminale dell’organello: quindi l’enzima catalizza una idrolisi intraluminale del substrato.
Il glucosio-6-fosfato, sia quello derivante dalla gluconeogenesi, rilasciato dalla reazione catalizzata dalla glucosio-6-fosfato isomerasi o fosfoglucosio isomerasi (EC 5.3.1.9), che dalla glicogenolisi, rilasciato dalla reazione catalizzata dalla fosfoglucomutasi (EC 5.4.2.2), è prodotto nel citosol, e dovrà entrare nel lume del reticolo endoplasmatico per essere defosforilato. Il suo passaggio è operato dalla glucosio-6-fosfato traslocasi.
La subunità catalitica della glucosio-6-fosfatasi, un enzima Mg2+-dipendente, catalizza la reazione corrisponde all’ultima tappa sia della gluconeogenesi che della glicogenolisi. E, al pari della reazione che porta alla sintesi del fruttosio-6-fosfato, anche questa è una semplice idrolisi di un estere fosforico.
Glucosio-6-fosfato + H2O → Glucosio + Pi
Va inoltre sottolineato che, grazie all’orientamento del sito attivo, la cellula separa questa reazione dal citosol, e dunque dalla glicolisi, che verrebbe bloccata dall’azione dell’enzima sul glucosio-6-fosfato.
Il ΔG°’ della reazione è pari a -13,8 kJ/mol (-3,3 kcal/mol), dunque una reazione irreversibile. Se invece la reazione fosse l’inverso di quella catalizzata dalla esochinasi/glucochinasi, comporterebbe il trasferimento di un gruppo fosforico all’ADP dal glucosio-6-fosfato, una reazione con ΔG pari a +33,4 kJ/mole (+8 kcal/mol), quindi fortemente endoergonica. Analoghe considerazioni possono essere estese alla reazione catalizzata dalla FBPasi-1.
Sembra che il glucosio e il gruppo Pi prodotti siano trasportati nel citosol da due distinti trasportatori, indicati rispettivamente come T2 e T3, quest’ultimo un trasportatore anionico.
Infine, grazie al trasportatore di membrana GLUT2, il glucosio potrà lasciare l’epatocita ed entrare in circolo per essere trasportato ai tessuti che lo richiedano. Come accennato in precedenza invece, il glucosio prodotto nel rene, in condizioni di normale efficienza epatica, viene utilizzato per la maggior parte dalla midollare del rene stesso.
La gluconeogenesi è energeticamente costosa
Al pari di quanto accade nella glicolisi, sono le tappe irreversibili della gluconeogenesi le responsabili del consumo della maggior parte dell’energia necessaria al processo.[5]
Sono consumati sei legami fosforici ad alta energia, due forniti dal GTP e quattro dall’ATP, cui vanno aggiunte due molecole di NADH per la riduzione di altrettante molecole di 1,3-bisfosfoglicerato nella reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi. Il consumo dei due NADH comporta la mancata produzione di 5 molecole di ATP che sarebbero potute essere sintetizzate nel caso in cui gli elettroni del coenzima ridotto fossero stati utilizzati per la sintesi di ATP nel mitocondrio attraverso la fosforilazione ossidativa.
Anche da queste considerazioni prettamente energetiche emerge che la gluconeogenesi non è semplicemente l’inverso della glicolisi, nel qual caso necessiterebbe del consumo di sole due molecole di ATP, come si evince dalla equazione glicolitica complessiva.
Almeno nel fegato, l’ATP necessario a sostenere la gluconeogenesi deriva di solito dall’ossidazione degli acidi grassi, o dall’ossidazione degli scheletri carboniosi degli amminoacidi, a seconda del “carburante” disponibile.
Regolazione coordinata della glicolisi e gluconeogenesi
Se la glicolisi e la gluconeogenesi procedessero simultaneamente ad alta velocità nella stessa cellula, ne risulterebbe solo il consumo di ATP e la produzione di calore, in particolare in corrispondenza delle tappe irreversibili dei due processi, e nulla di più.
Ad esempio, considerando PFK-1 e FBPasi-1:
ATP + Fruttosio-6-fosfato → ADP + Fruttosio-1,6-bisfosfato
Fruttosio 1,6-bisfosfato + H2O → Fruttosio 6-fosfato + Pi
E, dalla somma delle due reazioni:
ATP + H2O → ADP + Pi + Calore
Quando reazioni contrapposte di questo tipo procedono simultaneamente si parla di ciclo futile o ciclo del substrato.
La contemporanea attività ad alta velocità di enzimi che catalizzano reazioni contrapposte è evitata grazie al controllo dell’attività degli enzimi stessi, che può avvenire mediante:
meccanismi allosterici;
modificazioni covalenti, in particolare fosforilazioni e defosforilazioni;
modificazioni nella concentrazione degli enzimi coinvolti, conseguenti a variazioni del rapporto tra la loro sintesi e degradazione.
I meccanismi allosterici sono molto rapidi e istantaneamente reversibili, avvenendo in un arco temporale di millisecondi. Gli altri, innescati da segnali che provengono dall’esterno della cellula e trasportati da ormoni quali insulina, glucagone, o adrenalina, richiedono tempi più lunghi, dai secondi ai minuti per le modificazioni covalenti, e fino a ore per le modificazioni della concentrazione degli enzimi.
Grazie a questi meccanismi regolatori è possibile ottenere una regolazione coordinata delle due vie, tale da assicurare che quando il flusso di piruvato procede attraverso la gluconeogenesi, il flusso del glucosio attraverso la glicolisi rallenta, e viceversa.
Regolazione della gluconeogenesi
La regolazione della gluconeogenesi e della glicolisi avviene attraverso controlli esercitati sugli enzimi specifici delle singole vie, e non su quelli comuni.[1]
Mentre i principali punti di regolazione della glicolisi sono le reazioni catalizzate dagli enzimi PFK-1 e piruvato chinasi, i principali punti di regolazione della via gluconeogenetica sono le reazioni catalizzate dagli enzimi fruttosio-1,6-bisfosfatasi e piruvato carbossilasi. Anche gli altri due enzimi esclusivi della via gluconeogenetica, glucosio-6-fosfatasi e PEP carbossichinasi, sono soggetti a regolazione ma solo a livello trascrizionale.
Piruvato carbossilasi
Nel mitocondrio il piruvato può essere convertito in:
ossalacetato, nella reazione catalizzata dalla privato carbossilasi, l’enzima che catalizza la prima tappa della gluconeogenesi.
Il destino metabolico del piruvato dipende dai livelli dell’acetil-CoA e dunque dalla disponibilità di acidi grassi nel mitocondrio.
Quando gli acidi grassi sono disponibili, la loro beta-ossidazione porta alla liberazione di molecole di acetil-CoA, che entrano nel ciclo di Krebs per dar luogo alla formazione di GTP e NADH. Una volta che i fabbisogni energetici della cellula sono soddisfatti, la fosforilazione ossidativa rallenta, il rapporto NADH/NAD+ cresce, il NADH inibisce il ciclo dell’acido citrico e si verifica un accumulo di acetil-CoA nella matrice mitocondriale.[1][5]
L’acetil-CoA è un effettore allosterico positivo della piruvato carbossilasi e un effettore allosterico negativo della piruvato chinasi. Inoltre inibisce il complesso della piruvato deidrogenasi sia attraverso una inibizione da prodotto finale che mediante la sua fosforilazione, a seguito dell’attivazione di una specifica chinasi.
Destini del Piruvato
Dunque, quando la carica energetica della cellula è alta, quello che accade è che la formazione di acetil-CoA dal piruvato è rallentata, mentre viene stimolata la conversione del piruvato in glucosio. L’acetil-CoA è quindi un segnale metabolico che indica che un’ulteriore ossidazione del glucosio a scopo energetico non è necessaria e che i metaboliti carboniosi possono essere utilizzati per la sintesi e il deposito di glucosio.
Viceversa, quando i livelli di acetil-CoA si riducono, l’attività della piruvato chinasi e del complesso della piruvato deidrogenasi aumentano, e quindi anche il flusso di metaboliti attraverso il ciclo di Krebs, il tutto per rifornire di energia la cellula.
Si potrebbe riassumere dicendo che la piruvato carbossilasi è attiva quando la carica energetica della cellula è alta e che il primo punto di controllo della gluconeogenesi determina quello che sarà il destino del piruvato nel mitocondrio.
Fruttosio-1,6-bisfosfatasi
Il secondo punto principale di controllo della gluconeogenesi è rappresentato dalla reazione catalizzata dalla fruttosio-1,6-bisfosfatasi. L’enzima è inibito allostericamente dall’AMP. Quindi quando la concentrazione dell’AMP è alta, e di conseguenza quella dell’ATP è bassa, la gluconeogenesi rallenta. Ossia, come visto in precedenza, l’enzima è attivo quando la carica energetica della cellula è adeguata a sostenere la sintesi de novo del glucosio.
Di contro la PFK-1, l’enzima glicolitico corrispondente, è stimolata allostericamente dall’AMP e dall’ADP e inibita dall’ATP e dal citrato, quest’ultimo derivante dalla condensazione tra l’acetil-CoA e l’ossalacetato. Quindi riassumendo:
quando la concentrazione dell’AMP è alta la gluconeogenesi rallenta, mentre la glicolisi accelera;
quando la concentrazione dell’ATP è elevata, e quindi è bassa quella di ADP e AMP, o quando l’acetil-CoA o il citrato sono presenti in concentrazioni adeguate, viene promossa la gluconeogenesi mentre rallenta la glicolisi.
L’aumento della concentrazione del citrato segnala che l’attività del ciclo dell’acido citrico può rallentare; in questo modo il piruvato potrà essere utilizzato per la sintesi del glucosio.
Fruttosio-1,6-bisfosfatasi, PFK-1 e fruttosio-2,6-bisfosfato
Il fegato ha un ruolo centrale nel mantenimento della glicemia a valori costanti: questo richiede la presenza di meccanismi regolatori che coordinino il consumo e la produzione del glucosio. Due sono gli ormoni principalmente coinvolti: il glucagone e l’insulina.
Il glucagone, rilasciato in circolo quando la glicemia scende, segnala al fegato di ridurre il suo consumo di glucosio e di aumentarne la produzione de novo e il rilascio dal glicogeno.[6]
L’azione degli ormoni suddetti è mediata da un effettore allosterico della PFK-1 e della fruttosio-1,6-bisfosfatasi: il fruttosio-2,6-bisfosfato, una molecola strutturalmente correlata al fruttosio-1,6-bisfosfato ma che non è ne un intermedio della glicolisi nel della gluconeogenesi.[1]
La molecola fu scoperta nel 1980 da Emile Van Schaftingen and Henri-Gery Hers come un potente stimolatore della PFK-1; l’anno successive gli stessi ricercatori dimostrarono che è anche un potente inibitore dalla fruttosio-1,6-bisfosfatasi.[9][10]
A seguito del legame allo specifico sito allosterico sulla PFK-1, il fruttosio-2,6-bisfosfato svolge un duplice effetto: riduce l’affinità della proteina per i suoi inibitori allosterici ATP e citrato e ne aumenta l’affinità per il fruttosio-6-fosfato, il suo substrato. La PFK-1, in assenza di fruttosio-2,6-bisfosfato, e in presenza di concentrazioni fisiologiche di ATP, fruttosio-6-fosfato, e dei suoi effettori allosterici AMP, ATP e citrato, è praticamente inattiva. La presenza di fruttosio-2,6-bisfosfato ha invece l’effetto di attivare l’enzima quindi stimolare la glicolisi nell’epatocita. Nel contempo la molecola rallenta la gluconeogenesi, inibendo la fruttosio-1,6-bisfosfatasi, anche in assenza di AMP. Tuttavia gli effetti di AMP e fruttosio-2,6-bisfosfato sull’inibizione di FBPasi-1 sono sinergici.
F2,6BP e Regolazione della Glicolisi e Gluconeogenesi
La concentrazione di fruttosio-2,6-bisfosfato è regolata dalle velocità relative della sua sintesi e degradazione. Viene sintetizzato a partire dal fruttosio-6-fosfato nella reazione catalizzata dalla fosfofruttochinasi 2 o PFK-2 (EC 2.7.1.105), e idrolizzato a fruttosio-6-fosfato nella reazione catalizzata dalla fruttosio-2,6-bisfosfatasi o FBPasi-2 (EC 3.1.3.46). Le due attività enzimatiche sono presenti su una stessa proteina, che dunque è un enzima bifunzionale, anche detto enzima tandem, e nel fegato sono regolate dall’insulina e dal glucagone, nel modo di seguito descritto.
Il glucagone, a seguito del legame allo specifico recettore di membrana, stimola la adenilato ciclasi (EC 4.6.1.1) presente sulla membrana plasmatica a produrre 3’-5’ AMP ciclico o cAMP, che, a seguito del legame alla protein chinasi cAMP-dipendente o protein chinasi A o PKA (EC 2.7.11.11), la attiva. La chinasi attivata catalizza la fosforilazione, a spese di una molecola di ATP, di uno specifico residuo di serina (Ser32) di PFK-2/FBPasi-2. La fosforilazione comporta un aumento dell’attività fosfatasica a spese di quella chinasica, che si riduce in conseguenza di un aumento della Km per il fruttosio-6-fosfato. Tutto ciò porta a una riduzione dei livelli di fruttosio-2,6-bisfosfato, con conseguente stimolazione della gluconeogenesi e inibizione della glicolisi. Quindi, in risposta al segnale trasportato dal glucagone, aumenta la produzione epatica di glucosio attraverso la gluconeogenesi, il che rende l’organo capace di contrastare la riduzione della glicemia segnalata dall’ormone.
Nota: il glucagone, al pari dell’adrenalina, stimola la gluconeogenesi in parte anche aumentando la disponibilità di substrati quali il glicerolo e gli amminoacidi.
L’insulina, a seguito del legame agli specifici recettori di membrana dell’epatocita, va ad attivare una protein fosfatasi, la fosfoprotein fosfatasi 2A che catalizza la rimozione del gruppo fosforico dalla PFK-2/FBPasi-2, attivando così la PFK-2 e riducendo l’attività della FBPasi-2. (Nel contempo stimola anche una cAMP fosfodiesterasi che idrolizza il cAMP ad AMP). Il risultato è l’aumento dei livelli intracellulari di fruttosio-2,6-bisfosfato e la conseguente inibizione della gluconeogenesi e attivazione della glicolisi.
Inoltre il fruttosio-6-fosfato inibisce allostericamente la FBPasi-2 mentre attiva la PFK-2. Riguardo l’attività dell’enzima bifunzionale PFK-2/FBPasi-2 va sottolineato che entrambe le attività sono inibite dai rispettivi prodotti di reazione, e tuttavia i fattori predominanti sono la concentrazione del fruttosio-6-fosfato e lo stato di fosforilazione dell’enzima stesso.
Glucosio-6-fosfatasi
A differenza della piruvato carbossilasi e della fruttosio-1,6-bisfosfatasi, la subunità catalitica della glucosio-6-fosfatasi non è soggetta a regolazione allosterica o covalente, mentre viene regolata a livello trascrizionale.[7] La bassa glicemia e il glucagone, dunque fattori che determinano una maggiore produzione di glucosio, e i glucocorticoidi ne stimolano la sintesi, che al contrario è inibita dall’insulina.
Inoltre la sua Km per il glucosio-6-fosfato è decisamente più alta rispetto al normale intervallo di concentrazione della molecola stessa. Ne risulta che l’attività dell’enzima mostra una dipendenza quasi lineare rispetto alla concentrazione del substrato. Per questo si dice che l’enzima è sotto controllo da parte della concentrazione del substrato.
PEP carbossichinasi
La regolazione dell’enzima avviene principalmente a livello della sua sintesi e demolizione. Ad esempio elevati livelli di glucagone o il digiuno ne aumentano la produzione, a seguito della stabilizzazione del suo mRNA e dell’aumento della sua velocità di trascrizione. Glicemie elevate o l’insulina hanno effetto opposto.
Xilulosio-5-fosfato
Anche un altro meccanismo regolatorio scoperto di recente, attraverso l’azione dello xilulosio-5-fosfato, un intermedio della via del pentoso fosfato, stimola la glicolisi e inibisce la gluconeogenesi, intervenendo nel controllo della concentrazione del fruttosio-2,6-bisfosfato nell’epatocita.
Quando la concentrazione ematica del glucosio aumenta, come dopo un pasto ricco di carboidrati, nel fegato si verifica l’attivazione della glicolisi e della via dell’esoso monofosfato. In quest’ultima via metabolica viene prodotto anche xilulosio-5-fosfato che è in grado di attivare la protein fosfatasi 2A. Ciò porta alla defosforilazione della PFK-2/FBPasi-2, inibendo così la FBPasi-2 e stimolando la PFK-2. Ne risulta un aumento della concentrazione del fruttosio-2,6-bisfosfato, e quindi l’inibizione della gluconeogenesi e la stimolazione della glicolisi, con conseguente aumento della produzione di acetil-CoA, il principale substrato per la sintesi dei lipidi.[2]
Il concomitante aumento del flusso attraverso la via dell’esoso monofosfato produce NADPH, fonte di elettroni per la sintesi dei lipidi. Infine, la protein fosfatasi 2A defosforila anche ChREBP, un fattore di trascrizione che attiva l’espressione dei geni epatici per la sintesi dei lipidi. Quindi, in risposta a un aumento della glicemia, a livello epatico sarà stimolata la sintesi dei lipidi.
Risulta dunque evidente che lo xilulosio-5-fosfato è un regolatore chiave del metabolismo sia dei carboidrati che dei grassi.
Precursori
Oltre al piruvato, i principali precursori gluconeogenici sono il lattato, di cui si è parlato in precedenza, il glicerolo, la maggior parte degli amminoacidi, e comunque qualunque composto che possa essere convertito in piruvato od ossalacetato.[1]
Glicerolo
Il glicerolo deriva dalla lipolisi nel tessuto adiposo. Con l’esclusione del propionil-CoA, è l’unica parte delle molecole dei lipidi che negli animali possa essere utilizzata per la sintesi del glucosio.
Il suo punto di ingresso nella gluconeogenesi, o nella glicolisi, a seconda delle condizioni energetiche in cui si trova la cellula, è rappresentato dal diidrossiacetone fosfato, la cui sintesi avviene in due passaggi.
Nel primo il glicerolo è fosforilato a glicerolo-3-fosfato, nella reazione catalizzata dalla glicerolo chinasi (EC 2.7.1.30). La reazione consuma una molecola di ATP.
Glicerolo + ATP → Glicerolo-3-fosfato + ADP + Pi
L’enzima assente negli adipociti, ma presente nel fegato. Ciò significa che il glicerolo dovrà raggiungere il fegato prima di essere ulteriormente metabolizzato.
Il glicerolo-3-fosfato viene quindi ossidato a diidrossiacetone fosfato, nella reazione catalizzata dalla glicerolo-3-fosfato deidrogenasi (EC 1.1.1.8). Nella reazione il NAD+ viene ridotto a NADH.
Durante il digiuno prolungato il glicerolo è il principale precursore gluconeogenetico, essendo responsabile della produzione di circa il 20% del glucosio.[3]
Amminoacidi glucogenici
Piruvato e ossalacetato rappresentano i punti di ingresso per gli amminoacidi glucogenici, ossia quelli il cui scheletro carbonioso o parte di esso può essere utilizzato per la sintesi del glucosio.
Gli amminoacidi derivano dalla demolizione delle proteine, sia di origine alimentare che endogena, come quelle del muscolo scheletrico nel corso del digiuno o dell’attività fisica intensa e prolungata.
I processi catabolici a carico di ognuno dei venti amminoacidi che compongono le proteine convergono verso la sintesi di sette prodotti principali: acetil-CoA, acetoacetil-CoA, α-chetoglutarato, succinil-CoA, fumarato, ossalacetato e piruvato.
Con l’esclusione di acetil-CoA e acetoacetil-CoA, le altre cinque molecole possono essere utilizzate per la sintesi del glucosio; quindi gli amminoacidi glucogenici possono essere definiti anche come quelli il cui scheletro carbonioso, in parte o in toto, può essere convertito in una o più delle suddette molecole.
Solo leucina e lisina sono esclusivamente chetogenici, essendo il loro scheletro carbonioso catabolizzato ad acetil-CoA e/o acetoacetil-CoA.
Di seguito sono elencati gli amminoacidi glucogenici con i rispettivi punti di ingresso.
Piruvato: alanina, cisteina, glicina, serina, treonina e triptofano.
Ossalacetato: aspartato e asparagina.
α-Chetoglutarato: glutammato, arginina, glutammina, istidina e prolina.
Succinil-CoA: isoleucina, metionina, treonina e valina.
Fumarato: fenilalanina e tirosina.
α-Chetoglutarato, succinil-CoA e fumarato, tutti intermedi del ciclo dell’acido citrico, entrano nella via gluconeogenetica previa conversione in ossalacetato.
Propionato
Il propionato, che fa parte del gruppo degli acidi grassi a catena corta, in forma di propionil-CoA è un precursore gluconeogenetico in quanto può essere convertito in succinil-CoA.
Di seguito sono analizzate le diverse fonti di propionato.
Può derivare dalla beta-ossidazione di acidi grassi a catena dispari, come ad esempio l’acido margarico, acido grasso saturo con 17 atomi di carbonio. Tali acidi grassi sono molto rari rispetto a quelli a catena pari, e presenti in quantità significative nei lipidi di alcuni organismi marini, delle piante, e nel grasso dei ruminanti. Nell’ultimo passaggio del loro ciclo di ossidazione, il substrato è non a 4 ma a 5 atomi di carbonio, per cui una volta ossidato e scisso in due frammenti, darà origine a un acetil-CoA e un propionil-CoA.
Altra fonte è la ossidazione degli acidi grassi a catena ramificata, con ramificazioni costituite da gruppi alchilici con un numero dispari di atomi di carbonio. Un esempio è l’acido fitanico, prodotto nei ruminanti dall’ossidazione del fitolo, un derivato della degradazione della clorofilla.
Nei ruminanti, è prodotto anche a partire dal glucosio liberato dall’idrolisi della cellulosa grazie all’azione di batteri presenti nel rumine, una delle quattro camere che compongono lo stomaco di questi animali. Sempre nel rumine, gli stessi batteri convertono, attraverso fermentazione, il glucosio in propionato, che potrà, una volta assorbito, essere utilizzato per la gluconeogenesi, essere ossidato per la produzione di energia, o essere utilizzato per la sintesi degli acidi grassi.[5]
Nei ruminanti, dove la gluconeogenesi tende a essere un processo continuo, il propionato è il più importante precursore gluconeogenetico.
Il propionato può derivare anche catabolismo della valina, leucina e isoleucina.
Nel colon, il propionato viene prodotto dai batteri del microbiota intestinale attraverso la fermentazione anaerobica di carboidrati non digeribili, come l’amido resistente e le fibre. Assorbito dagli enterociti, per la maggior parte non è ivi metabolizzato, passa nel circolo portale e raggiunge il fegato, dove può entrare nella gluconeogenesi.
L’ossidazione del propionil-CoA a succinil-CoA avviene attraverso tre reazioni che si verificano nel fegato e in altri tessuti.
Nella prima reazione il propionil-CoA è carbossilato a dare D-metilmalonil-CoA, nella reazione catalizzata dalla propionil-CoA carbossilasi (EC 6.4.1.3), enzima che ha come cofattore la biotina. La reazione consuma un ATP.
Propionil-CoA + HCO3– + ATP → D-Metilmalonil-CoA+ ADP + Pi
Nella reazione successiva il D-metilmalonil-CoA viene epimerizzato nello stereoisomero L. La reazione è catalizzata dalla metilmalonil-CoA epimerasi (EC 5.1.99.1).
D-Metilmalonil-CoA ⇄ L-Metilmalonil-CoA
Infine l’L-metilmalonil-CoA, nella reazione catalizzata dalla metilmalonil-CoA mutasi (EC 5.4.99.2), enzima che richiede come coenzima la 5-deossiadenosilcobalamina, un derivato della cobalamina o vitamina B12, subisce un riarrangiamento intramolecolare a dare succinil-CoA.
^ Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003;100:5107-5112. doi:10.1073/pnas.0730817100
^ Kuriyama H. et all. Coordinated regulation of fat-specific and liver-specific glycerol channels, aquaporin adipose and aquaporin 9. Diabetes 2002;51(10):2915-2921. 10.2337/diabetes.51.10.2915
^ McCommis K.S. and Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J 2015;466(3):443-454. doi:10.1042/BJ20141171
^ abcdefghi Nelson D.L., M. M. Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ abcde Rosenthal M.D., Glew R.H. Medical biochemistry – Human metabolism in health and disease. John Wiley J. & Sons, Inc., Publication, 2009
^ Soty M., Chilloux J., Delalande F., Zitoun C., Bertile F., Mithieux G., and Gautier-Stein A. Post-Translational regulation of the glucose-6-phosphatase complex by cyclic adenosine monophosphate is a crucial determinant of endogenous glucose production and is controlled by the glucose-6-phosphate transporter. J Proteome Res 2016;15(4):1342-1349. doi:10.1021/acs.jproteome.6b00110
^ abc Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
^ Van Schaftingen E., and Hers H-G. Inhibition of fructose-1,6-bisphosphatase by fructose-2,6-bisphosphate. Proc Natl Acad Sci USA 1981;78(5):2861-2863. doi:10.1073/pnas.78.5.2861
^ Van Schaftingen E., Jett M-F., Hue L., and Hers H-G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA 1981;78(6):3483-3486. doi:10.1073/pnas.78.6.3483
Il ciclo glucosio-alanina, anche detto ciclo di Cahill, proposto per la prima volta tra il 1969 e il 1970 da Mallette, Exton e Park, e Felig e collaboratori, consiste in una serie di reazioni attraverso le quali i tessuti extraepatici, come ad esempio il muscolo scheletrico, esportano al fegato piruvato, la base coniugata dell’acido piruvico, e gruppi amminici in forma di alanina, e ricevono, attraverso il circolo sanguigno, glucosio prodotto nel fegato.[1][4]
Di seguito ne sono riassunte le tappe.[5][6]
Quando nei tessuti extraepatici gli amminoacidi sono utilizzati a fini energetici, il piruvato, prodotto dal glucosio attraverso la glicolisi, funge da accettore del loro gruppo amminico alfa, formando alanina, un aminoacido non essenziale.
L’alanina diffonde nel circolo sanguigno, grazie al quale raggiunge il fegato.
Nel fegato, il gruppo amminico dell’alanina viene trasferito all’alfa-chetoglutarato a dare rispettivamente piruvato e glutammato.
Il glutammato cede per la maggior parte il gruppo amminico al ciclo dell’urea, mentre in parte funge da donatore di azoto in molti processi biosintetici.
Il piruvato entra nella gluconeogenesi e viene utilizzato per la produzione di glucosio.
Il glucosio neoformato diffonde dall’epatocita nel circolo sanguigno e raggiunge i tessuti periferici dove, grazie alla glicolisi, può essere convertito in piruvato, di nuovo disponibile per accettare il gruppo amminico α degli amminoacidi liberi, chiudendo così il ciclo.
Il ciclo glucosio-alanina fornisce quindi un collegamento tra il metabolismo dei carboidrati e quello degli amminoacidi.[6]
In breve:
Il ciclo glucosio-alanina esiste non solo tra il muscolo scheletrico, il primo tra i tessuti in cui fu osservato, e il fegato, ma coinvolge anche altre cellule e tessuti extraepatici tra cui le cellule del sistema immunitario, ad esempio gli organi linfoidi.
L’analisi successiva verrà fatta considerando il ciclo tra muscolo scheletrico e fegato.
Le proteine, sia intracellulari che extracellulari, sono continuamente idrolizzate nei loro amminoacidi costituenti e risintetizzate; e la velocità con cui avvengono i due processi è tale da evitare una perdita netta di massa magra dall’organismo.[3]
Tuttavia, in condizioni cataboliche, come nel digiuno o nell’esercizio intenso e prolungato, la velocità con cui avviene l’idrolisi delle proteine muscolari supera quella della loro sintesi de novo. Questo porterà alla liberazione di amminoacidi, alcuni dei quali sono utilizzati a fini energetici, altri a fini glucogenetici. Infatti, l’ossidazione dello scheletro carbonioso degli amminoacidi, in particolare di quelli a catena ramificata, valina, leucina, e isoleucina, rappresenta una significativa fonte di energia per il muscolo. Ad esempio, dopo circa 90 minuti dall’inizio di un esercizio fisico intenso, l’ossidazione intramuscolare degli amminoacidi fornisce il 10-15% dell’energia necessaria alla contrazione.
L’utilizzazione degli scheletri carboniosi degli amminoacidi a fini energetici implica la rimozione del loro gruppo amminico α, e quindi il successivo smaltimento di tale azoto in una forma non tossica.[5][6]
La rimozione del gruppo amminico α avviene attraverso reazioni di transaminazione che possono essere schematizzate come segue:
alfa-Chetoacido + Aminoacido ⇄ Nuovo aminoacido + Nuovo alfa-chetoacido
Tali reazioni, catalizzate da enzimi detti amminotransferasi o transaminasi (EC 2.6.1-) sono liberamente reversibili.
Gli amminoacidi ramificati, ad esempio, trasferiscono il gruppo amminico α all’α-chetoglutarato o acido 2-ossoglutarico, a dare glutammato e l’α-chetoacido derivato dall’amminoacido stesso, in una reazione catalizzata dalla transaminasi specifica per tale gruppo di amminoacidi o BCAT (EC 2.6.1.42), acronimo dell’inglese branched chain aminotransferases.
Ciclo glucosio-alanina nel muscolo scheletrico
Nel muscolo scheletrico, il glutammato prodotto potrà accettare un altro gruppo amminico a dare glutammina, per molti tessuti e organi, come ad esempio il cervello, la principale forma di trasporto interorgano dell’azoto.[7] La reazione è catalizzata dall’enzima citosolico glutammina sintetasi (EC 6.3.1.2) e consuma un ATP.
Glutammato + NH4+ + ATP → Glutammina + ADP + Pi
Ciò tuttavia comporterebbe l’uscita dal ciclo glucosio-alanina.
In alternativa, e a differenza di quanto accade nella maggior parte degli altri tessuti, il glutammato prodotto potrà partecipare a una reazione di transaminazione catalizzata dalla alanina aminotransferasi o ALT (EC 2.6.1.2), enzima presente nella maggior parte dei tessuti animali e vegetali. In tale reazione il glutammato dona il gruppo amminico alfa al piruvato, derivante dalla glicolisi, a dare alanina e alfa-chetoglutarato:
L’alanina prodotta e quella derivante dalla degradazione delle proteine, e le proteine muscolari ne sono piuttosto ricche, può lasciare la cellula ed essere veicolata dal circolo ematico al fegato, trasportandovi quindi il gruppo amminico. La velocità con cui l’alanina formata per transaminazione dal piruvato viene trasferita in circolo è proporzionale alla produzione intracellulare del piruvato.
Nota: alanina e glutammina sono le principali fonti di azoto e carbonio nel metabolismo interorgano degli amminoacidi.
Ciclo glucosio-alanina nel fegato
Una volta nel fegato si verifica una transaminazione catalizzata dalla alanina aminotransferasi epatica, in cui l’alanina, il principale amminoacido gluconeogenico, funge da donatore del gruppo amminico alfa, e l’alfa-chetoglutarato da chetoacido accettore.[6] I prodotti della reazione sono il piruvato, ossia lo scheletro carbonioso dell’alanina, e il glutammato.
Il glutammato, nella reazione catalizzata dalla glutammato deidrogenasi (EC 1.4.1.2), enzima presente nella matrice mitocondriale, rilascia ione ammonio, che entra nel ciclo dell’urea, e una molecola di alfa-chetoglutarato, che può entrare nel ciclo dell’acido citrico. Questa reazione rappresenta processo anaplerotico che lega il metabolismo degli amminoacidi con il ciclo dell’acido citrico.[6]
Tuttavia il glutammato potrà entrare anche nella reazioni di transaminazione, catalizzata dalla aspartato amminotransferasi (EC 2.6.1.1), con l’ossalacetato a dare aspartato e alfa-chetoglutarato. L’aspartato è uno degli amminoacidi coinvolti nella produzione di urea attraverso il ciclo dell’urea, ma può essere utilizzato pure nella sintesi delle purine e pirimidine.
Anche il piruvato prodotto potrà seguire destini metabolici differenti: essere ossidato per la produzione di ATP, e quindi uscire dal ciclo glucosio-alanina, o entrare nella via gluconeogenetica, e dunque proseguire nel ciclo glucosio-alanina.
Il glucosio prodotto verrà rilasciato dall’epatocita e attraverso il circolo ematico distribuito ai vari tessuti che lo richiedono, tra cui il muscolo scheletrico, dove viene utilizzato per la produzione di piruvato, di nuovo disponibile per accettare il gruppo amminico α del glutammato, chiudendo così il ciclo.
Transaminasi
Come detto in precedenza, la rimozione del gruppo amminico α degli amminoacidi avviene in reazioni di transaminazione, catalizzate da enzimi detti amminotransferasi o transaminasi.[6]
Sono enzimi citosolici, presenti in tutte le cellule e particolarmente abbondanti nel fegato, rene, intestino e muscolo, la maggior parte dei quali richiede come coenzima il piridossal fosfato o PLP, acronimo dell’inglese pyridoxal phosphate, la forma attiva della vitamina B6 o piridossina. Il coenzima è legato strettamente al sito attivo dell’enzima.
Nelle reazioni di transaminazione il gruppo amminico α degli amminoacidi liberi, con l’esclusione della treonina e lisina, è incanalato verso un numero ristretto di chetoacidi, in particolare piruvato, ossalacetato e alfa-chetoglutarato.
Le cellule contengono diversi tipi di amminotransferasi: molte sono specifiche per l’alfa-chetoglutarato come alfa-chetoacido, ma differiscono nella specificità per l’amminoacido, da cui prendono parte del nome. Esempi sono le già citate alanina aminotransferasi, anche detta alanina transaminasi e glutammico piruvico transferasi (GPT), e l’aspartato aminotransferasi (AST) o glutammico ossalacetico transaminasi (GOT) (EC 2.6.1.1).
Va sottolineato che nelle reazioni di transaminazione non si verifica alcuna deaminazione netta, nessuna perdita di gruppi amminici, in quanto l’alfa-chetoacido accettore viene amminato e l’amminoacido deaminato.
Trasporta azoto in una forma non tossica dai tessuti periferici al fegato.
Trasporta al fegato piruvato, un substrato gluconeogenico.
Rimuove piruvato dai tessuti periferici nei quali è così possibile ottenere una maggior produzione di ATP dal glucosio. Infatti il NADH prodotto durante la glicolisi può entrare nei mitocondri ed essere ossidato attraverso la fosforilazione ossidativa.
Permette di mantenere nell’epatocita una concentrazione relativamente alta di alanina, tale da inibire la degradazione delle proteine.
Può avere un ruolo nella difesa dell’ospite nei confronti delle malattie infettive.
Infine è importante sottolineare che nel ciclo glucosio-alanina non c’è sintesi netta di glucosio.
Costo energetico
Al pari del ciclo di Cori, anche il ciclo glucosio-alanina ha un costo energetico netto, che corrisponde a 3-5 molecole di ATP.
La parte del ciclo che si svolge nei tessuti periferici comporta la produzione di 5-7 molecole di ATP per molecola di glucosio:
2 ATP sono prodotti dalla glicolisi;
3-5 ATP derivano dal trasferimento degli elettroni dal NADH/FADH2 alla catena di trasporto degli elettroni.
Nel fegato invece la gluconeogenesi e il ciclo dell’urea consumano 10 ATP:
6 ATP sono consumati nel corso della gluconeogenesi;
4 ATP sono necessari per il ciclo dell’urea per ogni molecola di urea prodotta.
Il ciclo glucosio-alanina, al pari del ciclo di Cori, sposta parte del carico metabolico dai tessuti extraepatici al fegato.[6] Tuttavia il prezzo pagato dal fegato è ampiamente giustificato dai vantaggi che il ciclo apporta all’intero organismo in quanto consente, in particolari condizioni, un efficiente catabolismo delle proteine nei tessuti extraepatici, il che a sua volta permette di ottenere substrati per la gluconeogenesi come anche l’utilizzazione a fini energetici degli aminoacidi nei tessuti extraepatici.
Analogie e differenze con il ciclo di Cori
Tra i due cicli esistono alcune analogie di seguito elencate.[6][7]
Il ciclo di Cahill in parte si sovrappone al ciclo di Cori quando il piruvato viene convertito in glucosio e lo stesso trasportato ai tessuti extraepatici, dove attraverso la via glicolitica rigenera piruvato.
L’ingresso nella gluconeogenesi epatica è simile per i due cicli: sia l’alanina che il lattato sono infatti convertiti in piruvato.
Al pari del ciclo di Cori, anche il ciclo glucosio-alanina si estende attraverso tipi cellulari differenti, al contrario di quanto accade con vie metaboliche come la glicolisi, il ciclo dell’acido citrico o la gluconeogenesi che sono confinate all’interno di singole cellule.
Ciclo di Cori vs Ciclo Glucosio-Alanina
Di seguito, alcune differenze tra i due cicli.
La principale riguarda l’intermedio a tre atomi di carbonio che dai tessuti periferici raggiunge il fegato: il lattato nel il ciclo di Cori e l’alanina nel ciclo glucosio-alanina.
Un’altra differenza riguarda il destino del NADH prodotto dalla glicolisi nei tessuti periferici.
Nel ciclo di Cori il coenzima funge da donatore di agenti riducenti nella riduzione del piruvato a lattato, nella reazione catalizzata dalla lattato deidrogenasi (EC 1.1.1.27).
Nel ciclo glucosio-alanina tale riduzione non si verifica e gli elettroni del NADH potranno essere trasportati all’interno del mitocondrio dai sistemi navetta del malato-aspartato o del glicerolo-3-fosfato, generando NADH la prima navetta e FADH2 l’altra, da cui si otterranno rispettivamente 2,5 e 1,5 molecole di ATP.
Infine, dal punto precedente emerge che, a differenza del ciclo di Cori, per il ciclo glucosio-alanina è richiesta nei tessuti periferici anche la presenza di ossigeno e mitocondri.
Bibliografia
^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
^ Gropper S.S., Smith J.L., Groff J.L. Advanced nutrition and human metabolism. Cengage Learning, 2009
^ Lecker S.H., Goldberg A.L. and Mitch W.E. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J Am Soc Nephrol 2006;17(7):1807-1819.doi:10.1681/ASN.2006010083
^ Mallette L. E., Exton J.H., and Park C.R. Control of gluconeogenesis from amino acids in the perfused rat liver. J Biol Chem 1969;244(20):5713-5723. doi:10.1016/S0021-9258(18)63618-X
^ abc Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
^ abcdefgh Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
^ abc Wu G. Amino acids: biochemistry and nutrition. CRC Press, 2010
Il ciclo di Cori fu scoperto negli anni 30 e 40 del secolo scorso dai coniugi Carl e Gerty Cori, i quali scoprirono l’esistenza di una cooperazione metabolica tra il muscolo scheletrico che lavora in condizioni di limitata disponibilità di ossigeno e il fegato.[1]
Il ciclo permette la conversione del lattato, la forma in cui l’acido lattico è quasi esclusivamente presente a pH fisiologico, in glucosio, assicurando così un continuo apporto del monosaccaride ai tessuti periferici. La sua importanza è testimoniata anche dal fatto che è responsabile di circa il 40% del turn over del glucosio plasmatico.[7][8]
Dal punto di vista biochimico, il ciclo di Cori connette la glicolisi anaerobica con la gluconeogenesi, utilizzando tessuti differenti per compartimentalizzare processi opposti.[10]
Questa cooperazione metabolica è stata dimostrata esistere anche tra il fegato e tessuti extraepatici diversi dal muscolo scheletrico. Si può infatti affermare che, come per il ciclo glucosio-alanina, al ciclo di Cori possono partecipare i tessuti che non ossidino completamente il glucosio a CO2 e H2O.[11]
Di seguito sono riassunte le tappe del ciclo di Cori, considerando il lattato prodotto dalle fibre muscolari o dal globulo rosso.[12]
Il ciclo ha inizio con la conversione del glucosio a lattato, attraverso la glicolisi anaerobica e la successiva azione della lattato deidrogenasi (EC 1.1.1.27), che catalizza la riduzione del piruvato, la base coniugata dell’acido piruvico.
Segue la diffusione del lattato dalla cellula al circolo sanguigno, grazie al quale raggiunge, tra gli altri, il fegato, che è il suo principale utilizzatore, e la corteccia renale, in particolare i tubuli prossimali, essendo questi un altro sito dove avviene la gluconeogenesi.
Nel fegato e nella corteccia renale si verifica l’ossidazione, catalizzata dalla lattato deidrogenasi, del lattato a piruvato, che è quindi convertito a glucosio a mezzo della via gluconeogenica.[8]
Infine il glucosio prodotto diffonde nel circolo sanguigno, grazie al quale potrà raggiungere i globuli rossi o le fibre muscolari, chiudendo il ciclo.[10][11]
Globulo rosso
I globuli rossi, essendo privi di nucleo, ribosomi e mitocondri, sono più piccoli rispetto a molte altre cellule, e le ridotte dimensioni permettono loro il passaggio attraverso gli stretti capillari. Tuttavia la mancanza dei mitocondri li rende completamente dipendenti dalla glicolisi anaerobica per la produzione di ATP, il che significa che queste cellule producono continuamente acido lattico.[8][10]
La possibilità di procedere della glicolisi, come la sua velocità, dipendono anche dalla disponibilità di NAD+. Il coenzima nella sua forma ossidata è infatti necessario per l’ossidazione della gliceraldeide-3-fosfato a 1,3-bisfosfoglicerato nella reazione catalizzata dalla gliceraldeide-3-fosfato deidrogenasi (EC 1.2.1.12).
L’accumulo di NADH è evitato dalla riduzione del piruvato a lattato, in una reazione catalizzata dalla lattato deidrogenasi nella quale il NADH funge da donatore di agenti riducenti, ossidandosi a NAD+.[4]
Fibre muscolari
Le fibre muscolari a contrazione rapida contengono un numero ridotto di mitocondri e, in condizioni di limitata disponibilità di ossigeno, come durante un lavoro intenso, producono notevoli quantità di acido lattico.[8] In queste condizioni infatti:
la velocità di produzione del piruvato attraverso la via glicolitica eccede la capacità del ciclo dell’acido citrico di ossidarlo, tanto che meno del 10% del piruvato prodotto entra nel ciclo stesso;
la velocità alla quale l’ossigeno è assunto dalle cellule non è sufficiente ad assicurare l’ossidazione aerobica di tutto il NADH formato.[3]
In tali condizioni la glicolisi anaerobica porta alla produzione di 2 molecole di ATP per molecola di glucosio, 3 se il glucosio proviene dal glicogeno muscolare, una quantità decisamente inferiore rispetto alle 29-30 molecole di ATP prodotte dalla completa ossidazione del glucosio.[9] Tuttavia la velocità con cui l’ATP è prodotto dalla glicolisi anaerobica è molto maggiore rispetto a quella ottenibile dalla completa ossidazione del glucosio.[10]
Infine, come nel globulo rosso, la reazione catalizzata dalla lattato deidrogenasi, rigenerando NAD+, permette alla glicolisi di procedere, ma produce lattato.[4]
Lattato
L’acido lattico è un prodotto finale del metabolismo, e per essere utilizzato dalla cellula deve essere convertito in piruvato.[3]
La membrana plasmatica della maggior parte delle cellule è liberamente permeabile sia al piruvato che al lattato, che possono quindi raggiungere il circolo ematico.[3] Considerando ad esempio la fibra muscolare, la quantità di lattato che lascia la cellula è maggiore rispetto a quella del piruvato grazie all’elevato rapporto NADH/NAD+ intracellulare e alle proprietà catalitiche dell’isoenzima muscolare della lattato deidrogenasi.[5]
Una volta in circolo il lattato raggiunge, tra gli altri, il fegato e la corteccia renale, dove viene ossidato a piruvato, nella reazione catalizzata dagli specifici isoenzimi della lattato deidrogenasi.
Nell’epatocita questa ossidazione è favorita dal basso rapporto NADH/NAD+ presente nel citosol. Il piruvato è quindi disponibile per entrare nella tappa successiva del ciclo di Cori, la gluconeogenesi.[3]
Il glucosio prodotto, tramite il circolo ematico raggiunge il muscolo e il globulo rosso, chiudendo così il ciclo. Ovviamente il monosaccaride raggiungerà anche tutti gli altri tessuti e cellule che lo richiedono.
Costo energetico
Il ciclo di Cori comporta un consumo netto di 4 molecole di ATP.
La parte del ciclo che comprende la gluconeogenesi consuma 6 equivalenti di ATP, nello specifico 4 ATP e 2 GTP, nelle reazioni catalizzate dagli enzimi:
piruvato carbossilasi (EC 6.4.1.1): un ATP;
fosfoenolpiruvato carbossichinasi (EC 4.1.1.32): un GTP;
gliceraldeide-3-fosfato deidrogenasi: un ATP.
Poiché sono utilizzate due molecole di lattato per la sintesi di ogni molecola di glucosio, il costo totale è di 2 x 3 = 6 legami ad alta energia per molecola di glucosio.[2]
Di contro, la parte del ciclo che comprende la glicolisi anaerobica ne produce 2.
In definitiva, è richiesta più energia per produrre glucosio dall’acido lattico nel fegato e nella corteccia renale rispetto a quella ottenuta dall’ossidazione anaerobica del glucosio nei tessuti extraepatici.
Il ciclo di Cori è un ciclo futile?
La demolizione e risintesi del glucosio caratteristica del ciclo di Cori può sembrare uno spreco di energia. In realtà questo ciclo permette l’efficace funzionamento di numerose cellule extraepatiche a spese del fegato e della corteccia renale, ed è quindi forse più corretto definirlo ciclo del substrato piuttosto che ciclo futile.[10] Di seguito alcuni esempi.
Il ciclo di Cori permette di smaltire parte dell’acido lattico che i globuli rossi producono.[8]
Se si considera il muscolo scheletrico, la glicolisi anaerobica rappresenta una efficiente sorgente di ATP nel corso di un esercizio intenso. Ma questo potrebbe portare a un accumulo intracellulare di lattato, e a una pericolosa diminuzione del pH intracellulare. Ovviamente tale accumulo non si verifica, grazie anche al ciclo di Cori, che scarica parte del lattato muscolare e del costo energetico per il suo smaltimento sui tessuti gluconeogenici.[3] Ad esempio, il debito di ossigeno, il “fiatone”, che sempre si presenta dopo un’attività fisica sostenuta, è in gran parte dovuto all’aumentata richiesta di ossigeno da parte degli epatociti per sostenere l’ossidazione degli acidi grassi, il loro principale carburante, ossidazione che porterà alla produzione dell’ATP necessario per la gluconeogenesi.[6][12]
Nel corso di traumi, sepsi, ustioni, o dopo grossi interventi chirurgici, si verifica un’intensa proliferazione cellulare nelle ferite, che sono tessuti ipossici, e nel midollo osseo. Questo a sua volta risulta in una maggiore produzione di acido lattico e un aumento del flusso attraverso il ciclo di Cori e, a livello epatico, in un consumo di ATP. Una situazione simile sembra presentarsi anche in quei pazienti oncologici che vanno incontro a una progressiva perdita di peso.[2]
Il ciclo di Cori è fondamentale anche nel digiuno.[6]
Ciclo di Cori e ciclo glucosio-alanina
Tra i due cicli esistono analogie e differenze.
Il ciclo glucosio-alanina e quello di Cori sono vie metaboliche che si estendono attraverso tipi cellulari differenti e contribuiscono ad assicurare un continuo rifornimento di glucosio ai tessuti.[12] In entrambe l’ingresso nella gluconeogenesi comporta la conversione del lattato e dell’alanina in piruvato. Infine, in entrambe, il glucosio prodotto è quindi trasportato ai tessuti periferici dove la via glicolitica rigenera piruvato.[13]
Invece, la loro principale differenza consiste nell’intermedio a tre atomi di carbonio che viene riciclato: nel ciclo di Cori il carbonio torna al fegato in forma di piruvato, mentre nel ciclo glucosio-alanina in forma di alanina.[8] I cicli differiscono anche per il destino metabolico del NADH: nel ciclo di Cori funge da agente riducente nella reazione catalizzata dalla lattato deidrogenasi, mentre nel ciclo glucosio-alanina gli elettroni del coenzima sono utilizzati per la sintesi di ATP nel mitocondrio. Pertanto, un’altra differenza è che il ciclo glucosio-alanina richiede la presenza di ossigeno, mentre non la richiede il ciclo di Cori.[8]
^ Gleeson T.T. Post-exercise lactate metabolism: a comparative review of sites, pathways, and regulation. Annu Rev Physiol 1996;58:565-81. doi:10.1146/annurev.ph.58.030196.003025
^ ab Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012
Gli acidi e i sali biliari sono derivati polari del colesterolo, e rappresentano la principale via per l’eliminazione dello steroide dal corpo.
Sono un gruppo di specie molecolari con struttura chimica simile ma non identica, proprietà fisiche differenti e caratteristiche biologiche anche più divergenti.
Sono sintetizzati nel fegato, immagazzinati nella cistifellea, secreti nel duodeno, e infine per la maggior parte riassorbiti nell’ileo.
Poiché a pH fisiologico sono presenti in forma di anioni, i termini acido biliare e sale biliare saranno di seguito utilizzati come sinonimi.
I sali biliari presentano analogia e differenze con la molecola del colesterolo.
Al pari dello steroide posseggono un nucleo formato da 4 anelli fusi, tre a sei atomi di carbonio, indicati come A, B e C, e uno, indicato come D, a 5; tale struttura è il ciclopentanoperidrofenantrene, più comunemente noto come nucleo steroideo.
Acidi BiIiari e Loro Coniugati
Negli vertebrati superiori sono formati da 24 atomi di carbonio, poiché hanno una coda idrocarburica più corta di tre atomi di carbonio rispetto a quella del colesterolo. Nei vertebrati inferiori sono formati a 25, 26 o 27 atomi di carbonio. La coda idrocarburica termina con un gruppo carbossilico, spesso ionizzato a pH 7, che può essere legato all’aminoacido glicina o taurina.
Oltre al gruppo ossidrilico in posizione 3, possono avere gruppi ossidrilici in posizione 7 e/o 12.
Tutto ciò rende queste molecole molto più polari del colesterolo.
Poiché gli anelli A e B sono fusi in configurazione cis, la struttura del nucleo steroideo risulta curva, ed è possibile individuarvi:
un lato concavo, verso cui sono orientati i gruppi idrossilici e il gruppo carbossilico della catena laterale, con o senza l’aminoacido a esso legato, e che risulta idrofilico;
un lato convesso, verso cui sono orientati i gruppi metilici presenti in posizione 18 e 19, che risulta idrofobico.
Rappresentazione dell’Acido Colico
Dunque, possedendo sia gruppi polari che non polari sono molecole anfifiliche e ottimi surfattanti. Tuttavia la loro struttura chimica li rende molto diversi rispetto ai surfattanti tradizionali, dove spesso si individua una testa polare e una lunga coda non polare.
Acidi e sali biliari primari, coniugati e secondari
Sono definiti acidi biliari primari le molecole sintetizzate negli epatociti direttamente dal colesterolo. Nell’uomo i principali sono l’acido colico e l’acido chenodesossicolico, che da soli formano fino all’80% di tutti gli acidi biliari.
Prima di essere secreti nell’albero biliare sono coniugati quasi per intero, sino al 98%, con gli amminoacidi glicina o taurina, a dare rispettivamente glicoconiugati e tauroconiugati. In particolare circa il 75% dell’acido colico e chenodesossicolico sono coniugati con la glicina, a dare acido glicocolico e acido glicochenodesossicolico, il restante 25% con la taurina, a dare acido taurocolico e acido taurochenodesossicolico.
Acidi Biliari Coniugati
Gli acidi biliari coniugati sono molecole dotate di gruppi maggiormente idrofilici rispetto a quelle di origine, dunque sono emulsionanti molto più efficaci. La coniugazione ha infatti l’effetto di ridurre il loro pKa facendo si che rimangano ionizzate in un intervallo più ampio di pH. Se infatti il pKa tipico dei non coniugati è di circa 6, si passa a circa 4 con l’acido glicocolico, e circa 2 con l’acido taurocolico.
L’idrofilicità dei comuni acidi e sali biliari decresce secondo il seguente ordine: coniugati della glicina < coniugati della taurina < acido litocolico < acido desossicolico < acido chenodesossicolico < acido colico < acido ursodesossicolico.
Infine la coniugazione ha anche l’effetto di ridurre la citotossicità delle molecole di origine.
Gli acidi biliari secondari derivano dai primari che non sono stati riassorbiti durante il loro passaggio nell’intestino tenue. Una volta raggiunto il colon, gli acidi biliari primari possono infatti subire diverse modificazioni ad opera del microbiota intestinale, che è parte del più ampio microbiota umano, a dare appunto gli acidi biliari secondari. Con gli acidi grassi a catena corta sono sono i due principali tipi di metaboliti batterici prodotto nel colon, e formano il il restante 20% del pool corporeo totale degli acidi biliari.
Un altro modo di suddividere i sali biliari fa riferimento alla loro coniugazione con aminoacidi e al grado di idrossilazione. Su queste base si individuano tre categorie.
I coniugati triidrossilati, quali l’acido taurocolico e il glicocolico.
I coniugati diidrossilati, come l’acido glicodesossicolico, glicochenodesossicolico, taurochenodesossicolico e taurodesossicolico. Nella bile rappresentano circa il 60% del totale dei sali biliari.
Forme non coniugate come l’acido colico, desossicolico, chenodesossicolico, e litocolico.
Funzione dei sali biliari
Premessa: tutte le funzioni fisiologiche sono portate a termine dai sali biliari in forma coniugata.
Rappresentano la via principale per l’eliminazione del colesterolo. Nell’uomo infatti non esiste il corredo enzimatico necessario per rompere nessuno dei quattro anelli del nucleo steroideo, ne per ossidare il colesterolo ad anidride carbonica e acqua.
L’altro modo per eliminare il colesterolo è con la bile, in forma libera.
I sali biliari sono potenti surfattanti. E in particolare, i coniugati di- e triidrossilati sono i surfattanti migliori, molto più efficaci rispetto ai corrispettivi non coniugati, avendo un numero maggiore di gruppi polari.
Una volta a contatto con i lipidi apolari nel lume del piccolo intestino, il lato convesso apolare va a interagire con i lipidi idrofobici, quali trigliceridi, esteri del colesterolo e delle vitamine liposolubili, mentre il lato concavo polare prende contatto con il mezzo acquoso circostante. Ciò incrementa la dispersione nel mezzo acquoso dei lipidi apolari, favorendo la formazione di minuscole goccioline lipidiche, che subiranno l’attacco delle lipasi, in particolare della lipasi pancreatica, nella cui attivazione i sali biliari hanno un ruolo diretto, e delle esterasi intestinali. In seguito facilitano l’assorbimento dei lipidi prodotti, nonché delle vitamine liposolubili, ad opera della mucosa intestinale grazie alla formazione di micelle miste.
Una funzione simile è svolta nella cistifellea dove, formando micelle miste con i fosfolipidi, prevengono la precipitazione del colesterolo.
Nota: grazie alla disposizione dei gruppi polari e non polari, una volta in soluzione acquosa, i sali biliari tendono a formare micelle, di solito composte da meno di 10 monomeri, purché la loro concentrazione sia superiore alla cosiddetta concentrazione micellare critica.
A livello intestinale modulano la secrezione degli enzimi pancreatici e della colecistochinina.
Sia nell’intestino tenue che nel colon hanno una potente attività antimicrobica, in primis l’acido desossicolico, in particolare contro i batteri Gram-positivi. Questa attività potrebbe essere dovuta a danno ossidativo al DNA e/o al danno alle membrane cellulari batteriche. Sono dunque importanti nella prevenzione della sovracrescita batterica, ma sembra abbiano anche un ruolo nella regolazione della composizione del microbiota intestinale.
Negli ultimi anni è divenuto evidente il loro ruolo regolatorio sul controllo del metabolismo energetico, in particolare per la “movimentazione” epatica del glucosio.
Circolo enteroepatico
A seguito del consumo di lipidi con la dieta, le cellule enteroendocrine del duodeno secernono in circolo la colecistochinina. Il successivo legame dell’ormone alle cellule muscolari lisce della parete della cistifellea ne promuove la contrazione; inoltre l’ormone causa anche il rilascio dello sfintere di Oddi. Da tutto ciò risulta la secrezione pulsatile della bile, e degli acidi biliari in essa contenuti, nel duodeno.
In condizioni fisiologiche il pool corporeo di acidi biliari è costante, e pari a circa 3-5 g; ciò è reso possibile da due processi:
il loro riassorbimento a livello intestinale;
la loro sintesi de novo.
Fino al 95% dei sali biliari secreti viene riassorbito a livello intestinale, non assieme ai prodotti della digestione dei lipidi, ma attraverso un processo definito circolo enteroepatico.
Si tratta di un sistema di recupero estremamente efficiente, che sembra avvenire almeno due volte per ogni pasto, cui partecipano il fegato, l’albero biliare, il duodeno, il colon, e il circolo portale attraverso cui le molecole riassorbite tornano al fegato. Tale ricircolo è reso necessario dal fatto che la capacità dell’epatocita di produrre acidi biliari è limitata e insufficiente a soddisfare le necessità fisiologiche intestinali se gli stessi sali andassero perduti in elevate quantità.
La maggior parte dei sali biliari è riassorbita una volta raggiunto l’ileo distale, la parte più bassa dell’intestino tenue, a mezzo di un trasportatore sodio-dipendente presente nell’orletto a spazzola degli enterociti, detto ASBT, acronimo dell’inglese apical sodium-dependent bile acid transporter, che opera un cotrasporto di due ioni sodio e un acido biliare.
Una volta nell’enterocita si ritiene che, a mezzo della proteina IBABP, acronimo dell’inglese ileal bile acid-binding protein, siano trasportati attraverso il citosol alla membrana basolaterale, che attraversano grazie all’intervento del trasportatore OSTalfa/OSTbeta, acronimo dell’inglese organic solute transporter alpha and beta. Tramite il circolo portale, veicolati dall’albumina, raggiungono il fegato.
Da notare che una piccola parte di acidi biliari raggiunge il fegato attraverso l’arteria epatica.
A livello epatico la loro estrazione dal circolo è molto efficiente, tanto che dal 50 al 90% sono rimossi al primo passaggio, percentuale che varia in funzione della struttura molecolare. Gli acidi biliari coniugati sono in gran parte rimossi attraverso un meccanismo di trasporto attivo sodio-dipendente, a mezzo del trasportatore NTCP, acronimo dell’inglese Na+-dependent taurocholate co-transport polipeptide. Tuttavia può avvenire anche un trasporto sodio-indipendente ad opera di proteine della famiglia OATP, acronimo dell’inglese organic anion transporting polypeptides, principalmente le isoforme OATP1B1 e OATP1B3.
Nel circolo enteroepatico il passaggio limitante è rappresentato dalla loro secrezione nei canalicoli biliari, in gran parte ad opera di BSEP, acronimo dell’inglese bile salt export pump, in un processo ATP-dipendente. Questa pompa trasporta gli acidi biliari monoanionici, che sono la maggior parte. Gli acidi biliari solforati o glucuronati, dianionici, sono secreti a mezzo di trasportatori differenti, quali MRP2 e BCRP.
Nota: il livello sierico degli acidi biliari varia sulla base della velocità di riassorbimento e quindi è più alto durante i pasti, quando il circolo enteroepatico è più attivo.
Metabolismo intestinale
Gli acidi biliari che sfuggono al riassorbimento ileale passano nel colon dove in parte subiscono l’azione di enzimi della flora batterica e sono trasformati in acidi biliari secondari.
Di seguito sono elencate le principali reazioni.
Deconiugazione
A livello della catena laterale si può verificare l’idrolisi del legame con l’aminoacido coniugato in posizione 24, con liberazione di acidi biliari non coniugati e glicina o taurina. Le reazioni sono catalizzate da idrolasi batteriche presenti sia nell’intestino tenue che nel colon.
7alfa-Deidrossilazione
E’ la reazione quantitativamente più importante, portata a termine da deidratasi batteriche coloniche che rimuovono il gruppo ossidrilico in posizione 7 dando origine ad un 7-deossi acido biliare. In particolare, dall’acido colico si formerà l’acido desossicolico, mentre dal chenodesossicolico il litocolico, due acidi biliari secondari tossici.
Da notare che la 7alfa-deidrossilazione, a differenza di quanto accade con l’ossidazione e l’epimerizzazione, può avvenire solamente sugli acidi biliari non coniugati, per cui la deconiugazione è un prerequisito essenziale.
Ossidazione ed epimerizzazione
Sono reazioni che interessano i gruppi idrossilici in posizione 3, 7 e 12, catalizzate da idrossisterolo deidrogenasi batteriche. Ad esempio, l’epimerizzazione dell’acido chenodesossicolico da origine all’acido ursodesossicolico.
Metabolismo Intestinale degli Acidi Biliari
Parte degli acidi biliari secondari sono poi riassorbiti e tornano al fegato, per essere riconiugati, se necessario, e secreti nuovamente. Quelli che invece sfuggono al riassorbimento saranno perduti con le feci.
Mentre le reazioni di deconiugazione e ossidazione sono portate a termine da un ampio spettro di batteri anaerobi, le 7alfa-deidrossilazioni sono effettuate da un numero ristretto di anaerobi.
La 7alfa-deidrossilazione e la deconiugazione hanno come effetto quello di aumentare il pKa e dunque l’idrofobicità degli acidi biliari, permettendone un certo grado di recupero passivo attraverso l’epitelio colonico.
L’aumento di idrofobicità è associato anche ad un aumento della loro citotossicità. E una elevata concentrazione di acidi biliari secondari nelle feci, sangue e bile è stata associata alla patogenesi del cancro al colon.
Riassorbimento e fibre solubili
Il riassorbimento degli sali biliari può essere ridotto dall’azione chelante delle fibre solubili, come quelle presenti nella frutta fresca, legumi, avena e crusca d’avena. Tutto questo ha come effetto quello di incrementare la loro sintesi de novo, up-regolando l’espressione della colesterolo 7alfa-idrossilasi e della sterolo 12alfa-idrossilasi, e quindi ridurre la concentrazione del colesterolo negli epatociti.
La deplezione del colesterolo epatico aumenta l’espressione del recettore per le LDL, e quindi abbassa la concentrazione plasmatica del colesterolo LDL. Di contro però stimola anche la sintesi della HMG-CoA reduttasi, l’enzima chiave nella sintesi dello steroide.
Anche diversi farmaci agiscono legando gli acidi biliari a livello intestinale, impedendone così il riassorbimento.
Sintesi
Dal punto di vista quantitativo, gli acidi biliari sono il prodotto principale del metabolismo del colesterolo.
Come detto in precedenza, il circolo enteroepatico e la loro sintesi de novo assicurano la costanza del pool corporeo. In particolare, la sintesi de novo permette la sostituzione di quel 5-10%, circa 0,5 g/die, che viene perduto con le feci.
Di seguito verrà presa in esame la sintesi dell’acido colico e dell’acido chenodesossicolico, e la loro coniugazione con gli aminoacidi taurina e glicina.
Esistono due vie principali per la sintesi dei suddetti acidi: la via classica e quella alternativa. A queste si aggiungono alcune vie minori.
Sintesi de Novo degli Acidi Biliari Primari e dei loro Coniugati
La via classica o neutra
Nell’uomo fino al 90% dei sali biliari sono sintetizzati attraverso la via classica, definita anche “neutra” in quanto i suoi intermedi sono steroli neutri.
E’ una via presente solo nel fegato, cui prendono parte numerosi enzimi localizzati nel citosol, nel reticolo endoplasmatico, nei mitocondriale e perossisomi, e i cui prodotti finali sono i coniugati degli acidi colico e chenodesossicolico.
La prima reazione è l’idrossilazione in posizione 7 del colesterolo, a dare il 7alfa-idrossicolesterolo. La reazione è catalizzata dalla colesterolo 7alfa-idrossilasi o CYP7A1 (E.C. 1.14.14.23). E’ un enzima localizzato sul reticolo endoplasmatico, e catalizza il passaggio limitante dell’intera via.
Il 7alfa-idrossicolesterolo subisce l’ossidazione del gruppo 3beta-ossidrilico e lo spostamento del doppio legame dalla posizione 5,6 alla posizione 4,5 a dare il 7alfa-idrossi-4-colesten-3-one. La reazione è catalizzata dalla 3beta-idrossi-Δ5-C27-steroide ossidoreduttasi o HSD3B7 (E.C. 1.1.1.181), un enzima del reticolo endoplasmatico.
Il 7alfa-idrossi-4-colesten-3-one può seguire due vie:
entrare nella via che porta alla sintesi dell’acido colico, attraverso la reazione catalizzata dalla 7alfa-idrossi-4-colesten-3-one 12alfa-monoossigenasi o sterolo 12alfa-idrossilasi o CYP8B1 (E.C. 1.14.18.8), enzima del reticolo endoplasmatico;
entrare nella via che porta la sintesi dell’acido chenodesossicolico, attraverso la reazione catalizzata dalla 3-osso-Δ4-steroide 5β-reduttasi o AKR1D1 (E.C. 1.3.1.3), enzima citosolico.
Ed è l’attività della sterolo 12alfa-idrossilasi che determinerà il rapporto tra gli acidi colico e chenodesossicolico prodotti, e, in definitiva, quella che sarà la potenza detergente del pool degli acidi biliari. E infatti la regolazione della trascrizione del gene corrispondente è uno dei punti di regolazione principali dell’intera via di sintesi.
Dunque, se il 7alfa-idrossi-4-colesten-3-one fluisce attraverso la reazione catalizzata dalla sterolo 12alfa-idrossilasi si avranno le seguenti reazioni.
La molecola è idrossilata in posizione 12 dall’enzima suddetto, a dare il 7alfa,12alfa-diidrossi-4-colesten-3-one.
Il 7alfa,12alfa-diidrossi-4-colesten-3-one subirà la riduzione del doppio legame in posizione 4,5, nella reazione catalizzata dalla 3-osso-Δ4-steroide 5beta-reduttasi, a dare 5beta-colestan-7alfa,12alfa-diol-3-one.
Il 5beta-colestan-7alfa,12alfa-diol-3-one subirà a sua volta la riduzione a gruppo ossidrilico del gruppo in posizione 4, nella reazione catalizzata dalla 3alfa-idrossisteroide deidrogenasi o AKR1C4 (EC 1.1.1.213), enzima citosolico, a dare 5beta-colestan-3alfa,7alfa,12alfa-triolo.
Il 5beta-colestan-3alfa,7alfa,12alfa-triolo subirà l’ossidazione della catena laterale in tre reazioni catalizzate dalla sterolo 27-idrossilasi o CYP27A1 (EC 1.14.15.15). Si tratta di un enzima mitocondriale presente anche in tessuti extraepatici e nei macrofagi, che introduce un gruppo ossidrilico in posizione 27. Il gruppo ossidrilico è poi ossidato ad aldeide e quindi ad acido carbossilico, con formazione di acido 3alfa,7alfa, 12alfa-triidrossi-5beta-colestanoico.
L’acido 3alfa,7alfa, 12alfa-triidrossi-5beta-colestanoico è attivato a seguito del legame con il coenzima A, nella reazione catalizzata dagli enzimi BACS, acronimo dell’inglese bile acid CoA synthetase (EC 6.2.1.7), o VLCS, acronimo dell’inglese very long chain acyl CoA synthetase (EC 6.2.1.-), entrambe localizzati nel reticolo endoplasmatico.
Il 3alfa,7alfa,12alfa-triidrossi-5beta-colestanoilCoA è trasportato nei perossisomi dove va incontro a 5 reazioni consecutive, catalizzate da altrettanti enzimi differenti. Nelle ultime due la catena laterale è accorciata a quattro atomi di carbonio e viene prodotto il colilCoA.
L’ultimo passaggio coinvolge la coniugazione, attraverso legame ammidico, del gruppo carbossilico terminale della catena laterale con l’aminoacido glicina o taurina, nella reazione catalizzata da BAAT, acronimo dell’inglese bile acid-CoA:amino acid N-acyltransferase (EC 2.3.1.65), enzima a localizzazione prevalentemente perossisomiale.
I prodotti della reazione saranno quindi gli acidi biliari coniugati glicocolico e taurocolico.
Nel caso in cui il 7alfa-idrossi-4-colesten-3-one non entri nella reazione catalizzata dalla sterolo 12alfa-idrossilasi prenderà la via che porta alla sintesi dei coniugati dell’acido chenodesossicolico attraverso le reazioni di seguito descritte.
Il 7alfa-idrossi-4-colesten-3-one viene convertito in 7alfa-idrossi-5β-colestan-3-one nella reazione catalizzata dall’enzima 3-osso-Δ4-steroide 5beta-reduttasi.
Il 7alfa-idrossi-5β-colestan-3-one nella reazioni dalla 3alfa-idrossisteroide deidrogenasi, viene convertito in 5beta-colestan-3alfa,7alfa-diolo.
Quindi, a seguito di modificazione analoghe a quelle viste per la sintesi dei coniugati dell’acido colico, e catalizzate per la maggior parte dagli stessi enzimi, si formeranno gli acidi biliari coniugati glicochenodesossicolico e taurochenodesossicolico.
Nota: gli acidi biliari deconiugati a livello intestinale dovranno raggiungere il fegato per essere riconiugati.
La via alternativa o acida
E’ prevalente nel feto e nel neonato, mentre nell’adulto porta alla sintesi di meno del 10% dei sali biliari.
Questa via si differenzia da quella classica, in quanto:
i prodotti intermedi sono acidi, per cui viene definita anche come “via acida”;
l’ossidazione della catena laterale precede le modificazioni del nucleo steroideo;
i prodotti finali sono i coniugati dell’acido chenodesossicolico.
Il primo passaggio comporta la conversione del colesterolo in 27-idrossicolesterolo nella reazione catalizzata dalla sterolo 27-idrossilasi.
A questo punto sono possibili due vie.
Via A
In una reazione catalizzata dalla sterolo 27-idrossilasi, il 27-idrossicolesterolo è convertito in acido 3beta-idrossi-5-colestenoico.
L’acido 3beta-idrossi-5-colestenoico nella reazione catalizzata dalla ossisterolo 7α-idrossilasi o CYP7B1 (EC 1.14.13.100), un enzima del reticolo endoplasmatico, viene idrossilato in posizione 7 a dare l’acido 3beta-7alfa-diidrossi-5-colestenoico.
L’acido 3beta-7alfa-diidrossi-5-colestenoico viene convertito in acido 3-osso-7alfa-idrossi-4-colestenoico, nella reazione catalizzata dalla 3beta-idrossi-Δ5-C27-steroide ossidoreduttasi.
L’acido 3-osso-7alfa-idrossi-4-colestenoico, a seguito di modificazioni della catena laterale, darà origine all’acido chenodesossicolico e quindi ai suoi coniugati.
Via B
Il 27-idrossicolesterolo è convertito in 7alfa,27-diidrossicolesterolo nella reazione catalizzata dalla ossisterolo 7alfa-idrossilasi e colesterolo 7alfa-idrossilasi.
Il 7alfa,27-diidrossicolesterolo è convertito in 7alfa,26-diidrossi-4-colesten-3-one nella reazione catalizzata dalla 3beta-idrossi-Δ5-C27-steroide ossidoreduttasi.
Il 7alfa,26-diidrossi-4-colesten-3-one può ora proseguire come tale o essere convertito in acido 3-osso-7alfa-idrossi-4-colestenoico e subire le modifiche alla catena laterale e le altre reazioni che portano alla sintesi dei coniugati dell’acido chenodesossicolico.
Le vie minori
Esistono anche vie minori che possono contribuire, sebbene in misura ridotta, alla sintesi degli acidi biliari.
Ad esempio:
Nel fegato viene espressa una colesterolo 25-idrossilasi (EC 1.14.99.38).
Nel cervello è espressa una colesterolo 24- idrossilasi o CYP46A1 (EC 1.14.14.25), e quindi, sebbene l’organo non sia in grado di esportare colesterolo, può esportare ossisteroli.
E’ stata scoperta anche una 7alfa-idrossilasi non specifica espressa in tutti i tessuti, che sembra essere coinvolta nella generazione di ossisteroli che possono poi essere trasportati al fegato per essere convertiti in acido chenodesossicolico.
Inoltre, la sterolo 27-idrossilasi è espressa in vari tessuti, per cui i suoi prodotti di reazione possono essere trasportati al fegato ed essere convertiti in sali biliari.
Regolazione della sintesi degli acidi biliari
La regolazione avviene attraverso un feedback negativo, in particolare sull’espressione degli enzimi colesterolo 7alfa-idrossilasi e sterolo 12alfa-idrossilasi.
In caso di eccesso di acidi biliari, sia coniugati che liberi, gli stessi si vanno a legare al recettore nucleare FRX, acronimo dell’inglese farnesoid X receptor, attivandolo. L’acido biliare più efficace nell’attivare FRX è l’acido chenodesossicolico, mentre altri, come l’acido ursodesossicolico non lo attivano.
FRX induce l’espressione del repressore trascrizionale SHP, acronimo dell’inglese small heterodimer partner, che a sua volta interagisce con altri fattori trascrizionali, come LRH-1, acronimo dell’inglese liver receptor homolog-1, e HNF-4alfa, acronimo dell’inglese hepatocyte nuclear factor-4alfa, che si legano ad una sequenza presente nella regione del promotore dei geni per la 7alfa-idrossilasi e 12alfa-idrossilasi, regione definita BAREs, acronimo dell’inglese bile acid response elements, inibendone la trascrizione.
Uno dei motivi per cui la sintesi dei sali biliari è strettamente regolata è perché molti dei loro metaboliti sono tossici.
Bibliografia
Chiang J.Y.L. Bile acids: regulation of synthesis. J Lipid Res 2009;50(10):1955-1966. doi:10.1194/jlr.R900010-JLR200
Gropper S.S., Smith J.L. Advanced nutrition and human metabolism. 6th Edition. Cengage Learning, 2012
Moghimipour E., Ameri A., and Handali S. Absorption-enhancing effects of bile salts. Molecules 2015;20(8); 14451-14473. doi:10.3390/molecules200814451
Monte M.J., Marin J.J.G., Antelo A., Vazquez-Tato J. Bile acids: Chemistry, physiology, and pathophysiology. World J Gastroenterol 2009;15(7):804-816. doi:10.3748/wjg.15.804
Rosenthal M.D., Glew R.H. Medical biochemistry – Human metabolism in health and disease. John Wiley J. & Sons, Inc., Publication, 2009
Sundaram S.S., Bove K.E., Lovell M.A. and Sokol R.J. Mechanisms of Disease: inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol 2008;5(8):456-468. doi:10.1038/ncpgasthep1179
Il tratto gastrointestinale dell’uomo è una delle più feroci e competitive nicchie ecologiche presenti in natura. Vi si ritrovano virus, eucarioti, batteri, e una sola specie di Archeobatteri, Methanobrevibacter smithii.
I batteri variano in proporzione e quantità lungo tutto il tratto gastrointestinale. La presenza maggiore si ha nel colon, con oltre 400 specie diverse appartenenti a 9 fila o divisioni, ed è a questi che ci si riferirà parlando di microbiota intestinale, che a sua volta è parte del più ampio microbiota umano. Di seguito, l’elenco dei phyla suddetti e di alcuni tra i loro generi maggiormente rappresentati.
Actinobacteria, Gram-positivi; Bifidobacterium, Collinsella, Eggerthella e Propionibacterium.
Bacteroidetes, Gram-negativi; oltre 20 generi tra cui Bacteroides, Prevotella e Corynebacterium.
Cyanobacteria, Gram-negativi.
Firmicutes, Gram-positivi; almeno 250 generi tra cui Mycoplasma, Bacillus, Clostridium, Dorea, Faecalibacterium, Ruminococcus, Eubacterium, Staphylococcus, Streptococcus, Lactobacillus, Lactococcus,Enterococcus, Sporobacter e Roseburia.
Fusobacteria, Gram-negativi; Sneathia.
Lentisphaerae, Gram-negativi.
Proteobacteria, Gram-negativi; Escherichia, Klebsiella, Shigella, Salmonella, Citrobacter, Helicobacter e Serratia.
Spirochaeates, Gram-negativi.
Verrucomicrobia, Gram-negativi.
La presenza nel colon di un piccolo sottoinsieme del mondo batterico, 9 phyla sui 30 esistenti nel dominio Bacteria, è il risultato di una forte pressione selettiva che nel corso dell’evoluzione ha agito sia sui colonizzatori microbici, selezionando organismi adattati eccezionalmente bene a questo ambiente e in grado di dominare il processo di colonizzazione, che sulla nicchia intestinale. E tuttavia, ciascun individuo possiede nel proprio intestino una comunità batterica unica.
Nonostante la grande variabilità esistente sia riguardo ai taxa presenti che tra gli individui, è stato proposto, ma non da tutti accettato, che nella maggior parte dei soggetti adulti il microbiota intestinale, nella sua componente batterica, possa essere classificato in varianti o “enterotipi” sulla base del rapporto tra l’abbondanza di Bacteroides e Prevotella. Questo sembra indicare che esista un numero limitato di stati simbiotici ben bilanciati, che potrebbe rispondere in maniera differente a fattori quali la dieta, l’età, la genetica e l’assunzione di farmaci.
L’intestino degli adulti ospita un’ampia e varia comunità di virus a DNA e RNA, formata da circa 2000 genotipi differenti, dove però non se ne individua uno dominante, considerando che il virus più abbondante rappresenta solo circa il 6% dell’intera comunità, al contrario di ciò che accade nei neonati dove il genotipo più abbondante rappresenta oltre il 40% dell’intera comunità. La maggior parte dei virus a DNA sono batteriofagi o fagi, ossia virus che infettano i batteri, che sono anche l’entità biologica più abbondante presente sulla terra, con un una popolazione stimata di circa 1031 unità, mentre la maggior parte di quelli a RNA sono virus vegetali.
Influenze sulla composizione e sviluppo del microbiota intestinale
La comunità batterica intestinale è regolata da diversi fattori, molti dei quali sono di seguito elencati.
La dieta
La dieta dell’ospite sembra essere il fattore più importante, a partire dal primo alimento assunto, il latte materno.
Sebbene considerato sterile, il latte materno contiene un ricco microbiota formato da oltre 700 specie, dominato da stafilococchi, streptococchi, bifidobatteri e batteri lattici. Dunque, nei i bambini allattati al seno rappresenta una fonte importante per la colonizzazione dell’intestino, ed è stato suggerito che questa modalità di colonizzazione giochi un ruolo cruciale per la salute, in quanto, tra le altre funzioni potrebbe proteggere il neonato dalle infezioni e contribuire alla maturazione del sistema immunitario. Il latte materno influenza il microbiota intestinale anche indirettamente, grazie alla presenza di oligosaccaridi con attività prebiotica che stimolano la crescita di gruppi batterici specifici quali stafilococchi e bifidobatteri.
Anche uno studio che ha confrontato il microbiota intestinale di bambini europei e africani, rispettivamente da Firenze e un villaggio rurale del Burkina Faso, di età compresa tra 1 e 6 anni, ha messo in evidenza il ruolo decisivo della dieta rispetto ad altre variabili quali il clima, la geografia, l’igiene e i servizi sanitari; è stata inoltre osservata un’assenza di differenze significative nell’espressione di geni chiave nel regolare la funzione immune, che suggerisce quindi una similarità funzionale tra i due gruppi. Infatti i bambini di entrambe i gruppi, fintanto che sono allattati al seno, presentano un microbiota intestinale con caratteristiche molto simili, ricco in Actinobacteria, principalmente Bifidobacterium.
La successiva introduzione di una dieta solida differente nei due gruppi, di tipo Occidentale negli europei e dunque ricca in grassi e proteine animali, povera in proteine animali ma ricca in carboidrati complessi nei bambini africani, porta a una differenziazione del rapporto Firmicutes/Bacteroidetes nei due gruppi. Nei bambini europei erano più abbondanti i Gram-positivi, principalmente Firmicutes, rispetto ai Gram-negativi, mentre nei bambini africani prevalevano i Gram-negativi, principalmente Bacteroidetes, rispetto ai Gram-positivi.
E la dieta a lungo termine è associata in modo molto stretto alla ripartizione in enterotipi. E’ stato infatti osservato che:
una dieta ricca in grassi e proteine animali, dunque di tipo occidentale, porta a un microbiota intestinale dominato da taxa dell’enterotipo Bacteroides;
una dieta ricca in carboidrati, tipica delle società agricole, vede la prevalenza dell’enterotipo Prevotella.
Analoghi risultati sono emersi dallo studio sopracitato sui bambini. Negli europei, il microbiota intestinale era dominato da taxa tipici dell’enterotipo Bacteroides, mentre in quelli del Burkina Faso, dominano taxa dell’enterotipo Prevotella.
Con cambiamenti a breve termine della dieta, 10 giorni, quali il passaggio da una povera in grassi e ricca in fibre a una ricca in grassi e povera in fibre e viceversa, sono stati osservati cambiamenti nella composizione del microbioma, già dopo 24 ore, ma nessuno scambio stabile nella suddivisione in enterotipi. E questo rimarca come per un cambiamento dell’enterotipo del microbiota intestinale sia necessaria una dieta a lungo termine.
Modifiche a carico della dieta si traducono anche in cambiamenti a carico del viroma intestinale, che si sposta verso un nuovo stato, ossia si osservano alterazioni delle proporzioni delle popolazioni preesistenti, verso il quale convergono individui che seguano la stessa dieta.
Il pH, sali biliari ed enzimi digestivi
Lo stomaco, a causa del pH estremamente acido del suo contenuto, è un ambiente ostile per i batteri, che non sono presenti in numero elevato, circa 102-103 cellule batteriche/grammo di tessuto. Oltre a Helicobacter pylori, capace di causare gastriti e ulcere gastriche, sono presenti anche batteri del genere Lactobacillus.
Dal duodeno si osserva un incremento nel numero di unità, 104-105 cellule batteriche/grammo di tessuto; e quantità simili si ritrovano nel digiuno e nelle prime parti dell’ileo. Il numero contenuto di microorganismi presente nell’intestino tenue è dovuto all’ambiente inospitale conseguente al fatto che nel tratto discendente del duodeno è presente l’apertura dell’ampolla di Vater dalla quale viene riversata la bile e il succo pancreatico ossia sali biliari ed enzimi pancreatici, entrambe in grado di causare danni ai microrganismi presenti.
Nella porzione terminale dell’ileo, dove l’attività dei sali biliari e degli enzimi pancreatici è meno intensa, la conta batterica è di circa 107 cellule batteriche/grammo di tessuto, fino ad arrivare nel colon a valori pari a 1012-1014 cellule batteriche/grammo di tessuto, tanto che le feci sono costituite per il 40% da batteri.
La distribuzione di batteri lungo l’intestino è strategica. Nel duodeno e nel digiuno la quantità di nutrienti ancora disponibile è molto più alta rispetto a quella presente nell’ileo terminale, dove sono rimasti acqua, fibre, ed elettroliti. Dunque non è un problema trovare nell’ileo terminale, e ancor più nel colon, un numero elevato di batteri. Il problema sarebbe trovarli in numero eccessivo nel duodeno, digiuno e prima parte dell’ileo; ed esiste una condizione patologia, definita sindrome da sovracrescita batterica nel tenue o SIBO, acronimo dell’inglese small intestinal bacterial overgrowth, nella quale il numero di batteri nel tenue aumenta di circa 10-15 volte, il che li mette in condizione di poter competere con l’ospite per i nutrienti e dare origine a disturbi gastrointestinali quali ad esempio diarrea.
La posizione geografica e le conseguenti differenze riguardo lo stile di vita, di alimentazione, di religione ecc.
E’ stato ad esempio osservata una sorta di gradiente geografico nel microbiota dei neonati europei, con un numero più elevato di specie di Bifidobacterium e alcune di Clostridium nei bambini delle zone settentrionali, mentre in quelli delle zone meridionali è stata trovata una maggiore abbondanza di Bacteroides, Lactobacillus ed Eubacterium.
La modalità di nascita.
L’assetto genetico dell’ospite.
Lo stato di salute, anche della madre nel corso della gravidanza.
Ad esempio, in pazienti con malattia infiammatoria intestinale o IBD, acronimo dell’inglese inflammatory bowel disease, risulta depleta Faecalibacterium prausnitzii, una tra le specie produttrici di acido butirrico, un’importante fonte di energia per le cellule intestinali, e dotata anche di un’azione antiinfiammatoria in vitro e nei topi, mentre si osserva un aumento nel numero di Escherichia coli aderenti.
L’assunzione di antibiotici.
Le infezioni batteriche e i predatori.
Le batteriocine, ossia proteine dotate di attività antibatterica, e i batteriofagi.
Questi ultimi sono una forza importante nel controllo dell’abbondanza e composizione del microbiota intestinale. In particolare potrebbero avere un ruolo di primo piano nel corso della colonizzazione dell’intestino del neonato, infettando via via gli ospiti dominanti e creando così l’opportunità per un altro ceppo di divenire abbondante. Questo modello di dinamiche preda-predatore, definito “kill the winner”, suggerisce che le fioriture di determinate specie batteriche porterebbero a fioriture dei loro corrispondenti fagi, seguite da riduzioni dell’abbondanza di entrambe. Di conseguenza il genotipo fagico più abbondante non sarà lo stesso in momenti differenti. E sebbene alcune sequenze virali presenti nell’intestino del neonato siano stabili nel corso dei primi tre mesi di vita, sono state osservate drammatiche variazioni nella composizione complessiva della comunità virale fecale tra la prima e la seconda settimana. Infine, anche la comunità batterica nel corso di questo periodo è estremamente dinamica.
La competizione per lo spazio e i nutrienti.
Composizione nel corso della vita
Lo sviluppo dell’ecosistema microbico intestinale è un evento complesso e cruciale nella vita dell’uomo, altamente variabile da individuo a individuo, e influenzato dai fattori visti in precedenza.
Sviluppo e Modificazioni della Flora Batterica Intestinale
Nell’utero materno i bambini sono considerati sterili, e dunque soggetti al momento del parto alla colonizzazione da parte dei microbi, grazie anche al fatto di nascere con una tolleranza immunitaria “insegnata” dalla madre.
Tuttavia alcuni lavori stanno evidenziando la presenza di batteri nel tessuto placentare, nel sangue del cordone ombelicale, nelle membrane fetali e nel liquido amniotico di neonati sani senza indicazioni di infezioni o infiammazioni. E ad esempio, il meconio di neonati prematuri nati da madri sane contiene un microbiota specifico, con i Firmicutes come phylum principale e predominanza di stafilococchi, mentre nelle prime feci i più abbondanti sono i Proteobacteria, in particolare specie quali Escherichia coli, Klebsiella pneumoniae, Serratia marcescens, ma anche gli enterococchi.
Nota: il meconio è privo di particelle virali rilevabili.
Sembra che sia i batteri vaginali che quelli intestinali possano avere accesso al feto, anche se attraverso vie differenti: per via ascendente i primi, tramite le cellule dendritiche del sistema immunitario i secondi. Dunque potrebbe esistere anche un microbiota fetale.
Al momento del parto si verifica la colonizzazione ad opera di un piccolo inoculo di origine materna, formato in genere da aerobi obbligati e facoltativi (inizialmente l’intestino contiene ossigeno), poi sostituiti da anaerobi obbligati, i batteri tipici presenti nell’età adulta, cui i primi colonizzatori hanno preparato l’ambiente idoneo.
Inoltre è presente un basso numero di taxa differenti, con dominio relativo dei phyla Proteobacteria e Actinobacteria, che rimane tale anche durante il primo mese di vita, ma non nei successivi dove si verifica un grande aumento della variabilità, parallelo a quello di nuove varianti genetiche. Si ritiene che l’esposizione microbica iniziale sia importante nel definire le “traiettorie” che porteranno agli ecosistemi adulti. Infine, le comunità iniziali possono fungere da fonte diretta di batteri protettivi o patogeni molto presto nel corso della vita.
Nei bambini nati con parto naturale le più importanti fonti di inoculo sono il microbiota vaginale e fecale della madre. Infatti i neonati ospitano comunità microbiche dominate dai specie dei generi Lactobacillus, i più abbondanti nel microbiota vaginale e anche nell’intestino neonatale nei primi giorni, Bifidobacterium, Prevotella, o Sneathia. E sembra probabile che gli anaerobi, quali i membri dei phyla Bacteroidetes e Firmicutes, non potendo vivere all’esterno dei loro ospiti, facciano affidamento allo stretto contatto tra genitore e figli per la trasmissione. Infine la trasmissione degli anerobi stretti, data la presenza nell’intestino del neonato di ossigeno, potrebbe avvenire in seguito per mezzo di spore e non direttamente al momento del parto.
In caso di parto cesareo, i primi batteri incontrati sono quelli della pelle e dell’ambiente ospedaliero, e i neonati ospitano un microbiota dominato da specie appartenenti ai generi Corynebacterium, Staphylococcus e Propionibacterium, con una conta batterica intestinale più bassa e una minore diversità nelle prime settimane di vita rispetto ai bimbi nati con parto naturale.
Ulteriore prova che avvalora l’ipotesi di una trasmissione verticale è il fatto che esista una somiglianza tra il microbiota intestinale presente nel meconio, e campioni prelevati dai possibili siti di contaminazione. Queste “firme” materne non persistono indefinitamente, ma vengono sostituite da altre popolazioni microbiche entro il primo anno di vita.
Fonti successive di inoculo sono la bocca e la pelle dei parenti, oggetti, animali, ma sembra essere il latte materno ad avere il ruolo primario nel determinare la successione microbica a livello intestinale.
La variazione e la diversità presente invece tra i bambini rifletterebbe l’individualità di queste esposizioni accidentali.
Nota: il tipo di parto sembra influenzare anche le funzioni immunologiche nel corso del primo anno di vita, forse attraverso l’influenza esercitata sullo sviluppo del microbiota intestinale. I bambini nati con il cesareo avrebbero:
una più bassa conta di cellule batteriche nei campioni di feci a un mese dalla nascita, dovuta soprattutto alla più elevata presenza di Bifidobacterium nei bambini nati con parto naturale;
un più alto numero di cellule secretrici anticorpi, IgA, IgG e IgM, il che potrebbe riflettere un’eccessiva esposizione agli antigeni che riuscirebbero ad attraversare la più vulnerabile barriera intestinale.
Entro giorni dalla nascita si viene a creare una fiorente comunità la cui popolazione è molto più variabile in composizione e meno stabile nel tempo rispetto a quella dell’adulto, e che ben presto supererà in numero quella delle cellule del neonato stesso, evolvendo secondo un pattern temporale notevolmente variabile da individuo a individuo.
I virus, assenti alla nascita, dalla fine della prima settimana di vita raggiungono un numero di circa le 108 unità/grammo di peso fresco di feci, e quindi rappresentano una componente abbondante e dinamica del microbiota intestinale in via di sviluppo, ma con una diversità estremamente bassa, al pari di quella batterica. La comunità virale è dominata dai fagi, che probabilmente influenzano anche la diversità e abbondanza dei batteri concomitanti come visto in precedenza. La sorgente iniziale di virus non è nota; tra le possibili fonti ci sono ovviamente quelle materne e ambientali. D’altro canto i primi virus potrebbero essere il risultato dell’induzione dei profagi della “neonata” flora batterica, ipotesi avvalorata dall’osservazione che oltre il 25% delle sequenze fagiche sembrano essere molto simili a quelle dei fagi che infettano batterici come Lactobacillus, Lactococcus, Enterococcus, e Streptococcus, abbondanti nel latte materno.
Dal termine del primo mese di vita si ritiene che la fase iniziale di rapida acquisizione di microbi seguente alla nascita sia terminata.
Dal punto di vista tassonomico nei bambini di un mese i microrganismi più abbondanti sembrano essere quelli appartenenti ai generi Bacteroides ed Escherichia, mentre i Bifidobacterium compaiono e crescono fino a dominare, assieme a Ruminococcus, il tratto gastrointestinale del bambino allattato al seno tra il primo e l’undicesimo mese di vita.
I bifidobatteri, come Bifidobacterium longum subspecies infantis, sono:
strettamente correlati all’allattamento al seno;
tra i meglio caratterizzati fra i batteri commensali benefici;
probiotici, ossia microrganismi in grado di apportare effetti benefici sulla salute dell’ospite.
La loro abbondanza conferisce benefici anche attraverso un’esclusione competitiva con cui sono di ostacolo alla colonizzazione da parte di patogeni. E infatti Escherichia e Bacteroides possono divenire preponderanti se i Bifidobacterium non colonizzano in modo adeguato.
Al contrario, nei bambini allattati artificialmente batteri dei generi Escherichia, come E. coli, Clostridium, come C. difficile, Bacteroides, come B. fragilis e Lactobacillus sono presenti in modo significativamente maggiore rispetto a quanto osservato nei bambini allattati al seno.
Anche se prima dello svezzamento la dieta del neonato allattato al seno è piuttosto costante, il suo microbiota non lo è altrettanto. Si osservano infatti ampie fluttuazioni nell’abbondanza dei taxa batterici, con differenze tra soggetti che riguardano anche i pattern temporali di variazione. Le variazioni osservate potrebbero essere dovute a eventi casuali di colonizzazione, malattie, assunzione di antibiotici, cambiamenti nel comportamento dell’ospite o ad altri aspetti legati allo stile di vita, come anche a differenze nelle risposte immunitarie ai microbi colonizzatori. Ma come questi fattori contribuiscano a plasmare il microbiota intestinale del bambino non è ancora chiaro.
Sembra che anche il viroma si modifichi molto rapidamente dopo la nascita, in quanto la maggior parte delle sequenze virali presenti nella prima settimana di vita non si ritrova dopo la seconda, e il repertorio si espande rapidamente in diversità e numero nel corso dei primi tre mesi. E questo contrasta con la stabilità che si osserva nell’adulto dove il 95% delle sequenze si conserva nel tempo.
In condizioni normali, verso la fine del primo anno di vita, il neonato è stato esposto a una dieta complessa per un periodo significativo e dovrebbe aver sviluppato una comunità microbica con caratteristiche analoghe a quelle del microbiota intestinale adulto, quali:
una composizione più stabile, filogeneticamente più complessa, e progressivamente più simile tra soggetti diversi;
una preponderanze di Bacteroidetes e Firmicutes, una comune presenza di Verrucomicrobia e un’abbondanza molto bassa di Proteobacteria;
un aumento del carico batterico come dei livelli di acidi grassi a catena corta o SCFA, principalmente l’acido acetico, l’acido propionico e l’acido butirrico, nelle feci;
un aumento dei geni associati all’utilizzo dei carboidrati, alla biosintesi delle vitamine e alla degradazione degli xenobiotici.
Interessante notare che il significativo turn-over di taxa che si verifica dalla nascita al termine del primo anno è accompagnato da una notevole costanza nelle capacità funzionali complessive.
Verso la fine del primo anno di vita anche i colonizzatori virali sono oramai stati sostituiti da virus specifici del bambino.
Verso i due anni e mezzo di vita il microbiota intestinale raggiunge un suo equilibrio, somigliando in modo completo a quello dell’adulto.
La selezione dei batteri più altamente adattati è conseguenza di vari fattori.
Il passaggio a una dieta solida.
Una maggiore idoneità all’ambiente intestinale dei taxa che tipicamente dominano il microbiota colonico adulto rispetto agli opportunisti iniziali.
I cambiamenti progressivi nell’ambiente intestinale, dovuti ai cambiamenti legati allo sviluppo della mucosa stessa.
Gli effetti del microbiota medesimo.
Quindi i primi 2-3 anni di vita sono il periodo più critico in cui intervenire al fine di ottenere il miglior microbiota possibile così da ottimizzare la crescita e lo sviluppo del bambino.
Tutto questo, partendo da un inizio caotico, porta alla costituzione dell’ecosistema intestinale tipico del giovane adulto, che è relativamente stabile nel tempo, comprese le componenti virale, eucariotica e gli Archeobatteri, sino alla vecchiaia, dominato almeno nella popolazione occidentale da specie dei phyla Firmicutes, che rappresentano circa il 60% della comunità batterica intestinale, Bacteroidetes e Actinobacteria, principalmente con il genere Bifidobacterium, ognuno circa il 10% della comunità batterica, seguiti da Proteobacteria e Verrucomicrobia. I generi Bacteroides, Clostridium, Faecalibacterium, Ruminococcus ed Eubacterium, costituiscono, assieme a Methanobrevibacter smithii, la grande maggioranza della comunità batterica intestinale dell’adulto.
Risultati contrastanti con i dati esposti stanno emergendo dall’analisi di popolazioni ad esempio di villaggi rurali dell’Africa, come visto sopra.
E il microbiota intestinale è sufficientemente simile tra gli individui da permettere l’individuazione di un nucleo microbiomico condiviso.
La stabilità e resilienza è però soggetta a numerose variabili tra cui, come detto, la dieta sembra essere una delle più importanti, variabili che quindi dovranno essere mantenute costanti, o nel caso della malattie evitate, ad esempio attraverso le vaccinazioni, al fine di mantenere la stabilità del microbiota. Va comunque sottolineato che stabilità e resilienza potrebbero essere dannose nel caso in cui la comunità intestinale dominante sia patogena.
Negli anziani, come già visto nei neonati, il microbiota intestinale va incontro a sostanziali cambiamenti. In uno studio condotto in Irlanda su 161 persone sane di 65 o più anni, nella maggior parte dei soggetti il microbiota intestinale è distinto da quello degli adulti più giovani, con una composizione che sembra essere dominato dai phyla Bacteroidetes, i principali, seguiti dai Firmicutes, con percentuali quasi inverse rispetto a quelle trovate negli adulti più giovani, anche se sono state osservate notevoli variazioni tra i soggetti. E tra i generi più abbondanti si trovano Faecalibacterium, che rappresenta circa il 6% dei 15 generi principali, seguiti dalle specie dei generi Ruminococcus, Roseburia e Bifidobacterium, quest’ultimo intorno allo 0,4%.
Anche la variabilità nella composizione delle comunità è più grande rispetto ai giovani adulti, il che potrebbe essere dovuto alla maggior morbilità associata con l’età, e quindi il successivo utilizzo di medicinali, nonché a variazioni della dieta.
Bibliografia
Breitbart M., Haynes M., Kelley S., Angly F., Edwards R.A., Felts B., Mahaffy J.M., Mueller J., Nulton J., Rayhawk S., Rodriguez-Brito B., Salamon P., Rohwer F. Viral diversity and dynamics in an infant gut. Res Microbiol 2008;159:367-373. doi:10.1016/j.resmic.2008.04.006
Claesson M.J., Cusack S., O’Sullivan O., Greene-Diniz R., de Weerd H., Flannery E., Marchesi J.R., Falush D., Dinan T., Fitzgerald G., et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA 2011;108(Suppl 1);4586-4591. doi:10.1073/pnas.1000097107
Clemente J.C., Ursell L.K., Wegener Parfrey L., and Knight R. The impact of the gut microbiota on human health: an integrative view. Cell 2012;148:1258-1270. doi:10.1016/j.cell.2012.01.035
De Filippo c., Cavalieri D., Di Paola M., Ramazzotti M., Poullet J.B., Massart S., Collini S., Pieraccini G., and Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci 2010;107(33):14691-14696. doi:10.1073/pnas.1005963107
Dominguez-Bello M.G., Costello E.K., Contreras M., Magris M., Hidalgo G., Fierer N., and Knight R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci 2010;107:11971-11975. doi:10.1073/pnas.1002601107
Fernández L., Langa S., Martín V., Maldonado A., Jiménez E., Martín R., Rodríguez J.M. The human milk microbiota: origin and potential roles in health and disease. Pharmacol Res 2013;69(1):1-10. doi:10.1073/pnas.1002601107
Huurre A., Kalliomäki M., Rautava S., Rinne M., Salminen S., and Isolauri E. Mode of delivery-effects on gut microbiota and humoral immunity. Neonatology 2008;93:236-240. doi:10.1159/000111102
Koenig J.E., Spor A., Scalfone N., Fricker A.D., Stombaugh J., Knight R., Angenent L.T., and Ley R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci 2011;108(1):4578-4585. doi:10.1073/pnas.1000081107
Ley R.E., Peterson D.A., and Gordon J.I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006;124(4):837-848. doi:10.1016/j.cell.2006.02.017
Minot S., Sinha R., Chen J., Li H., Keilbaugh S.A., Wu G.D., Lewis J.D., and Bushman F.D. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res 2011;21:1616-1625. doi:10.1101/gr.122705.111
Moreno-Indias I.M., Cardona F., Tinahones F.J. and Queipo-Ortuño M.I. Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front Microbiol 2014;5(190):1-10. doi:10.3389/fmicb.2014.00190
Newburg D.S. & Morelli L. Human milk and infant intestinal mucosal glycans guide succession of the neonatal intestinal microbiota. Pediatr Res 2015;77:115-120. doi:10.1038/pr.2014.178
Palmer C., Bik E.M., DiGiulio D.B., Relman D.A., and Brown P.O. Development of the human infant intestinal microbiota. PLoS Biol 2007;5(7):e177. doi:10.1371/journal.pbio.0050177
Rodrıguez J.M., Murphy K., Stanton C., Ross R.P., I. Kober O.I., Juge N., Avershina E., Rudi K., Narbad A., Jenmalm M.C., Marchesi J.R. and Collado M.C. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb Ecol Health Dis 2015;26:26050. doi:10.3402/mehd.v26.26050
Wu G.D., Chen J., Hoffmann C., Bittinger K., Chen Y.Y., Keilbaugh S.A., Bewtra M., Knights D., Walters W.A., Knight R., et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011;334:105-108. doi:10.1126/science.1208344
E’ noto da quasi un secolo che gli esseri umani ospitano un ecosistema microbico, definito microbiota umano, straordinariamente denso e diversificato, formato da un numero di virus e cellule molto superiore a quello che compone il corpo umano, e che rappresenta dall’uno al tre per cento del peso corporeo.
I geni che i microrganismi componenti il microbiota umano codificano, che sono circa 1000 volte più numerosi rispetto a quelli del nostro genoma, formano il microbioma umano.
I microrganismi colonizzano tutte le superfici del corpo esposte all’ambiente. Distinte comunità microbiche sono infatti presenti sulla pelle, nella vagina, nelle vie aeree, e lungo tutto il tubo digerente, a partire dalla bocca, passando attraverso lo stomaco sino a raggiungere le parti terminali dell’intestino.
II microbiota umano è costituito da organismi provenienti da tutti i taxa. Vi si ritrovano infatti batteri, virus, archeobatteri ed eucarioti.
Batteri
I batteri sono presenti con almeno cento trilioni (1014) di cellule, un numero dieci volte maggiore rispetto a quelle che compongono il corpo umano.
Si ritrovano per la maggior parte a livello del tratto intestinale, dove, con concentrazioni sino a 1012-1014/grammo di tessuto, formano uno degli habitat più densamente popolati esistenti sulla terra. In questa sede sono particolarmente abbondanti membri dei phyla Firmicutes, ma anche di Bacteroidetes e Actinobacteria.
Nota: le comunità microbiche di una data sede si “somigliano” tra di loro molto di più di quanto le stesse non somiglino a quelle presenti in altri siti dello stesso soggetto; ad esempio, le comunità delle vie aeree superiori sono molto più simili tra individui differenti che non a quelle della pelle o dell’intestino di uno stesso soggetto.
Virus
I virus sono in assoluto i componenti più numerosi essendo presenti con quadrilioni di unità. I genomi di tutti i virus ospitati costituiscono il viroma umano.
In passato i virus e gli eucarioti del microbiota umano sono stati studiati focalizzandoci sui microrganismi patogeni, ma negli ultimi anni l’attenzione si è spostata anche sui numerosissimi membri non patogeni di questi gruppi. E riguardo ai virus, molte delle sequenze geniche trovate sono nuove, il che suggerisce che ci sia ancora molto da conoscere sul viroma umano.
Infine, al pari dei batteri, anche per i virus esiste una notevole variabilità interpersonale.
Archeobatteri
Gli Archeobatteri o archei, nome scientifico Archaea, sono rappresentati in particolare dai microrganismi appartenenti all’ordine dei Methanobacteriales. Tra questi Methanobrevibacter smithii predomina nell’intestino umano, rappresentando sino al 10% di tutti gli anaerobi.
Eucarioti
Anche gli eucarioti sono presenti, e tra i primi ad essere identificati probabilmente ci sono i parassiti del genere Giardia e Entamoeba. Ma vi è anche una grande abbondanza e diversità di funghi, appartenenti ad esempio a generi quali Candida, Penicillium, Aspergillus, Hemispora, Fusarium, Geotrichum, Cryptococcus, Hormodendrum, Saccharomyces e Blastocystis.
Candida albicans
Funzione del microbiota umano
Talvolta definito “l’organo dimenticato”, il microbiota umano, e in particolare la sua componente batterica intestinale, svolge numerose ed importanti funzioni che possono portare a benefici nutrizionali, immunologici, e di sviluppo, ma può anche essere causa di malattie per l’ospite.
Di seguito alcuni esempi.
E’ coinvolto nello sviluppo del sistema gastrointestinale, come dimostrato da esperimenti condotti su animali germ-free nei quali, ad esempio, lo spessore della mucosa intestinale è più sottile rispetto a quella di animali colonizzati, dunque più facilmente soggetto ad insulti che ne causino la rottura.
Concorre all’estrazione dell’energia dai nutrienti, grazie alla sua capacità di fermentare carboidrati per noi indigeribili; inoltre promuove l’assorbimento dei monosaccaridi ed il deposito dell’energia ricavata. Tutto ciò con molta probabilità ha rappresentato una forza evolutiva molto forte che ha giocato a favore del fatto che questi batteri siano diventati nostri simbionti.
Concorre al mantenimento del pH acido della pelle e del contenuto del colon.
E’ coinvolto nel metabolismo degli xenobiotici e di molti polifenoli.
Migliora l’assorbimento di acqua e sali minerali (ferro, calcio e magnesio) nel colon.
Aumenta la velocità di transito intestinale, più lenta negli animali germ-free.
Ha un ruolo importante nella resistenza alla colonizzazione da parte di microrganismi patogeni, in particolare nella vagina e nell’intestino.
E’ coinvolto nella biosintesi di isoprenoidi e vitamine attraverso la via del metileritritolo fosfato.
Stimola l’angiogenesi.
Interagisce con il sistema immunitario, fornendo segnali per promuovere la maturazione delle cellule immunitarie ed il normale sviluppo delle funzioni immuni. E questo è forse l’effetto più importante derivante dalla simbiosi tra uomo e microrganismi. Esperimenti condotti su animali germ-free hanno infatti evidenziato quanto segue.
I macrofagi, le cellule che hanno il compito di fagocitare i patogeni e poi presentarne gli antigeni al sistema immunitario, sono presenti in scarsissima quantità rispetto all’intestino colonizzato, e se messi in presenza di batteri non riescono a trovarli e quindi a fagocitarli, a differenza dei macrofagi estratti da un intestino colonizzato.
Manca la flogosi cronica aspecifica, una condizione normale del nostro intestino dovuta alla presenza di un tessuto immunitario molto sviluppato e allenato, proprio grazie alla presenza dei batteri nel lume intestinale (e anche di ciò che mangiamo).
Cambiamenti nella sua composizione possono contribuire allo sviluppo di obesità e sindrome metabolica.
Protegge dallo sviluppo del diabete di tipo I.
Molte malattie, sia del bambino che dell’adulto, tra cui il tumore dello stomaco e del colon, i linfomi del tessuto linfoide associato alla mucosa, l’enterocolite necrotizzante, che è un’importante causa di morbilità e mortalità nei prematuri, o le malattie croniche intestinali, sono e altre appaiono essere collegate al microbiota intestinale.
Sembra quindi molto probabile che l’organismo umano rappresenti un superorganismo frutto di tanti anni di evoluzione, composto oltre che dalle proprie cellule e capacità metaboliche e fisiologiche che ne derivano, anche da un altro organo aggiunto, il microbiota.
Commensali e patogeni
Sulla base dei rapporti che stabiliscono con l’ospite, i differenti microrganismi componenti il microbiota umano possono essere quindi suddivisi in due categorie: commensali e patogeni.
I commensali non causano danno all’ospite, con cui anzi instaurano una simbiosi di tipo mutualistico che in genere porta benefici ad entrambe.
I patogeni sono al contrario in grado di causare malattie, ma fortunatamente rappresentano una minima percentuale della nostra flora microbica. Si tratta di microrganismi che stabiliscono una simbiosi con l’ospite traendone beneficio a suo svantaggio.
In genere causano malattia se si verifica:
un “cambio di sede”, ossia se si spostano dalla loro normale nicchia, ad esempio l’intestino, ad un’altra impropria, quale la vagina o la vescica, come nel caso del fungo Candida albicans presente normalmente, ma in piccolissima quantità, nell’intestino;
un indebolimento delle difese immunitarie dell’ospite, come dopo un trapianto o comunque una terapia immunosoppressiva.
Human Microbiome Project
La componente batterica del microbiota umano è l’oggetto di vari studi tra i quali un progetto molto ampio partito nel 2008 chiamato “Human Microbiome Project”, che ne analizza il microbioma associato a vari habitat del corpo, quali pelle, bocca, naso, vagina ed intestino, in una popolazione sana di 242 adulti.
Questi studi hanno evidenziato l’esistenza di una grande variabilità nella composizione del microbiota umano, con i gemelli che condividono meno del 50% dei loro taxa batterici a livello di specie, ed una percentuale anche minore riguardo i virus.
I fattori che modellano la composizione delle comunità microbiche iniziano ad essere compresi. Ad esempio le caratteristiche genetiche dell’ospite hanno un ruolo importante nel creare e plasmare le comunità batteriche presenti, anche se questo non è vero per quelle virali. E studi di metagenomica hanno evidenziato che, nonostante la grande variabilità interpersonale nella composizione delle comunità microbiche, esiste un ampio nucleo condiviso di geni codificante vie del segnale e metaboliche. Sembra cioè che l’assemblaggio e la struttura delle comunità microbiche non avvenga in base alle specie quanto al gruppo più funzionale di geni.
E da questo ne deriverebbe che stati di malattia di queste comunità possano essere meglio identificati da distribuzione atipiche di classi di geni funzionali.
Effetti degli antibiotici
l microbiota di un individuo adulto sano è generalmente stabile nel tempo. Tuttavia la sua composizione può essere alterata ad opera di fattori esterni come l’urbanizzazione, i viaggi , le modifiche della dieta, ma soprattutto l’uso di antibiotici a largo spettro.
Gli antibiotici hanno un profondo effetto.
Vi è una riduzione a lungo termine nella diversità batterica.
I taxa colpiti variano da individuo ad individuo, e posso essere interessati fino ad un terzo di quelli presenti.
Alcuni taxa non recuperano anche dopo 6 mesi dal trattamento.
Una volta che la comunità batterica si è rimodellata dopo il trattamento con il farmaco, si osserva una ridotta resistenza alla colonizzazione. Ciò permette a microbi estranei e/o patogeni in grado di crescere più dei commensali di causare cambiamenti permanenti nella struttura del microbiota umano, come anche malattie sia acute, ad esempio la pericolosa colite pseudomembranosa, che croniche, come si sospetta per l’asma a seguito dell’uso ed abuso di antibiotici nell’infanzia.
Inoltre il loro ripetuto uso sembra aumentare la riserva di geni per la resistenza agli antibiotici nel nostro microbioma. A supporto di questa ipotesi in alcune nazioni europee è stata osservata una riduzione nel numero dei patogeni antibiotico-resistenti a seguito di un calo nel numero degli antibiotici prescritti.
Infine non va sottovalutato il fatto che la microflora batterica intestinale è implicata in molte trasformazioni chimiche, per cui una sua alterazione potrebbe avere implicazioni nello sviluppo del cancro come dell’obesità.
Tuttavia, riguardo all’uso degli antibiotici va sottolineato che se abbiamo un’aspettativa di vita molto superiore al passato è anche perché non moriamo più di malattie infettive!
Bibliografia
Burke C., Steinberg P., Rusch D., Kjelleberg S., and Thomas T. Bacterial community assembly based on functional genes rather than species. Proc Natl Acad Sci USA 2011;108:14288-14293. doi:10.1073/pnas.1101591108
Clemente J.C., Ursell L.K., Wegener Parfrey L., and Knight R. The impact of the gut microbiota on human health: an integrative view. Cell 2012;148:1258-1270. doi:10.1016/j.cell.2012.01.035
Gill S.R., Pop M., Deboy R.T., Eckburg P.B., Turnbaugh P.J., Samuel B.S., Gordon J.I., Relman D.A., Fraser-Liggett C.M., and Nelson K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006;312:1355-1359. doi:10.1126/science.1124234
Palmer C., Bik E.M., DiGiulio D.B., Relman D.A., and Brown P.O. Development of the human infant intestinal microbiota. PLoS Biol 2007;5(7):e177. doi:10.1371/journal.pbio.0050177
Turnbaugh P.J., Gordon J.I. The core gut microbiome, energy balance and obesity. J Physiol 2009;587:4153-4158. doi:10.1113/jphysiol.2009.174136
Zhang, T., Breitbart, M., Lee, W., Run, J.-Q., Wei, C., Soh, S., Hibberd, M., Liu, E., Rohwer, F., Ruan, Y. Prevalence of plant viruses in the RNA viral community of human feces. PLoS Biol 2006;4(1):e3. doi:10.1371/journal.pbio.0040003
La biosintesi dei flavonoidi, probabilmente la via metabolica meglio caratterizzata tra quelle del metabolismo secondario delle piante, fa parte della via biosintetica dei fenilpropanoidi, che porta alla formazione, oltre che dei flavonoidi, anche di un’ampia gamma di composti fenolici quali gli acidi idrossicinnamici, gli stilbeni, i lignani e le lignine.
La biosintesi dei flavonoidi è legata al metabolismo primario da intermedi di derivazione sia mitocondriale che plastidica. Queste molecole dovranno essere trasportate nel citoplasma per essere utilizzate, in quanto sembra che la maggior parte degli enzimi coinvolti sino ad ora caratterizzati lavorino uniti in complessi localizzati nel citosol della cellula.
I prodotti finali raggiungono i vari distretti intra- od extracellulari, con i flavonoidi coinvolti nella pigmentazione in genere trasportati all’interno dei vacuoli.
La biosintesi di questo ampio gruppo di polifenoli richiede come substrati iniziali una molecola di p-cumaril-CoA e tre di malonil-CoA.
Il p-cumaril-CoA rappresenta il più importante punto di ramificazione della via dei fenilpropanoidi, essendo il precursore di un’ampia varietà di prodotti fenolici, di natura sia flavonoide che non flavonoide.
E’ prodotto a partire dalla fenilalanina attraverso un nucleo di tre reazioni catalizzate da enzimi ad azione citosolica detti enzimi del gruppo I od ad azione precoce, in ordine di azione:
fenilalanina ammoniaca liasi (EC 4.3.1.24);
trans-cinnamato 4-monoossigenasi (EC:1.14.14.91);
4-cumarato:CoA ligasi (EC 6.2.1.12).
Biosintesi del p-Cumaril-CoA
Sembra che questi enzimi si associno a formare un complesso multienzimatico ancorato al reticolo endoplasmatico, ancoraggio probabilmente assicurato dalla cinnamato 4-idrossilasi che inserisce il proprio dominio N-terminale nella membrana del reticolo stesso. Queste strutture, definite “metaboloni”, fanno si che il prodotto di una reazione sia incanalato direttamente verso il sito attivo dell’enzima che catalizza la reazione successiva.
Con l’esclusione della cinnamato 4-idrossilasi, in tutte le specie analizzate gli enzimi che intervengono a valle della fenilalanina ammoniaca liasi sono codificati da piccole famiglie di geni.
Le diverse forme isoenzimatiche mostrano pattern di espressione temporali, tissutali e indotti da elicitori, distinti; sembra infatti che ogni membro di ciascuna famiglia possa essere utilizzato soprattutto per la sintesi di uno specifico composto, agendo in questo modo come punto di controllo del flusso del carbonio verso le vie di biosintesi dei flavonoidi, lignani e lignine.
Nota: la fenilalanina, che da il nome alla via dei fenilpropanoidi, è un prodotto della via dell’acido shikimico, via che converte semplici precursori derivanti dal metabolismo dei carboidrati, fosfoenolpiruvato e eritrosio-4-fosfato, intermedi rispettivamente dalla glicolisi e della via del pentoso fosfato, negli aminoacidi aromatici fenilalanina, tirosina e triptofano. Poiché gli animali non posseggono questa via metabolica, presente invece nelle piante e nei microorganismi, non sono in grado di sintetizzare i tre aminoacidi suddetti, che dunque risultano essenziali.
Fenilalanina ammonica liasi
E’ uno degli enzimi meglio studiati e caratterizzati del metabolismo secondario delle piante. Non richiede cofattori e catalizza la reazione che lega il metabolismo primario e quello secondario: la deaminazione della fenilalanina in acido trans-cinnamico, con liberazione dell’azoto in forma di ammoniaca e inserzione di un doppio legame trans tra il C7 e il C8 della catena laterale.
Fenilalanina ⇄ Acido trans-Cinnamico + NH3
Quindi dirige il flusso di carbonio dalla via dello shikimato a quella delle varie branche del metabolismo fenilpropanoico. L’ammoniaca rilasciata viene probabilmente fissata nella reazione catalizzata dalla glutammina sintetasi.
L’enzima presente nelle monocotiledoni è anche in grado di agire come tirosina ammoniaca liasi (EC 4.3.1.25), convertendo direttamente la tirosina in acido p-cumarico (saltando quindi la idrossilazione in posizione 4), anche se con efficienza minore rispetto all’attività sulla fenilalanina.
In tutte le specie sono state trovate numerose copie dei geni per la fenilalanina ammoniaca liasi, copie che probabilmente rispondono in maniera differente a stimoli interni ed esterni. La trascrizione del gene e dunque l’attività dell’enzima è infatti sottoposta a regolazione da parte di fattori interni legati allo sviluppo e fattori esterni.
Di seguito alcuni esempi che richiedono un aumento dell’attività dell’enzima.
La fioritura.
La produzione di lignina per il rafforzamento della parete cellulare secondaria delle cellule dello xilema.
La produzione di pigmenti per attrarre gli impollinatori.
L’attacco di patogeni che richieda la produzione di fitoalessine di natura fenilpropanoica, o l’esposizione a raggi UV.
trans-Cinnamato 4-monoossigenasi
Enzima della famiglia del citocromo P450 (EC 1.14.-.-), è una monossigenasi a localizzazione microsomiale, contenente un gruppo eme come cofattore, e dipendente dal NADPH e dall’ossigeno molecolare.
Catalizza l’introduzione del gruppo ossidrilico in posizione 4 dell’acido trans-cinnamico (gruppo ossidrilico che si osserva nella maggior parte dei flavonoidi conosciuti) e formazione di acido p-cumarico.
Questa reazione fa parte anche della via di sintesi degli acidi idrossicinnamici.
Aumenti nei livelli di trascrizione del gene e di attività dell’enzima sono stati osservati in correlazione con la sintesi di fitoalessine in risposta a infezioni fungine, la lignificazione o lesioni alla pianta.
4-Cumarato:CoA ligasi
In presenza di Mg2+ che agisce da cofattore, permette l’attivazione ATP-dipendente del gruppo carbossilico dell’acido p-cumarico e degli altri acidi idrossicinnamici, di per se piuttosto inerti, attraverso la formazione dei corrispondenti CoA-tioestere.
Acido p-Cumarico + ATP + CoA ⇄ p-Cumaril-CoA + AMP + PPi
In genere l’acido p-cumarico e l’acido caffeico sono i substrati preferiti, seguiti dall’acido ferulico e dall’acido 5-idrossiferulico, mentre si osserva una bassa attività nei confronti dell’acido trans-cinnamico e nessuna nei confronti dell’acido sinapico. I CoA-tioesteri prodotti sono in grado di entrare in differenti vie metaboliche come:
la riduzione ad aldeidi e alcol (monolignoli);
l’addizione di unità di acetato fornite dal malonil-CoA, nel caso della biosintesi dei flavonoidi e degli stilbeni;
il trasferimento a molecole accettrici.
Va infine sottolineato che l’attivazione del gruppo carbossilico può essere ottenuta anche attraverso il trasferimento al glucosio dipendente non dall’ATP ma dall’UDP-glucosio.
Biosintesi del malonil-CoA
Il malonil-CoA non deriva dalla via dei fenilpropanoidi, ma si forma nella reazione catalizzata dall’acetil-CoA carbossilasi (EC 6.4.1.2, la forma citosolica).
L’enzima, che ha come cofattori la biotina e il Mg2+, catalizza la carbossilazione ATP-dipendente dell’acetil-CoA, utilizzando lo ione bicarbonato come donatore di anidride carbonica (CO2).
Acetil-CoA + HCO3– + ATP → Malonil-CoA + ADP + Pi
Si trova sia nei plastidi, dove interviene nella sintesi degli acidi grassi, che nel citoplasma, ed è quest’ultimo che catalizza la formazione del malonil-CoA che sarà utilizzato nella biosintesi dei flavonoidi e di altri composti.
Aumenti dei livelli di trascrizione del gene e dell’attività dell’enzima sono indotti in risposta a stimoli che aumentano la biosintesi di questi polifenoli, come l’esposizione a funghi patogeni o raggi UV.
A sua volta l’acetil-CoA può essere prodotto nei mitocondri, plastidi, perossisomi e nel citosol attraverso differenti vie metaboliche. Quello che è utilizzato nella biosintesi del malonil-CoA e quindi dei flavonoidi è di derivazione citosolica, prodotto nella reazione catalizzata dalla ATP-citrato liasi (EC 2.3.3.8) che converte citrato, ATP e CoA in acetil-CoA, ossalacetato, ADP e fosfato inorganico.
Passaggi iniziali della biosintesi dei flavonoidi
Il primo passo nella biosintesi dei flavonoidi è catalizzato dalla calcone sintasi (EC 2.3.1.74), un enzima ancorato al reticolo endoplasmatico e privo di cofattori noti.
In presenza di una molecola di p-cumaril-CoA e tre di malonil-CoA, catalizza una serie di decarbossilazioni e condensazioni sequenziali, nel corso delle quali si forma un intermedio polichetide che subisce ciclizzazioni e aromatizzazioni che portano alla formazione dell’anello A, e la risultante struttura dei calconi. Il prodotto delle reazioni suddette è la naringenina calcone (2’,4,4′,6′-tetraidrossicalcone), un 6’-idrossicalcone e il primo flavonoide prodotto.
p-Cumaril-CoA + 3 Malonil-CoA → Naringenina Calcone + 4 CoA + 3 CO2
La reazione, citosolica, è irreversibile grazie al rilascio di tre molecole di CO2 e 4 di CoA.
L’anello B e il ponte centrale a 3 atomi di carbonio della molecole derivano dal p-cumaril-CoA (e quindi dalla fenilalanina), mentre l’anello A dalle tre unità di malonil-CoA.
Origine dello Scheletro di Base dei Flavonoidi
Altro possibile prodotto della reazione è il 6’-deossicalcone, la cui sintesi sembra coinvolgere un passaggio addizionale di riduzione catalizzata da una polichetide reduttasi (EC. 1.1.1.-).
Le calcone sintasi di alcune specie, ad esempio l’orzo (Hordeum vulgare), possono accettare come substrati anche il caffeoil-CoA, il feruloil-CoA e il cinnamoil-CoA. E’ l’enzima più abbondante della via dei fenilpropanoidi, probabilmente perché ha una bassa attività catalitica, ed è infatti considerato l’enzima limitante della via di biosintesi dei flavonoidi.
Come per la fenilalanina ammoniaca liasi, anche la sua sintesi è soggetta a controlli multipli dell’espressione genica ad opera di fattori interni ed esterni. In alcune piante, sono state trovate una o due isoforme, mentre in altre fino a 9.
Appartiene al gruppo delle polichetide sintasi, presenti nei batteri, funghi e piante. Sono enzimi in grado di formare catene polichetidiche attraverso la condensazione sequenziale di unità di acetato provenienti da malonil-CoA. Questa classe di enzimi comprende anche la stilbene sintasi (EC 2.3.1.146), che catalizza la formazione di resveratrolo, un polifenolo non flavonoide difensivo delle piante che ha suscitato molto interesse per la salute umana.
Di solito le piante non accumulano calconi, e infatti dopo la sua formazione la naringenina calcone, nella reazione catalizzata dalla calcone isomerasi (EC 5.5.1.6) viene convertita in (2S)-naringenina, un flavanone.
L’enzima, il primo della via di biosintesi dei flavonoidi a essere scoperto, catalizza una isomerizzazione stereospecifica, chiudendo l’anello C. Sono state ritrovate due tipi di calcone isomerasi, indicate come tipo I e II. Il tipo I può utilizzare come substrati solo i 6’-idrossicalconi, come la naringenina calcone, mentre il tipo II, prevalente nelle leguminose, può catalizzare l’isomerizzazione sia dei 6’-idrossi- che dei 6’-deossicalconi.
Da notare che con i 6’-idrossicalconi l’isomerizzazione può avvenire anche non enzimaticamente a formare una miscela racemica, sia in vitro che in vivo; addirittura sembra con un grado tale da permettere una moderata sintesi di antociani. Al contrario in condizioni fisiologiche i 6’-deossicalconi sono stabili, per cui è richiesta la catalisi ad opera della isomerasi di tipo II per convertirli in flavanoni.
L’enzima assicura un aumento della velocità di reazione di 107 volte rispetto alla reazione spontanea, ma con una cinetica decisamente più lenta con i 6’-deossicalconi rispetto ai 6’-idrossicalconi. Infine permette l’ottenimento dei (2S)-flavanoni, che sono quelli biosinteticamente necessari.
Al pari di altri enzimi della biosintesi dei flavonoidi, anche la sua sintesi è soggetta a uno stretto controllo. E come la fenilalanina ammoniaca liasi e la calcone sintasi, è indotta dagli elicitori.
Nella reazione catalizzata dalla flavanone-3β-idrossilasi (EC 1.14.11.9) i (2S)-flavanoni vanno incontro a una isomerizzazione stereospecifica che li converte nei rispettivi (2R,3R)-diidroflavonoli. In particolare la naringenina è convertita in diidrocampferolo.
L’enzima, citosolico, è una non-eme diossigenasi dipendente dal Fe2+ e dal 2-ossiglutarato, e dunque appartenente alla famiglia delle diossigenasi 2-ossiglutarato-dipendenti (il che le distingue delle altre idrossilasi della via di biosintesi dei flavonoidi che sono enzimi citocromo P450).
La naringenina calcone, la (2S)-naringenina e il suo derivato diidrocampferolo (diidroflavonolo) sono intermedi centrali nella biosintesi dei flavonoidi, essendo punti di ramificazione da cui si diparte la sintesi di distinte sottoclassi di flavonoidi. Ad esempio, direttamente o indirettamente:
dai diidroflavanoli i flavan-3,4-dioli, le antocianidine, gli antociani, le catechine, le proantocianidine o tannini condensati, e i flavonoli.
Non tutte queste vie di sintesi sono presenti in tutte le piante, o sono attive all’interno di ogni tessuto di una data pianta.
Al pari degli enzimi visti in precedenza, anche l’attività di quelli coinvolti in queste vie metaboliche “collaterali” è soggetta a uno stretto controllo, che risulta in un profilo di metaboliti di natura flavonoide tessuto specifico. L’esempio è il chicco d’uva, dove la buccia, la polpa e i semi hanno un profilo ben distinto riguardo al contenuto in antociani, catechine, tannini condensati e flavonoli, la cui sintesi e accumulo è strettamente e temporalmente coordinata nel corso dello sviluppo.
Bibliografia
Andersen Ø.M., Markham K.R. Flavonoids: chemistry, biochemistry, and applications. CRC Press Taylor & Francis Group, 2006
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
Sono ampiamente distribuiti nel regno vegetale, essendo presenti in oltre 55 famiglie di piante, dove svolgono funzioni difensive nei confronti di attacchi da parte di funghi e batteri patogeni, e agiscono anche come antiossidanti.
Nell’uomo, studi epidemiologici e fisiologici hanno dimostrato che sono in grado di esercitare effetti positivi nella prevenzione di patologie correlate allo stile di vita, quali il diabete di tipo II e il cancro. Ad esempio, un aumento del loro consumo nella dieta si correla con una riduzione dell’insorgenza di alcuni tipi di tumori estrogeno-dipendenti, come il tumore al seno in donne in postmenopausa.
Inoltre alcuni lignani hanno suscitato anche interesse farmacologico. Esempi o sono:
la podofillotossina, ottenuta da piante del genere Podophyllum (famiglia Berberidaceae), una tossina mitotica i cui derivati sono stati utilizzati come chemioterapici;
l’arctigenina e la tracheologina, ottenute da piante rampicanti tropicali, che posseggono proprietà antivirali e sono state testate nella ricerca di un farmaco per la cura dell’AIDS.
La loro struttura chimica di base si compone di due unità di fenilpropano legate attraverso un legame carbonio-carbonio che si stabilisce principalmente tra gli atomi centrali delle rispettive catene laterali (posizione 8 o β), legame anche detto β-β’. Meno frequentemente si osservano legami 3-3’, 8-O-4’, o 8-3’; in questi casi i dimeri sono definiti neolignani.
Dunque la loro struttura chimica può essere indicata come (C6-C3)2 e pertanto, al pari degli acidi idrossicinnamici da cui derivano, appartengono alla classe dei fenilpropanoidi.
Unità di Fenilpropano
Sulla base dello scheletro carbonioso, del pattern di ciclizzazione e del modo in cui l’ossigeno è incorporato nello scheletro della molecola, possono essere suddivisi in 8 sottogruppi: furani, furofurani, dibenzilbutani, dibenzilbutirrolattoni, dibenzocicloottadieni, dibenzilbutirrolattoli, ariltetraline e arilnafatleni. In aggiunta esiste una notevole variabilità riguardo i livelli di ossidazione delle catene laterali propiliche e di entrambe gli anelli aromatici.
In natura non sono presenti in forma libera ma legati ad altre molecole, in genere glicosilati.
Tra i più comuni si ritrovano il secoisolariciresinolo, il più abbondante, ma in buone quantità anche lariciresinolo, pinoresinolo, matairesinolo e 7-idrossimatairesinolo.
Nota: i lignani si possono presentare non solo in forma di dimeri ma anche di oligomeri più complessi, come i dilignani e i sesquilignani.
Biosintesi
Di seguito verrà presa in esame la biosintesi di alcuni tra i lignani più comuni.
La via metabolica ha inizio a partire da 3 dei 4 acidi idrossicinnamici alimentari più comuni: l’acido p-cumarico, l’acido sinapico e l’acido ferulico (l’acido caffeico non è un precursore dei questo sottogruppo di polifenoli). Quindi in ultima analisi derivano dalla fenilalanina e dunque dalla via dell’acido shikimico.
Sintesi dei Lignani
Le prime tre reazioni riducono i gruppi carbossilici degli idrossicinnamati a gruppi alcolici, con formazione di alcol detti monolignoli, ossia l’alcol p-cumarilico, l’alcol sinapilico e l’alcol coniferilico, molecole che entrano anche nella biosintesi della lignina.
La prima reazione, che porta all’attivazione degli acidi idrossicinnamici, è catalizzata dalla idrossicinnamato:CoA ligasi o 4-cumarato:CoA ligasi (EC 6.2.1.12), con formazione del corrispettivo idrossicinnamato-CoA, e dunque feruloil-CoA, p-cumaril-CoA e sinapil-CoA.
Di seguito intervengono le cinnamoil-CoA ossidoreduttasi NADPH-dipendenti o cinnamoil-CoA reduttasi (EC1.2.1.44), che catalizzano la formazione dell’aldeide corrispondente, con liberazione del coenzima A.
Nell’ultima delle tre tappe suddette le cinnamil alcol deidrogenasi o monolignolo deidrogenasi NADPH-dipendenti (EC 1.1.1.195) riducono ulteriormente il gruppo aldeidico ad alcol, con formazione di alcol coniferilico, alcol p-cumarilico e alcol sinapilico.
Il passaggio successivo, la dimerizzazione, comporta l’intervento di meccanismi stereoselettivi, o più precisamente enantioselettivi. Infatti la maggior parte dei lignani delle piante esiste in forma di (+)- o (-)-enantiomeri, ossia isomeri dotati di almeno un centro di chiralità, le cui quantità relative possono variare da specie a specie, ma anche all’interno di organi differenti della stessa pianta, a seconda del tipo di reazioni coinvolte.
La dimerizzazione può essere ottenuta attraverso reazioni catalizzate da laccasi (EC 1.10.3.2). Questi enzimi di per se catalizzano la formazione di radicali che dimerizzando creano una miscela racemica, il che dunque non spiega come si formino le miscele racemiche presenti nelle piante. Il meccanismo più accreditato per spiegare la sintesi stereospecifica chiama in causa l’azione degli enzimi suddetti e di una proteina in grado di dirigere la sintesi verso una o l’altra delle due forme enantiomeriche: la proteina dirigente. Lo schema di reazione potrebbe essere il seguente: l’enzima forma i radicali che sono orientati in modo da ottenere l’accoppiamento stereospecifico desiderato dalla proteina dirigente.
(-)-Matairesinolo
Ad esempio, la pinoresinolo sintetasi, composta da laccasi e proteina dirigente, catalizza la sintesi stereospecifica del (+)-pinoresinolo a partire da due residui di alcol coniferilico. Di seguito il (+)-pinoresinolo, in due reazione stereospecifiche consecutive catalizzate dalla pinoresinolo/lariciresinolo reduttasi NADPH-dipendente (EC 1.23.1.2), viene dapprima ridotto a (+)-lariciresinolo e poi a (-)-secoisolariciresinolo. Il (-)-secoisolariciresinolo, nella reazione catalizzata dalla secoisolariciresinolo deidrogenasi NAD(P)-dipendente (EC 1.1.1.331), è ossidato a (-)-matairesinolo.
Metabolismo intestinale
La loro importanza per la salute dell’uomo deriva in larga misura dalla metabolizzazione che subiscono nel colon da parte del microbiota intestinale, che è parte del più ampio microbiota umano, e che opera deglicosilazioni, para-deidrossilazioni e meta-demetilazioni senza inversione enantiomerica.
Le trasformazioni batteriche infatti portano alla formazione di metaboliti dotati di una modesta attività simil-estrogenica, una situazione analoga a quella alcuni isoflavoni, come quelli della soia, di alcuni stilbeni e alcune cumarine. Si parla pertanto di fitoestrogeni. Il prodotto delle reazioni suddette sono i cosiddetti lignani dei mammiferi o enterolignani, come gli agliconi dell’enterodiolo e dell’enterolattone, prodotti rispettivamente a partire dal secoisolariciresinolo e dal matairesinolo.
Osservazioni condotte in animali alimentati con diete ricche di lignani hanno evidenziato la loro presenza in forma non modificata, in basse concentrazioni, nel siero, dimostrando così che possono essere assorbite anche intatti a livello intestinale. Queste molecole esercitano azioni estrogeno-indipendenti, sia in vivo che in vitro, quali l’inibizione dell’angiogenesi, la riduzione del diabete e la soppressione della crescita tumorale.
Nota: con il termine di “fitoestrogeno” si intende una molecola dotata di attività estrogenica o antiandrogenica, almeno in vitro.
Dopo essere stati assorbiti, entrano nel circolo enteroepatico, e a livello epatico possono subire le reazione di fase II ed essere solforati o glucoronidati, per essere infine escreti nelle urine.
Fonti alimentari
La fonte più ricca è rappresentata dai semi di lino, che contengono in prevalenza secoisolariciresinolo, ma in buone quantità anche lariciresinolo, pinoresinolo e matairesinolo (in totale oltre 3,7 mg/100 g di prodotto secco). Si ritrovano anche nei semi di sesamo.
(-)-Secoisolariciresinolo
Un’altra fonte importante è rappresentata dai cereali integrali.
Sono presenti anche in altri alimenti, ma in concentrazioni tra le cento e le mille volte inferiori rispetto a quelle osservate nei semi di lino. Esempi sono:
le bevande, dove in genere sono più abbondanti nel vino rosso, seguito in ordine decrescente dal tè nero, latte di soia e caffè;
la frutta come albicocche, pere, pesche e fragole;
le verdure, come le Brassicaceae, l’aglio, gli asparagi e le carote;
lenticchie e fagioli.
La loro presenza nei cereali integrali e, in misura minore, nel vino rosso e nella frutta fa si che, almeno nella popolazione che segua una dieta mediterranea, rappresentino la principale fonte di fitoestrogeni.
Bibliografia
Andersen Ø.M., Markham K.R. Flavonoids: chemistry, biochemistry, and applications. CRC Press Taylor & Francis Group, 2006
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
Manach C., Scalbert A., Morand C., Rémésy C., and Jime´nez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr 2004;79(5):727-747. doi:10.1093/ajcn/79.5.727
Satake H, Koyama T., Bahabadi S.E., Matsumoto E., Ono E. and Murata J. Essences in metabolic engineering of lignan biosynthesis. Metabolites 2015;5:270-290. doi:10.3390/metabo5020270
Tsao R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010;2:1231-1246. doi:10.3390/nu2121231
van Duynhoven J., Vaughan E.E., Jacobs D.M., Kemperman R.A., van Velzen E.J.J, Gross G., Roger L.C., Possemiers S., Smilde A.K., Doré J., Westerhuis J.A.,and Van de Wiele T. Metabolic fate of polyphenols in the human superorganism. PNAS 2011;108(suppl. 1):4531-4538. doi:10.1073/pnas.1000098107
Wink M. Biochemistry of plant secondary metabolism – 2nd Edition. Annual plant reviews (v. 40), Wiley J. & Sons, Inc., Publication, 2010
Gli acidi idrossicinnamici o idrossicinnamati sono composti fenolici che fanno parte del gruppo dei polifenoli non flavonoidi.
Sono presenti praticamente in tutte le parti della frutta e verdura anche se le concentrazioni maggiori si ritrovano nelle porzioni esterne dei frutti maturi, concentrazioni che si riducono nel corso della maturazione, mentre il contenuto totale aumenta grazie all’aumentare delle dimensioni del frutto.
Il loro consumo con i cibi è stato associato a un effetto di prevenzione dello sviluppo di malattie croniche come:
le malattie cardiovascolari;
il cancro;
il diabete di tipo 2.
Questi effetti sembra non siano dovuti solamente al loro notevole potere antiossidante, potere che variare in base al pattern metilazione, e soprattutto di idrossilazione dell’anello aromatico, ma anche ad altri meccanismi d’azione come la riduzione dell’assorbimento intestinale del glucosio o la modulazione della secrezione di alcuni ormoni intestinali.
La struttura chimica di base è composta da un anello benzenico cui è legata una catena di tre atomi di carbonio, struttura che è indicata anche come C6-C3. Pertanto possono essere inseriti nel gruppo dei fenilpropanoidi.
Scheletro di Base degli Acidi Idrossicinnamici
Gli idrossicinnamati più comuni sono:
l’acido caffeico o acido 3,4-diidrossicinnamico;
l’acido ferulico o acido 4-idrossi-3-metossicinnamico;
l’acido sinapico o acido 4-idrossi-3,5-dimetossicinnamic
l’acido p-cumarico o acido 4-cumarico o acido 4-idrossicinnamico.
In natura si trovano associati ad altre molecole, in genere in forma di derivati glicosilati o di esteri dell’acido chinico, tartarico e shikimico o acido scichimico. Inoltre sono state identificate diverse centinaia di antociani acilati con gli idrossicinnamati sopracitati (in ordine decrescente con l’acido p-cumarico, oltre 150, acido caffeico, circa 100, acido ferulico, circa 60, e acido sinapico, circa 25).
Raramente sono presenti in forma libera, tranne che nei cibi lavorati che abbiano subito congelamento, fermentazione o sterilizzazione. Ad esempio, una conservazione eccessivamente lunga delle arance rosse provoca una idrolisi massiva dei derivati idrossicinnamici a dare acidi liberi, e questo a sua volta potrebbe portare alla formazione di composti maleodoranti quali i vinil-fenoli, indicatori di una senescenza troppo avanzata del frutto.
Biosintesi
La biosintesi degli idrossicinnamati consiste in una serie di reazioni successive a quella catalizzata dalla fenilalanina ammonio liasi (PAL, acronimo dell’inglese phenylalanine ammonia lyase).
Fenilalanina ⇄ Acido trans-Cinnamico + NH3
La reazione, deaminando la fenilalanina a dare acido trans-cinnamico, lega l’aminoacido aromatico agli acidi idrossicinnamici e alle loro forme attivate.
Biosintesi degli Idrossicinnamati
Nel primo passaggio viene introdotto un gruppo ossidrilico in posizione 4 dell’anello aromatico dell’acido trans-cinnamico a dare l’acido p-cumarico. La reazione catalizzata dalla trans-cinnamato 4-monoossigenasi (EC 1.14.14.91).
L’addizione di un secondo gruppo ossidrilico in posizione 3 dell’anello dell’acido p-cumarico porta alla formazione di acido caffeico. La reazione catalizzata dalla p-cumarato 3-idrossilasi (EC 1.14.13.-).
La O-metilazione del gruppo ossidrilico in posizione 3 dell’acido caffeico produce acido ferulico. La reazione catalizzata dalla caffeato 3-O-metiltransferasi (EC:2.1.1.68).
Acido caffeico + SAM ⇄ Acido ferulico + SAH
L’acido ferulico a sua volta è convertito in acido sinapico attraverso due reazione: una idrossilazione in posizione 5 a dare l’acido 5-idrossiferulico, reazione catalizzata dalla ferulato 5-idrossilasi (EC:1.14.-.-), e la successiva O-metilazione dello stesso ossidrile, reazione catalizzata ancora dalla caffeato 3-O-metiltransferasi.
Acido-5-idrossiferulico + SAM ⇄ Acido sinapico + SAH
Gli idrossicinnamati non sono presenti in quantità elevate in quanto sono rapidamente convertiti in esteri del coenzima A (CoA) o in esteri del glucosio, nelle reazioni catalizzate da idrossicinnamato:CoA ligasi o da O-glucosiltransferasi. Questi intermedi attivati rappresentano punti di ramificazione in quanto in grado di partecipare a un’ampia gamma di reazioni successive, quali la condensazione con il malonil-CoA a dare flavonoidi, o la riduzione NADPH-dipendente a dare lignani, che saranno di seguito utilizzati nella sintesi della lignina.
Fonti alimentari
Tra le fonti più ricche si ritrovano kiwi, mirtilli, prugne, ciliegie, mele, pere, cicoria, carciofi, carote, lattuga, melanzane, grano e caffè.
Acido caffeico
In genere, sia in forma libera che legata ad altre molecole, è l’acido idrossicinnamico più abbondante nella verdura e nella maggior parte della frutta, dove rappresenta il 75-100% del totale degli idrossicinnamati.
Le fonti più ricche sono il caffè, inteso come bevanda, le carote, la lattuga, le patate, anche quelle dolci, ma anche frutti di bosco quali mirtilli, mirtilli rossi e more.
Fonti minori sono rappresentate da uva e prodotti derivati, succo d’arancia, mele, prugne, pesche, e pomodori.
L’acido caffeico e il chinico si legano a formare l’acido clorogenico, presente in molti tipi di frutta e in concentrazione elevata nel caffè.
Acido ferulico
E’ l’acido idrossicinnamico più abbondante nei cereali, che ne sono anche la fonte alimentare principale.
Nel grano il contenuto è compreso tra 0,8 e 2 g/kg di peso secco, che rappresenta fino al 90% del totale dei polifenoli. Si ritrova quasi esclusivamente, fino al 98% del totale, nelle parti più esterne del chicco, ossia lo strato aleuronico e il pericarpo, e quindi il suo contenuto nelle farine dipende dal loro livello di raffinazione, mentre la principale fonte è ovviamente rappresentata dalla crusca. La molecola è presente principalmente nella forma trans, esterificata con arabinoxilani e emicellulose. Infatti solamente il 10% si ritrova in forma libera solubile nella crusca.
Nei cereali sono state ritrovati anche dimeri che formano strutture a ponte tra le catene di emicellulosa.
Nei frutti e nella verdura è molto meno comune dell’acido caffeico. Le principali fonti sono asparagi, melanzane e broccoli, mentre concentrazioni più basse sono state ritrovate nelle more, mirtilli, mirtilli rossi, mele, carote, patate, barbabietole, caffè e succo d’arancia.
Acido sinapico
Le quantità più elevate si ritrovano nella buccia e nei semi degli agrumi (il contenuto del succo d’arancia è decisamente più basso); buoni valori sono presenti anche nel cavolo cinese (o cavolo di Pechino), e in alcune varietà di mirtilli rossi.
Acido p-cumarico
Elevate quantità sono presenti nelle melanzane, le più ricche, nei broccoli e asparagi; altre fonti sono le ciliegie dolci, le prugne, i mirtilli, anche rossi, la buccia e i semi degli agrumi, e il succo d’arancia.
Bibliografia
Andersen Ø.M., Markham K.R. Flavonoids: chemistry, biochemistry, and applications. CRC Press Taylor & Francis Group, 2006
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
Manach C., Scalbert A., Morand C., Rémésy C., and Jime´nez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr 2004;79(5):727-747. doi:10.1093/ajcn/79.5.727
Preedy V.R. Coffee in health and disease prevention. Academic Press, 2014
Zhao Z., Moghadasian M.H. Bioavailability of hydroxycinnamates: a brief review of in vivo and in vitro studies. Phytochem Rev 2010;9(1):133-145. doi:10.1007/s11101-009-9145-5
Il consumo di uva e prodotti derivati, in primis il vino rosso ma solo durante i pasti, è stato associato a numerosi effetti positivi sulla salute, che non si limitano al solo effetto antiossidante/antiradicalico, ma includono anche un’azione:
antiinfiammatoria;
cardioprotettiva;
anticancerosa;
antimicrobica;
neuroprotettiva
Nell’uva sono presenti numerosi nutrienti quali zuccheri, vitamine, sali minerali, fibre e fitochimici. Tra questi ultimi, i polifenoli si sono dimostrati i composti più importanti nel determinare gli effetti positivi del frutto e dei prodotti derivati. L’uva è infatti uno dei frutti più ricchi in polifenoli, la cui composizione è fortemente influenzata da diversi fattori quali la varietà o cultivar, le condizioni ambientali in cui avviene la maturazione, eventuali malattie quali infezioni fungine, come anche la lavorazione che subisce.
Al momento le specie di vite principalmente coltivate a livello mondiali sono: l’europea, Vitis vinifera, le nordamericane, Vitis labrusca e Vitis rotundifolia, e ibridi francesi.
Nota: l’uva in realtà non è un frutto ma un’infruttescenza ossia un raggruppamento di frutti: il grappolo. A sua volta il grappolo è composto dal peduncolo, dal raspo o graspo, dai pedicelli, e dalle bacche o acini o chicchi.
I polifenoli sono presenti sia in quantità che in varietà decisamente maggiori nell’uva rossa, e quindi nel vino rosso, rispetto a quella bianca. Questo, secondo molti ricercatori, sarebbe alla base dei maggiori benefici sulla salute derivanti al consumo di uva/vino rosso rispetto a quella bianca e i suoi derivati.
I polifenoli dell’uva e del vino sono una complessa miscela di composti flavonoidi, il gruppo più abbondante, e non flavonoidi.
Tra i flavonoidi si ritrovano:
La maggior parte dei flavonoidi presenti nel vino derivano dallo strato epidermico della buccia, mentre il 60-70% del totale dei polifenoli è presente nel vinacciolo. Da notare che oltre il 70% dei polifenoli dell’uva non sono estratti e rimangono nella vinaccia.
Le complesse interazioni chimiche che si stabiliscono tra questi composti, e tra di loro e gli altri composti di natura differente presenti nell’uva e nel vino, sono probabilmente essenziali nel determinare sia la qualità delle uve e del vino che l’ampio spettro di effetti terapeutici propri di questi alimenti.
Nel vino la miscela di polifenoli svolge importanti funzioni essendo in gradi di influenzare:
il gusto amaro;
l’astringenza;
il colore rosso, di cui sono tra i maggiori responsabili;
la sensibilità all’ossidazione, essendo sostanze facilmente ossidabili quando esposte all’aria.
Infine sono un conservante importante per il vino stesso e la base per un lungo invecchiamento.
Antociani
Sono flavonoidi ampiamente presenti nella frutta e verdura.
Nell’uva si accumulano in modo principale nella buccia (nei primi strati esterni del tessuto ipodermico), cui conferiscono il colore, avendo tonalità che variano dal rosso al blu. In alcune varietà, dette “teinturier”, si accumulano anche nella polpa dell’acino.
Esiste una stretta correlazione tra la sintesi degli antociani e lo sviluppo dell’acino. Quando l’acino raggiunge l’invaiatura, ossia il momento in cui termina la sua crescita, ha inizio la loro sintesi, che determina anche il cambiamento di colore dell’acino stesso che diventa viola. La sintesi raggiunge il massimo livello alla maturazione completa dell’acino.
Tra i flavonoidi del vino sono uno degli antiossidanti più potenti.
Ogni specie e varietà di uva ha una composizione unica in antociani. Inoltre nelle uve di Vitis vinifera, a seguito di una mutazione a carico del gene che codifica per 5-O-glucosiltransferasi, mutazione che determina la sintesi di un enzima inattivo, sono prodotti solo 3-monoglucosidi, mentre nelle uve derivanti da altre specie avviene anche la glicosilazione in posizione 5. Interessante notare che i derivati 3-glucosidici sono colorati più intensamente dei 3,5-diglucosidi.
Malvidina-3-glucoside
Nell’uva e nel vino rosso i più abbondanti sono i 3-monoglucosidi della malvidina, la più abbondante sia nell’uva che nel vino, e della petunidina, delfinidina, peonidina, cianidina.
L’idrossile in posizione 6 del glucosio può a sua volta essere acilato con un gruppo acetilico, caffeico o cumarico, acilazione che ne aumenta ulteriormente la stabilità.
Le antocianidine, ossia le forme non coniugate, non sono presenti ne nell’uva ne nel vino, se non in tracce.
Gli antociani sono scarsamente presenti nelle uve bianche, e dunque nel vino bianco.
La composizione in antociani del vino è fortemente influenzata sia dal tipo di cultivar che dalle tecniche di vinificazione, ritrovandosi nel vino in conseguenza di processi di estrazione dalla buccia dovuti alla macerazione delle uve. Di conseguenza vini derivanti da varietà simili di uve possono avere composizioni in antocianine molto diverse.
Insieme alla proantocianidine, sono i polifenoli dell’uva più importanti nel determinare alcune importanti proprietà organolettiche del vino rosso, in quanto sono i principali responsabili dell’astringenza, amarezza, stabilità chimica nei confronti dell’ossidazione, come anche del colore del vino giovane.
Riguardo al colore va sottolineato che con il tempo la loro concentrazione si riduce, mentre il colore è dovuto sempre più alla formazione di pigmenti polimerici prodotti della condensazione degli antociani sia tra di loro che con altre molecole.
Nel corso dell’invecchiamento del vino gli antociani e le proantocianidine possono interagire a dare molecole con struttura complessa che possono parzialmente precipitare.
Catechine
Sono, insieme ai tannini condensati, i flavonoidi più abbondanti, rappresentando fino al 50% del totale dei polifenoli nelle uve bianche e dal 13% al 30% in quelle rosse.
Il loro livello nel vino dipende dal tipo di cultivar.
Catechina
In genere il flavanolo più abbondante nel vino è la catechina, ma si ritrovano anche epicatechina ed epicatechina-3-gallato.
Proantocianidine
Formate da unità di catechine, sono presenti nella buccia, nel vinacciolo e nel raspo del grappolo d’uva in forma di:
dimeri, di cui i più comuni sono le procianidine B1-B4, ma possono essere presenti anche le procianidine B5-B8;
trimeri, e tra questi la procianidina C1 è la più abbondante;
tetrameri;
polimeri, formati fino da 8 monomeri.
Procianidina C1
Il loro livello nel vino dipende dalle tecniche di vinificazione e dalla varietà dell’uva e, al pari degli antociani, sono molto più abbondanti nei vini rossi, in particolare in quelli invecchiati, rispetto ai bianchi.
Inoltre, come detto in precedenza, insieme agli antociani, i tannini condensati sono importanti nel determinare alcune proprietà organolettiche del vino.
Flavonoli
Sono presenti in una grande varietà di frutta e verdura, anche se in basse concentrazioni.
Nell’uva sono il terzo gruppo di flavonoidi più abbondanti, dopo proantocianidine e catechine.
Si ritrovano principalmente nell’epidermide esterna della buccia, dove agiscono come agenti protettivi nei confronti della radiazione UV-A e UV-B, e hanno un ruolo di copigmentazione insieme agli antociani.
La loro sintesi inizia nel germoglio; la concentrazione più elevata è raggiunta poche settimane dopo l’invaiatura, per poi ridursi quando il chicco aumenta di dimensioni.
Il loro contenuto totale è molto variabile, con le varietà rosse spesso più ricche rispetto a quelle bianche.
Nell’uva sono presenti come 3-glucosidi. Il loro profilo dipende dal tipo di uva e cultivar:
nell’uva bianca si ritrovano i derivati della quercetina, campferolo e isoramnetina;
i derivati della miricetina, laricitrina e siringetina si ritrovano, insieme ai precedenti, solo in quella rossa, a causa della mancata espressione nell’uva bianca del gene che codifica per la flavonoide-3’,5’-idrossilasi.
Quercetina-3-glucoside
In generale i 3-glucosidi e i 3-glucoronidi della quercetina sono i principali flavonoli nella maggior parte delle uve. Nelle uve moscate invece i più rappresentati sono la quercetina-3-ramnoside e la quercetina aglicone.
A differenza dell’uva, nel vino e nel succo d’uva i flavonoli sono presenti anche come agliconi, in conseguenza dell’idrolisi acida che si verifica durante la lavorazione e la conservazione. Si ritrovano nel vino in quantità variabile, e i principali sono i glicosidi della miricetina e quercetina, che da soli rappresentano il 20-50% del totale dei flavonoli del vino rosso.
Acidi idrossicinnamici
Gli acidi idrossicinnamici sono la principale classe di polifenoli dell’uva non flavonoidi e i principali polifenoli del vino bianco.
I più importanti sono gli acidi p-cumarico, caffeico, sinapico e ferulico, presenti nel vino in forma di esteri con l’acido tartarico.
Sono molecole dotate di attività antiossidante e in alcune cultivar bianche di Vitis vinifera, assieme ai flavonoli, sono i principali polifenoli responsabili dell’assorbimento della radiazione ultravioletta a livello dell’acino.
Stilbeni
Sono fitoalessine che, al contrario dei flavonoidi che sono presenti in tutte le piante superiori, sono prodotti in basse concentrazioni solo da poche specie edibili, tra cui la vite.
Insieme agli altri polifenoli dell’uva e del vino anche gli stilbeni, e in particolare il resveratrolo, sono stati associati agli effetti benefici sulla salute conseguenti al consumo della bevanda.
trans-Resveratrolo
Il loro contenuto aumenta dall’invaiatura sino alla maturazione del chicco, ed è influenzato dal tipo di cultivar, dal clima, dalle tecniche di vinificazione e dalla pressione fungina.
I principali stilbeni presenti nell’uva e nel vino sono:
cis– e trans-resveratrolo (3,5,4’-triidrossistilbene);
piceide o resveratrolo-3-glucopiranoside e astringina o 3’-idrossi trans-piceide;
piceatannolo;
dimeri e oligomeri del resveratrolo, detti viniferine, di cui le più importanti sono:
α-viniferina, un trimero;
β-viniferina, un tetramero ciclico;
γ-viniferina, un oligomero altamente polimerizzato;
ε-viniferina, un dimero ciclico.
Nell’uva sono state identificati in tracce anche altri isomeri e forme glicosilate del resveratrolo e del piceatannolo, come il resveratroloside, l’opeafenolo, il resveratrolo di- e triglucoside.
La glicosilazione degli stilbeni è importante per la conservazione, il trasporto, la modulazione dell’attività antifungina e la protezione dalla degradazione ossidativa del vino.
La sintesi di dimeri ed oligomeri del resveratrolo, prodotti sia nell’uva che nel vino, rappresenta un meccanismo di difesa nei confronti di attacchi esogeni, o al contrario è il risultato dell’azione di enzimi extracellulari rilasciati da patogeni nel tentativo di eliminare composti tossici indesiderati.
Idrossibenzoati
I derivati dell’acido idrossibenzoico sono componenti minori dell’uva e del vino.
Nell’uva i principali sono gli acidi gentisico, gallico, p-idrossibenzoico e protocatechico.
Acido Gallico
A differenza degli idrossicinnamati, che nel vino sono presenti come esteri con l’acido tartarico, si ritrovano in forma libera.
Insieme ai flavonoli, proantocianidine, catechine e idrossicinnamati sono tra i responsabili dell’astringenza del vino.
Bibliografia
Andersen Ø.M., Markham K.R. Flavonoids: chemistry, biochemistry, and applications. CRC Press Taylor & Francis Group, 2006
Basli A, Soulet S., Chaher N., Mérillon J.M., Chibane M., Monti J.P.,1 and Richard T. Wine polyphenols: potential agents in neuroprotection. Oxid Med Cell Longev 2012. doi:10.1155/2012/805762
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
Flamini R., Mattivi F., De Rosso M., Arapitsas P. and Bavaresco L. Advanced knowledge of three important classes of grape phenolics: anthocyanins, stilbenes and flavonols. Int J Mol Sci 2013;14:19651-19669. doi:10.3390/ijms141019651
Georgiev V., Ananga A. and Tsolova V. Recent advances and uses of grape flavonoids as nutraceuticals. Nutrients 2014;6: 391-415. doi:10.3390/nu6010391
Guilford J.M. and Pezzuto J.M. Wine and health: a review. Am J Enol Vitic 2011;62(4):471-486. doi:10.5344/ajev.2011.11013
He S., Sun C. and Pan Y. Red wine polyphenols for cancer prevention. Int J Mol Sci 2008;9:842-853. doi:10.3390/ijms9050842
Xia E-Q., Deng G-F., Guo Y-J. and Li H-B. Biological activities of polyphenols from grapes. Int J Mol Sci 2010;11:622-646. doi:10.3390/ijms11020622
L’olio di oliva è un grasso alimentare ottenuto dalla spremitura delle olive, i frutti dell’olivo (Olea europaea).
È un elemento fondamentale della dieta mediterranea e un prodotto tipico dell’area mediterranea, con una storia millenaria e un grande valore economico.
Dal punto di vista chimico nell’olio di oliva è possibile individuare, sulla base del comportamento in presenza di soluzioni fortemente alcaline e calore, due frazioni:
la frazione saponificabile, che rappresenta il 98-99% del peso totale, ed è formata da lipidi che, nelle condizioni suddette, formano saponi;
la frazione insaponificabile, che costituisce il restante 1-2% del peso, e che nelle condizioni suddette non forma saponi.
E’ composta da acidi grassi saturi e acidi grassi insaturi, quasi per intero legati al glicerolo a formare trigliceridi o triacilgliceroli. In misura molto minore si ritrovano anche digliceridi o diacilgliceroli, monogliceridi o monoacilgliceroli, e acidi grassi liberi.
L’acido oleico è l’acido grasso più abbondante dell’olio di oliva, con una concentrazione che, in base alle norme stabilite dall’International Olive Oil Council (IOOC) deve essere compresa tra il 55% e 83% del totale degli acidi grassi.
L’acido linoleico è l’acido grasso polinsaturo più abbondante, con una concentrazione che deve essere comprese tra il 2,5% e il 21%. A causa del suo elevato grado di insaturazione è soggetto facilmente a ossidazione; questo significa che un olio che ne contenga quantità elevate irrancidisce con più facilità, e dunque può essere conservato per un tempo minore rispetto a un olio che ne contenga meno.
Nella cucina mediterranea l’olio di oliva è la principale fonte di grassi: dunque l’acido oleico, tra gli acidi grassi monoinsaturi, e il linoleico, tra i polinsaturi, sono i principali acidi grassi presenti.
L’acido alfa-linolenico deve essere presente in quantità molto bassa, secondo gli standard IOOC ≤1%. Si tratta di un acido grasso polinsaturo della famiglia degli acidi grassi polinsaturi omega-3, che può apportare benefici dal punto di vista nutrizionale, ma, a causa della forte insaturazione, al pari dell’acido linoleico, è molto suscettibile a ossidazione, e quindi favorisce l’irrancidimento dell’olio che lo contiene.
La presenza di acidi grassi che non dovrebbero essere presenti, o che dovrebbero essere presenti in percentuali diverse rispetto a quelle osservate, sono indici di adulterazione dell’olio, che è stato “tagliato” con altri oli di semi. A questo riguardo particolare attenzione viene rivolta agli acidi miristico, arachidico, beenico, lignocerico, gadoleico e alfa-linolenico, le cui percentuali ammissibili sono specificate dal regolamento IOOC.
La composizione in acidi grassi è influenzata da diversi fattori.
Il clima.
La latitudine.
La zona di produzione.
Gli oli prodotti in Italia, Spagna e Grecia sono ricchi in acido oleico e poveri in acido linoleico e palmitico, mentre l’olio tunisino è ricco in acido linoleico e palmitico e più povero in acido oleico. Dunque, gli oli d’oliva possono essere suddivisi in due gruppi:
uno con elevato contenuto di acido oleico e basso di acido linoleico e palmitico;
l’altro con un elevato contenuto in acido linoleico e palmitico e basso di acido oleico.
Il tipo di cultivar.
Il grado di maturazione delle olive al momento dell’estrazione dell’olio.
Va notato che l’acido oleico è formato per primo nel frutto, e i dati sembrano indicare una forte rapporto antagonistico tra l’acido oleico e gli acidi palmitico, palmitoleico e linoleico.
Trigliceridi
Come detto, gli acidi grassi dell’olio di oliva sono quasi per intero legati a formare molecole di trigliceridi.
In piccola percentuale si ritrovano anche legati in molecole di digliceridi, monogliceridi, oppure in forma libera.
Numerazione Stereospecifica o sn
Durante la biosintesi dei trigliceridi, grazie alla presenza di specifici enzimi, solo circa il 2% delle molecole del glicerolo legherà in posizione sn-2 una molecola di acido palmitico; anche la percentuale di acido stearico in posizione 2 è molto bassa. Per la maggior parte la posizione sn-2 sarà invece occupata da acido oleico.
Al contrario, se si considerano oli che abbiano subito processi di esterificazione artificiali, viene a mancare la specificità dell’azione enzimatica, per cui la percentuale di acido palmitico in posizione sn-2 aumenta in modo significativo.
Nota: sn- è l’acronimo dell’inglese sterospecific numbering, che significa numerazione sterospecifica.
Tra i trigliceridi presenti in proporzioni più significative si ritrovano:
OOO (40-59%);
POO (12-20%);
OOL (12,5-20%);
POL (5,5-7%);
SOO (3-7%).
Si ritrovano anche quantità minori di POP, POS, OLnL, OLnO, PLL, PLnO.
La trilinoleina (LLL) è un trigliceride contenente tre molecole di acido linoleico. Un suo basso livello è indice di buona qualità dell’olio.
Non sono stati osservati trigliceridi con soli acidi grassi saturi e neppure contenenti solo residui di acido alfa-linolenico.
Digliceridi e monogliceridi
La loro presenza è dovuta sia ad una incompleta sintesi dei trigliceridi che a parziali reazioni di idrolisi dei trigliceridi stessi.
La concentrazione dei digliceridi nell’olio di oliva vergine varia tra 1% e il 2,8%. Nell’olio fresco prevalgono gli 1,2-digliceridi, rappresentando oltre l’80% dei digliceridi. Nel corso della conservazione dell’olio si verifica una isomerizzazione con un progressivo incremento della forma 1,3, più stabile, che dopo circa 10 mesi diviene la forma principale. Dunque il rapporto tra 1,2- e 1,3-digliceridi può essere utilizzato come indicatore del grado di invecchiamento dell’olio.
I monogliceridi sono presenti in quantità inferiori rispetto ai digliceridi, meno dello 0,25%, con gli 1-monogliceridi ben più abbondanti dei 2-monogliceridi.
Frazioni insaponificabile
E’ composta da un elevato numero di molecole differenti molto importanti dal punto di vista nutrizionale, in quanto contribuiscono in modo considerevole a quelli che sono gli effetti salutari dell’olio di oliva. Inoltre sono responsabili della stabilità e del sapore dell’olio di oliva, e sono utilizzati anche per determinarne una eventuale adulterazione con altri oli.
Vi si ritrovano tocoferoli, steroli, polifenoli, pigmenti, idrocarburi, alcol aromatici e alifatici, acidi triterpenici, cere, e composti minori.
Il loro contenuto è influenzato da fattori in parte analoghi a quelli visti per la composizione in acidi grassi, quali:
il tipo di cultivar;
il grado di maturazione dell’oliva;
la zona di produzione;
l’anno di raccolta e le modalità di raccolta delle olive;
il tempo di stoccaggio delle olive;
il tipo di lavorazione per l’estrazione dell’olio;
le condizioni di conservazione dell’olio.
Da sottolineare che negli oli di oliva raffinati molti di questi composti non sono presenti in quanto rimossi durante la lavorazione.
Polifenoli
I polifenoli rappresentano il 18-37% della frazione insaponificabile.
Sono un gruppo molto eterogeneo di molecole con proprietà sia nutrizionali che organolettiche, come l’oleuropeina e l’idrossitirosolo che danno all’olio un gusto amaro e pungente.
Per una loro più ampia trattazione si rimanda all’articolo: Polifenoli dell’olio di oliva.
Idrocarburi
Rappresentano il 30-50% della frazione insaponificabile.
I principali sono lo squalene e il beta-carotene.
Lo squalene, isolato per la prima volta dal fegato di squalo, è il principale componente della frazione insaponificabile, e rappresenta oltre il 90% della frazione idrocarburica; la sua concentrazione oscilla tra i 200 e i 7500 mg/kg di olio.
E’ un intermedio nella via di sintesi del nucleo a 4 anelli degli steroidi. Sembra essere responsabile, almeno in parte, degli effetti benefici dell’olio di oliva.
Nella frazione idrocarburica dell’olio vergine di oliva, oltre allo squalene, si ritrovano anche idrocarburi diterpenici e triterpenici, poliolefine isoprenoidi e n-paraffine.
Il beta-carotene agisce sia come antiossidante, proteggendo l’olio durante la conservazione, che come colorante.
Steroli
Sono un’importante gruppo di lipidi dell’olio di oliva che hanno:
diversi effetti salutari;
si correlano con la qualità dell’olio;
sono ampiamente utilizzati per testarne la genuinità.
A questo riguardo c’è da sottolineare il fatto che sono molecole specie specifiche, per cui trovare ad esempio nell’olio di oliva elevate concentrazioni di brassicasterolo, uno sterolo tipico delle Brassicaceae, come la colza, indica una sua adulterazione con olio di canola.
Si individuano 4 classi di steroli: steroli comuni, 4-metilsteroli, alcol triterpenici, e dialcol triterpenici. Il loro livello nell’olio di oliva oscilla tra 1000 mg/kg, il valore minimo richiesto dagli standard IOOC, e 2000 mg/kg. I valori più bassi si osservano negli oli raffinati, dove il processo di raffinazione può causare perdite anche del 25%.
Steroli comuni o 4-alfa-desmetilsteroli
Gli steroli comuni sono presenti per la maggior parte in forma libera ed esterificata, sebbene siano stati rinvenuti anche come lipoproteine e steril glucosidi.
I principali sono il beta-sitosterolo, che rappresenta dal 75 al 90% del totale degli steroli, il Δ5-avenasterolo, tra il 5 e il 20%, e il campesterolo, 4%.Altre molecole presenti in quantità inferiori o tracce sono ad esempio il stigmasterolo, 2%, colesterolo, brassicasterolo, ergosterolo.
4-Metilsteroli
Sono intermedi nella biosintesi degli steroli, e si ritrovano sia in forma libera che esterificata.
Sono presenti in piccole quantità, molto minori rispetto a quelle degli steroli comuni e degli alcol triterpenici, oscillando tra 50 e 360 mg/kg. Le molecole principali sono l’obtusifoliolo, il gramisterolo, cicloeucalenolo, e il citrostadienolo.
Alcol triterpenici o 4,4-dimetilsteroli
Sono una classe molto complessa di steroli, e si ritrovano sia in forma libera che esterificata.
Sono presenti in quantità variabili tra 350 e 1500 mg/kg.
I principali sono β-amirina, butirrospermolo, 24 metilen cicloartanolo, e cicloartenolo; altre molecole presenti in quantità inferiori sono ad esempio il ciclobranolo, il ciclosadolo, dammaradienolo, e il germanicolo.
Dialcol triterpenici
I principali dialcol triterpenici dell’olio di oliva sono l’eritrodiolo e l’uvaolo.
L’eritrodiolo è presente sia in forma libera che esterificata, in quantità totale, nell’olio vergine di oliva, oscillante tra i 19 e i 69 mg/kg, mentre la forma libera è in genere inferiore a 50 mg/kg.
Tocoferoli
I tocoferoli rappresentano il 2-3% della frazione insaponificabile, e includono la vitamina E.
Degli otto vitameri della vitamina E, nell’olio di oliva vergine l’alfa-tocoferolo costituisce circa il 90% dei tocoferoli. E’ presente in forma libera e in quantità molto variabile, ma comunque in media superiore ai 100 mg/kg d’olio. Grazie alle sue proprietà antiossidanti in vivo, la sua abbondante presenza è un fattore molto positivo per la salute. La concentrazione dell’alfa-tocoferolo sembra essere correlata agli alti livelli di clorofille e alla concomitante richiesta di disattivazione dell’ossigeno singoletto.
Beta-tocoferolo, delta-tocoferolo e gamma-tocoferolo sono in genere presenti in basse quantità.
Pigmenti
In questo gruppo si ritrova le clorofille e i carotenoidi.
Le clorofille sono presenti come feofitine, in particolare nella forma alfa, ossia una clorofilla cui è stato rimosso il magnesio, e conferiscono all’olio di oliva il caratteristico colore verde. Sono molecole fotosensibilizzanti per cui contribuiscono alla foto-ossidazione dell’olio stesso.
I principali carotenoidi sono il beta-carotene e la luteina; sono presenti anche xantofille, quali anteraxantina, beta-criptoxantina, luteoxantina, mutatoxantina, neoxantina, violaxantina.
Il colore dell’olio di oliva è la risultante della presenza di clorofille e carotenoidi e delle loro tonalità verde e giallo. La loro presenza è strettamente correlata.
Acidi triterpenici
Gli acidi triterpenici sono componenti importanti dell’oliva dove sono presenti in tracce.
I principali acidi triterpenici presenti nell’olio vergine di oliva sono l’acido oleanolico e l’acido maslinico, ritrovandosi nella sansa da cui possono essere estratti in piccola quantità durante la lavorazione.
Alcol alifatici e aromatici
I più importanti sono gli alcol grassi e gli alcol diterpenici.
Gli alcol alifatici hanno un numero di atomi di carbonio pari, compreso tra 20 e 30, e per la maggior parte sono localizzati all’interno del nocciolo, da dove sono parzialmente estratti attraverso processi meccanici.
Alcol grassi
Sono alcol saturi lineari con più di 16 atomi di carbonio.
Sono presenti nell’olio vergine di oliva in quantità di solito non superiori a 250 mg/kg. Possono essere presenti sia in forma libera che esterificata.
I principali sono il docosanolo, tetracosanolo, esacosanolo, e l’octacosanolo, mentre le molecole con un numero dispari di atomi di carbonio sono presenti in tracce. Tetracosanolo ed esacosanolo sono quelli presenti in quantità maggiore.
Le cere, componenti minori dell’olio di oliva, sono esteri degli alcol grassi con acidi grassi. Le principali cere presenti sono esteri dell’acido palmitico e oleico. Possono essere utilizzate come criterio per differenziare i vari tipi di olio di oliva; ad esempio nell’olio vergine ed extravergine di oliva devono essere presenti in quantità inferiori a 150 mg/kg, in accordo con gli standard stabiliti dalla IOOC.
Alcol diterpenici
Fitolo e geranilgeraniolo sono due alcol diterpenici aciclici, presenti sia in forma libera che esterificata. Tra gli esteri nella frazione cerosa dell’olio extravergine di oliva si ritrovano quelli con l’oleato, eicosanoato, eicosenoato, docosanoato e tetracosanoato, principalmente in forma di fitil derivati.
Composti volatili
Nell’olio di oliva sono stati identificati oltre 280 composti volatili, tra cui idrocarburi, i più abbondanti, alcol, aldeidi, chetoni, esteri, acidi, eteri e altri. Solo circa 70 di questi composti sono presenti a livelli superiori a quella che è la soglia di percezione oltre la quale le molecole contribuiscono all’aroma dell’olio vergine di oliva.
Componenti minori
Tra i componenti minori si ritrovano fosfolipidi, di cui i principali sono la fosfatidilcolina, fosfatidiletanolamina, fosfatidilinositolo e fosfatidilserina.
Possono essere presenti, negli oli non filtrati, quantità minime di proteine.
Bibliografia
Gunstone F.D. Vegetable oils in food technology: composition, properties and uses. 2th Edition. Wiley J. & Sons, Inc., Publication, 2011
Caponio F., Bilancia M.T., Pasqualone A., Sikorska E., Gomes T. Influence of the exposure to light on extra virgin olive oil quality during storage. Eur Food Res Technol 2005;221:92-98. doi:10.1007/s00217-004-1126-8
Servili M., Sordini B., Esposto S., Urbani S., Veneziani G., Di Maio I., Selvaggini R. and Taticchi A. Biological activities of phenolic compounds of extra virgin olive oil. Antioxidants 2014;3:1-23. doi:10.3390/antiox3010001
Il glutine non è una semplice proteina, ma è una miscela composta da proteine dei cereali, per circa l’80% del suo peso secco, ad esempio gliadine e glutenine per il frumento, lipidi, 5-7%, amido, 5-10%, acqua, 5-8%, e sostanze minerali, <2%.
Si forma quando componenti naturalmente presenti nel chicco del cereale, la cariosside, e nella farina derivata, si uniscono tra di loro, in ambiente acquoso e sotto l’azione di sollecitazioni meccaniche, ossia durante la formazione dell’impasto.
Il termine è associato anche alla famiglia di proteine che causa problemi ai soggetti affetti da celiachia.
Isolato per la prima volta dal chimico italiano Jacopo Bartolomeo Beccari nel 1745 dalla farina di frumento, può essere estratto dall’impasto lavando lo stesso in modo delicato sotto acqua corrente: una volta allontanato l’amido, le albumine e le globuline, tutti solubili in acqua, rimane una massa appiccicosa ed elastica, appunto il glutine, termine che deriva dal latino gluten, che significa colla.
grano duro (Triticum durum), da cui si ottengono semole e semolati per pasta alimentare secca;
grano tenero (Triticum aestivum), da cui si ottengono farine per pane, paste fresche e prodotti da forno;
segale (Secale cereale);
orzo (Hordeum vulgare);
farro, nelle tre specie:
piccolo o monococco (Triticun monococcum);
medio (Triticum dicoccum Schrank);
rande o granfarro o spelta (Triticum spelta)
grano khorasan, di cui il Kamut® ne è una varietà;
triticale, che è un ibrido tra la segale e il grano tenero (× Triticosecale Wittmack);
bulgur o grano spezzato, che è grano duro integrale germogliato e successivamente lavorato;
seitan, che non è un cereale ma un derivato del frumento, da alcuni definito anche “bistecca di glutine”.
Dato che la maggior parte del glutine assunto con l’alimentazione proviene dalle farine di frumento, di cui se ne raccoglie circa 700 milioni di tonnellate annue, che rappresentano circa il 30% della produzione mondiale dei cereali, la discussione seguente sarà incentrata sul glutine di frumento, e in particolare sulle sue proteine.
Nota: il termine glutine viene anche utilizzato per indicare il residuo proteico che rimane dopo aver allontanato l’amido e le proteine solubili dall’impasto ottenuto con farina di mais o granturco: questo “glutine di mais” è tuttavia “funzionalmente” differente rispetto a quello ottenuto dal frumento.
Le proteine dei cereali
Lo studio delle proteine dei grani, come visto, ebbe inizio con il lavoro di Beccari.
In seguito, nel 1924, quindi ben 150 anni dopo, l’inglese Osborne T.B., che a ragione può essere considerato il padre della chimica delle proteine vegetali, ne sviluppò una classificazione sulla base della loro solubilità in vari solventi.
La classificazione, ancora in uso, suddivide le proteine vegetali in 4 famiglie.
Albumine, solubili in acqua.
Globuline, solubili in soluzioni saline, come l’avenalina dell’avena.
Prolamine, solubili in soluzione alcolica al 70%, ma non in acqua o alcol assoluto.
Comprendono:
gliadine del frumento;
zeina del mais;
avenina dell’avena;
ordeina dell’orzo;
secalina della segale.
Sono le responsabili dell’effetto tossico del glutine per il celiaco.
Gluteline, insolubili in acqua e soluzioni saline neutre, ma solubili in soluzioni acide e basiche.
Comprendono le glutenine del frumento.
Cereale
Albumine
Globuline
Prolamine*
Gluteline**
Frumento
9
5
40
46
Mais
4
2
55
39
Orzo
13
12
52
23
Avena
11
56
9
23
Riso
5
10
5
80
* Nel frumento gliadine
** Nel frumento glutenine
Albumine e globuline sono proteine citoplasmatiche, spesso di natura enzimatica, ricche di aminoacidi essenziali, quali lisina, triptofano e metionina. Si ritrovano nell’aleurone e nell’embrione della cariosside.
Prolamine e gluteline sono le proteine di riserva dei cereali. Sono ricche in asparagina, glutammina, arginina e prolina, ma molto povere in lisina, triptofano e metionina. Si ritrovano nell’endosperma, e rappresentano la grande maggioranza delle proteine presenti nel frumento, mais, orzo, avena e segale.
Sebbene la classificazione di Osborne sia ancora ampiamente utilizzata, sarebbe più corretto suddividere le proteine dei grani in tre gruppi: di riserva, strutturali e metaboliche, e con funzioni difensive.
Le proteine del glutine di frumento
Nel frumento le proteine rappresentano il 10-14% del peso della cariosside, mentre circa l’80% del peso è costituito da carboidrati.
Seguendo la classificazione di Osborne, il 15-20% delle proteine sono rappresentate dalla albumine e globuline, mentre il restante 80-85%, è costituito da prolamine e gluteline, rispettivamente gliadine, 30-40%, e glutenine, 40-50%. Quindi, e a differenza delle prolamine e gluteline degli altri cereali, gliadine e gluteine sono presenti in quantità simili, circa il 40%.
Proteine del Grano
Gliadine e glutenine hanno una notevole importanza dal punto di vista tecnologico. Perché?
Le proteine appartenenti alle due classi sono insolubili in acqua, e nell’impasto, dunque in un ambiente ricco d’acqua, si legano tra loro attraverso legami quali:
legami covalenti, ossia ponti disolfuro;
legami non covalenti, quali interazioni idrofobiche, forze di van der Waals, legami idrogeno e legami ionici.
Grazie alla formazione di questi legami intermolecolari, si crea un reticolo tridimensionale, che intrappola i granuli di amido e le bolle di anidride carbonica che si formano durante la lievitazione, e conferisce resistenza ed elasticità all’impasto di farina e acqua, due proprietà del glutine ampiamente sfruttate industrialmente.
Nella dieta abituale della popolazione europea adulta, e in particolare di quella italiana che è molto ricca di derivati del frumento, gliadine e glutenine sono le proteine maggiormente rappresentate, circa 15 g al giorno. Che significa? Che la dieta gluten-free è una dieta che impegna sia sotto l’aspetto psicologico che sociale la persona affetta da celiachia.
Nota: i lipidi componenti il glutine sono strettamente legati alle zone idrofobiche di gliadine e glutenine e, rispetto a quanto è possibile fare con la farina originale, sono rimossi con maggiore difficoltà (il contenuto in lipidi del glutine dipende dal contenuto in lipidi della farina da cui è stato ottenuto).
Gliadine: estensibilità e viscosità
Le gliadine sono prolamine idrofobiche monomeriche, cioè formate da una sola subunità, di natura globulare e con basso peso molecolare. Sulla base della mobilità elettroforetica in condizioni di basso pH, sono state suddivise nei seguenti gruppi:
alfa/beta, e gamma, ricche di zolfo, contenendo residui di cisteina, coinvolti nella formazione di ponti disolfuro intramolecolari, e di metionina;
omega, povere di zolfo, data l’assenza o quasi di cisteina e metionina.
Hanno uno scarso valore nutrizionale e un’altissima tossicità per il celiaco per la presenza di particolari sequenza aminoacidiche nella struttura primaria, in particolare prolina-serina-glutammina-glutammina e glutammina-glutammina-glutammina-prolina.
Le gliadine si associano tra di loro e con le glutenine attraverso legami non covalenti; grazie a ciò, nella formazione dell’impasto agiscono come “plasticizzanti”. Infatti, a loro si deve la viscosità e l’estensibilità proprie del glutine, il cui reticolo tridimensionale proteico si può deformare permettendo l’aumento di volume della massa a seguito della produzione di gas con la lievitazione. Questa proprietà è importante nella panificazione.
Un loro eccesso comporta la formazione di un impasto assai estensibile.
Glutenine: elasticità e tenacità
Le glutenine sono proteine polimeriche, ossia formate da più subunità, di natura filamentosa legate insieme da ponti disolfuro intermolecolari. Dopo riduzione dei suddetti legami, tramite SDS-PAGE possono essere suddivise in due gruppi.
Glutenine a elevato peso molecolare o HMW, acronimo dell’inglese high molecolar weight.
Povere di zolfo, rappresentano circa il 12% del totale delle proteine del glutine. I legami non covalenti che si stabiliscono tra le subunità di questo gruppo sono responsabili dell’elasticità e tenacità delle network di proteine del glutine, ossia delle proprietà viscoelastiche del glutine stesso e quindi anche dell’impasto che lo contiene.
Glutenine a basso peso molecolare o LMW, acronimo dell’inglese low molecolar weight.
Ricche di zolfo (cisteina), formano ponti disolfuro tra di loro e con le subunità HMW, formando così un macropolimero di glutenina.
Le glutenine fanno si che l’impasto mantenga la sua forma durante gli stress meccanici, come l’impastamento, e non meccanici, come l’aumento di volume dovuto alla lievitazione e all’aumento di volume dei gas che intrappola in conseguenza del riscaldamento dovuto alla cottura, cui è sottoposto. Questa proprietà è importante nella pastificazione.
Se in eccesso, le glutenine portano alla formazione di un impasto forte e rigido.
Proprietà del glutine di frumento
Dal punto di vista nutrizionale le proteine che compongono il glutine non hanno un elevato valore biologico, essendo povere di lisina, un aminoacido essenziale. Dunque una dieta senza glutine non comporta alcuna carenza significativa di nutrienti importanti.
Di contro, il glutine ha un grande valore per l’industria alimentare: la matrice proteica tridimensionale derivante dalla combinazione in soluzione acquosa di gliadine e glutenine, conferisce proprietà viscoelastiche, ossia di estensibilità-viscosità ed elasticità-tenacità, all’impasto di cui fa parte, e quindi una struttura ben definita al pane, alla pasta, e in generale a tutti gli alimenti che si fanno con la farina di frumento.
Ha un alto grado di palatabilità.
Ha un elevato potere fermentante a livello dell’intestino tenue.
E’ un’esorfina: alcuni peptidi derivati dalla digestione delle proteine del glutine possono andare ad agire a livello del sistema nervoso centrale.
Cereali senza glutine
Di seguito una lista di cereali, cereali minori, e pseudocereali gluten-free utilizzati a fini alimentari.
Cereali
Mais o granturco (Zea mais)
Riso (Oryza sativa)
Cereali minori
Definiti “minori” non perché di scarsa importanza nutrizionale, quanto perché coltivati in piccole aree e in quantità inferiori rispetto a frumento, riso e mais.
Fonio (Digitaria exilis)
Miglio (Panicum miliaceum)
Panico (Panicum italicum)
Sorgo (Sorghum vulgare)
Teff (Eragrostis tef)
Teosinte; gruppo composto da 4 specie appartenenti al genere Zea. Sono piante che crescono in Messico (Sierra Madre), Guatemala e Venezuela.
Pseudocereali
Così definiti perché associano nella loro botanica e nei loro aspetti nutrizionali caratteristiche peculiari dei cereali e dei legumi, quindi di un’altra famiglia di piante.
Grano saraceno (Fagopyrum esculentum)
Quinoa (Chenopodium quinoa), uno pseudocereale con ottime proprietà nutritive, contenendo fibre, ferro, zinco e magnesio, che fa parte della famiglia delle Chenopodiaceee, come le barbabietole.
Manioca, anche nota come tapioca, yuca e cassava (Manihot utilissima). Coltivata principalmente nel sud del Sahara e nell’America del Sud, è una radice tubero commestibile da cui si origina la fecola di tapioca.
Nota: non sempre i prodotti naturalmente privi di glutine al momento della commercializzazione sono effettivamente gluten-free. Infatti per gli alimenti preparati a livello industriale ci può essere o l’utilizzo di derivati contenenti gliadina o una contaminazione nella filiera produttiva, e questo è ovviamente importante in quanto anche tracce di glutine nella dieta possono causare problemi al celiaco.
Avena e glutine
Discorso a parte merita l’avena (Avena sativa), che è tra i cereali concessi ai celiaci. Studi condotti negli ultimi anni hanno evidenziato che è tollerata dal celiaco, adulto e bambino, anche nel soggetto con dermatite erpetiforme. Ovviamente, deve essere certificata per l’assenza di glutine da contaminazione.
Bibliografia
Beccari J.B. De Frumento. De bononiensi scientiarum et artium instituto atque Academia Commentarii, II. 1745:Part I.,122-127
Bender D.A. “Benders’ dictionary of nutrition and food technology”. 8th Edition. Woodhead Publishing. Oxford, 2006
Berdanier C.D., Dwyer J., Feldman E.B. Handbook of nutrition and food. 2th Edition. CRC Press. Taylor & Francis Group, 2007
Phillips G.O., Williams P.A. Handbook of food proteins. 1th Edition. Woodhead Publishing, 2011
Shewry P.R. and Halford N.G. Cereal seed storage proteins: structures, properties and role in grain utilization. J Exp Bot 2002:53(370);947-958. doi:10.1093/jexbot/53.370.947
Yildiz F. Advances in food biochemistry. CRC Press, 2009
L’olio di oliva, che si ottiene dalla spremitura delle olive o drupe, il frutto della pianta dell’olivo (Olea europaea), è la principale fonte di lipidi della dieta mediterranea e un’ottima fonte di polifenoli.
I polifenoli, molecole con proprietà antiossidanti, sono presenti nella polpa dell’oliva e, a seguito della spremitura, passano nell’olio.
La concentrazione dei polifenoli dell’olio di oliva è il risultato di una complessa interazione tra vari fattori, sia ambientali che legati al processo di estrazione dell’olio stesso, quali:
il luogo di coltivazione;
il cultivar (la varietà);
il grado di maturazione delle olive al momento del raccolto.
Il loro livello di solito si riduce con l’eccessiva maturazione delle drupe, anche se ci sono delle eccezioni a queste regole. Ad esempio le olive coltivate nei climi più caldi, a dispetto della loro maturazione più veloce, producono oli più ricchi in polifenoli.
il clima;
il processo di estrazione. A questo riguardo c’è da sottolineare il fatto che nell’olio d’oliva raffinato il contenuto in polifenoli non è significativo.
Ogni variazione nella concentrazione dei differenti polifenoli dell’olio influenza il gusto, le proprietà nutrizionali e la stabilità dell’olio di oliva stesso. Ad esempio, l’idrossitirosolo e l’oleuropeina, conferiscono all’olio extravergine di oliva un sapore pungente e amaro.
Tra i polifenoli dell’olio d’oliva sono presenti sia molecole con struttura semplice, come gli acidi fenolici e gli alcool fenolici, che complessa, quali i flavonoidi, i secoiridoidi e i lignani.
Flavonoidi
I flavonoidi comprendono glicosidi dei flavonoli (rutina o quercetina-3-rutinoside), dei flavoni (luteolina-7-glicoside), e degli antociani (glicosidi della delfinidina).
Acidi fenolici e alcol fenolici
Tra gli acidi fenolici, i primi polifenoli con struttura semplice a essere osservati nell’olio d’oliva, si ritrovano:
gli acidi idrossibenzoici, come l’acido gallico, l’acido protocatecuico, e l’acido 4-idrossibenzoico (tutti con struttura C6-C1);
gli acidi idrossicinnamici, come gli acidi caffeico, vanillico, siringico, p-cumarico e o-cumarico (tutti con struttura C6-C3).
Tra gli alcoli fenolici, i più abbondanti sono l’idrossitirosolo o 3,4-diidrossifeniletanolo, e il tirosolo o 2-(4-idrossifenil)-etanolo.
Idrossitirosolo
L’idrossitirosolo può essere presente sia libero che esterificato con l’acido elenoico a dare oleuropeina e il suo aglicone, sia come componente della molecola verbascoside. Inoltre si può ritrovare in diverse forme glicosidiche, a seconda del gruppo ossidrile cui si va a legare il glucoside.
Idrossitirosolo
E’ uno dei principali composti fenolici presente nelle olive, nell’olio extravergine di oliva e nelle acque di vegetazione.
In natura, la sua concentrazione, come quella del tirosolo, aumenta durante la maturazione del frutto, in parallelo con l’idrolisi di composti con peso molecolare più elevato, mentre il contenuto totale dei composti fenolici e dell’alfa-tocoferolo diminuisce. Può quindi essere considerato come un indicatore del grado di maturazione delle olive.
Nell’olio extravergine di oliva fresco l’idrossitirosolo per la maggior parte si trova impegnato in un legame in forma esterifica, mentre con il passare del tempo, grazie a reazioni di idrolisi, la forma non esterificata diventa quella prevalente.
Infine, la sua concentrazione si correla con la stabilità dell’olio.
Secoiridoidi
Sono i polifenoli dell’olio d’oliva con struttura più complessa, e sono il prodotto del metabolismo secondario dei terpeni.
Sono composti che legano uno zucchero e sono caratterizzati dalla presenza nella loro struttura di acido elenolico (sia nella sua forma glucosidica che agliconica), la molecola comune ai glicosidi secoiroidi della famiglia delle Oleaceae.
A differenza dei tocoferoli, flavonoidi, e acidi e alcol fenolici che di ritrovano in molta frutta e verdura appartenente a famiglie botaniche differenti, i secoiridoidi sono presenti soltanto nelle piante della famiglia delle Oleaceae.
I principali secoiridoidi sono l’oleuropeina, la demetiloleuropeina, il ligstroside e la nuzenide.
In particolare, l’oleuropeina e la demetiloleuropeina, come la verbascoside, sono abbondanti nella polpa, ma si ritrovano anche nelle altre parti del frutto. La nuzenide è presente solo nei semi.
Oleuropeina
L’oleuropeina, il secoiridoide più importante e il principale tra i polifenoli dell’olio d’oliva, è l’estere tra l’idrossitirosolo e l’acido elenoico.
Oleuropeina
E’ presente in quantità molto elevate nelle foglie dell’olivo, come anche in tutte le parti del frutto, buccia, polpa e nocciolo compreso.
Si accumula nell’oliva durante la fase di crescita, sino a raggiungere il 14% del peso netto; quando il frutto diventa più verde, la sua quantità si riduce. Infine, quando la drupa vira verso il marrone scuro, colore dovuto alla presenza di antociani, la riduzione nella concentrazione della oleuropeina diventa più evidente. E’ stato inoltre dimostrato che nelle cultivar verdi il suo contenuto è maggiore rispetto alle cultivar nere.
Nel corso della riduzione dei livelli di oleuropeina e di altri secoiridoidi, è possibile osservare un aumento di composti come i flavonoidi, i verbascosidi, e i fenoli semplici. La riduzione del suo contenuto è accompagnata anche da un aumento dei suoi prodotti secondari glicosilati, che raggiungono i valori massimi nelle olive nere.
Lignani
Un altro gruppo di polifenoli dell’olio d’oliva sono i lignani, in particolare (+)-1-acetossipinoresinolo e (+)-pinoresinolo.
Il (+)-pinoresinolo è un composto comune della frazione lignana di diverse piante come il sesamo (Sesamun indicum) e i semi della specie Forsithia, appartenente alla famiglia delle Oleaceae. E’stato ritrovato anche nel nocciolo delle olive.
Il (+)-1-acetossipinoresinolo e (+)-1-idrossipinoresinolo, e i loro glicosidi, sono stati ritrovati nella corteccia dell’oliva (Olea europeae).
Lignani
I lignani non sono presenti nel pericarpo delle drupe, ne nei rametti e foglie che possono accidentalmente essere spremuti insieme alle olive. Pertanto, come riescano a passare nell’olio divenendone una delle frazioni fenoliche più importanti non è ancora noto.
(+)-1-acetossipinoresinolo e (+)-pinoresinolo non sono presenti negli oli di semi, e sono virtualmente assenti dagli oli di oliva vergini raffinati, mentre nell’olio extravergine di oliva possono raggiungere una concentrazione di 100 mg/kg.
Come per i fenoli semplici e i secoiridoidi, esiste una notevole variazione nella loro concentrazione tra gli oli di oliva di varia origine, variabilità probabilmente legata alle differenze tra le zone di produzione, clima, varietà di olive e tecniche di produzione dell’olio.
Bibliografia
Cicerale S., Lucas L. and Keast R. Biological activities of phenolic compounds present in virgin olive oil. Int. J. Mol. Sci. 2010;11: 458-479. doi:10.3390/ijms11020458
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
Manach C., Scalbert A., Morand C., Rémésy C., and Jime´nez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr 2004;79(5):727-747. doi:10.1093/ajcn/79.5.727
Owen R.W., Mier W., Giacosa A., Hull W.E., Spiegelhalder B. and Bartsch H. Identification of lignans as major components in the phenolic fraction. Clin Chem 2000;46:976-988.
Tripoli E., Giammanco M., Tabacchi G., Di Majo D., Giammanco S. and La Guardia M. The phenolic compounds of olive oil: structure, biological activity and beneficial effects on human health. Nutr Res Rev 2005:18;98-112. doi:10.1079/NRR200495
Nella corsa il runner va incontro a un consumo calorico e a una perdita di acqua e sali minerali che dipendono da diversi fattori quali la tecnica, il grado di allenamento, le condizioni ambientali e le caratteristiche fisiologiche proprie di ciascun atleta. Ne consegue che la loro conoscenza permette di impostare un’alimentazione adeguata sia nella fase di allenamento che in quella di recupero tra un allenamento e il successivo, con l’obbiettivo di ottimizzare la performance.
Di seguito verrà analizzato il consumo calorico del runner impegnato in allenamenti su diverse distanze, e, nel dettaglio la quantità di carboidrati, lipidi, e proteine ossidati per ricavare l’energia necessaria a sostenere il lavoro muscolare, e quali sono i sali minerali maggiormente persi con la sudorazione.
Nella corsa il consumo calorico è pari a 0,85-1,05 kcal per kg di peso corporeo al chilometro.
Il range è conseguenza del fatto che gli atleti dotati di un’ottima tecnica consumano meno energia rispetto a quelli con una tecnica meno raffinata.
Un runner di 70 kg avrà un consumo calorico al chilometro compreso tra:
70 x 0,85 x 1 = 59,5 kcal
e
70 x 1,05 x 1 = 73,5 kcal
Nella tabella sono mostrati i calcoli per derivare la spesa calorica sostenuta dall’atleta per correre 10, 20, 30, e 40 km.
Distanza
Consumo calorico
10 km
0,85 x 70 x 10 = 595 kcal
1,05 x 70 x 10 = 735 kcal
20 km
0,85 x 70 x 20 = 1190 kcal
1,05 x 70 x 20 = 1470 kcal
30 km
0,85 x 70 x 30 = 1785 kcal
1,05 x 70 x 30 = 2205 kcal
40 km
0.85 x 70 x 40 = 2380 kcal
1.05 x 70 x 40 = 2940 kcal
Nota: chi ha cominciato a correre da poco avrà spese maggiori di 1,05 Kcal per kg di peso corporeo al chilometro.
L’energia richiesta per sostenere il lavoro muscolare nella corsa deriva dall’ossidazione di carboidrati, lipidi, e proteine. Carboidrati e lipidi rappresentano la principale fonte di energia, e il loro utilizzo è influenzato dall’intensità dell’esercizio: al suo aumentare si riduce la percentuale dei lipidi ossidati mentre aumenta quella dei carboidrati, come riassunto di seguito.
Intensità
Carburante
<30% VO2max
Principalmente grassi
40-60% VO2max
Grassi e carboidrati egualmente
75% VO2max
Principalmente carboidrati
>80% VO2max
Circa 100% carboidrati
Nota: L’impossibilità di utilizzare il carburante metabolico adeguato può promuovere la fatica e portare al sovrallenamento.
Quindi per andature maggiori della soglia anerobica, l’ossidazione dei carboidrati può arrivare a soddisfare l’intera richiesta energetica. Nell’andatura tipica della maratona, i carboidrati forniscono il 60-70% dell’energia necessaria, mentre per andature inferiori la percentuale si riduce a valori inferiori al 50%.
Di seguito verrà analizzato il consumo di carboidrati, lipidi e proteine nell’allenamento, nel corso del quale il dispendio energetico è sostenuto per circa il 60% dai carboidrati, per circa il 40% dai lipidi, mentre la percentuale residua, compresa tra il 3% e il 5%, dalle proteine
Ossidazione dei carboidrati nell’allenamento
Per un runner di 70 kg, il consumo di carboidrati al chilometro sarà compreso tra:
(0,6 x 59,5) / 4 = 8,9 g/km
e
(0,6 x 73,5) / 4 = 11 g/km
Nota: un grammo di carboidrati apporta circa 4 kcal.
Nella tabella sono mostrati i calcoli per derivare il consumo di carboidrati per correre 10, 20, 30, e 40 km.
Distanza
Consumo di carboidrati
10 km
[(0,85 x 70 x 10) x 0,6] / 4 = 89 g
[(1,05 x 70 x 10) x 0,6] / 4 = 110 g
20 km
[(0,85 x 70 x 20) x 0,6] / 4 = 179 g
[(1,05 x 70 x 20) x 0,6] / 4 = 221 g
30 km
[(0,85 x 70 x 30) x 0,6] / 4 = 268 g
[(1,05 x 70 x 30) x 0,6] / 4 = 331 g
40 km
[(0,85 x 70 x 40) x 0,6] / 4 = 357 g
[(1,05 x 70 x 40) x 0,6] / 4 = 441 g
Ossidazione dei lipidi nell’allenamento
Con calcoli simili a quelli fatti per i carboidrati è possibile derivare il consumo di lipidi al chilometro, che sarà compreso tra:
(0,4 x 59,5)/9 = 2,6 g/km
e
(0,4 x 73,5)/9 = 3,3 g/km
Nota: un grammo di lipidi apporta circa 9 kcal.
Nella tabella sono mostrati i calcoli per derivare il consumo di lipidi per correre 10, 20, 30, e 40 km.
Distanza
Consumo di lipidi
10 km
[(0,85 x 70 x 10) x 0,4] / 9 = 26 g
[(1,05 x 70 x 10) x 0,4] / 9 = 33 g
20 km
[(0,85 x 70 x 20) x 0,4] / 9 = 53 g
[(1,05 x 70 x 20) x 0,4] / 9 = 65 g
30 km
[(0,85 x 70 x 30) x 0,4] / 9 = 79 g
[(1,05 x 70 x 30) x 0,4] / 9 = 98 g
40 km
[(0,85 x 70 x 40) x 0,4] / 9 = 106 g
[(1,05 x 70 x 40) x 0,4] / 9 = 131 g
Ossidazione delle proteine nell’allenamento
Il fabbisogno proteico giornaliero di un soggetto adulto è pari a 0,9 grammi per kg di peso corporeo, e, per un atleta di 70 kg corrisponde a:
70 x 0,9 = 63 g
Durante la corsa circa il 3-5% dell’energia consumata per sostenere il lavoro muscolare deriva dall’ossidazione delle proteine.
Nella tabella sono mostrati i calcoli per derivare la quantità di proteine ossidate quando l’atleta corre per 10, 20, 30 e 40 km, considerando che le proteine forniscano il 3% dell’energia necessaria..
Distanza
Consumo di proteine al 3%
10 km
[(0,85 x 70 x 10) x 0,03)] / 4 = 4,5 g
[(1,05 x 70 x 10) x 0,03)] / 4 = 5,5 g
20 km
[(0,85 x 70 x 20) x 0,03)] / 4 = 8,9 g
[(1,05 x 70 x 20) x 0,03)] / 4 = 11 g
30 km
[(0,85 x 70 x 30) x 0,03)] / 4 = 13,4 g
[(1,05 x 70 x 30) x 0,03)] / 4 = 16,5 g
40 km
[(0,85 x 70 x 40) x 0,03)] / 4 = 17,9 g
[(1,05 x 70 x 40) x 0,03)] / 4 = 22,1 g
Nota: un grammo di proteine apporta circa 4 kcal.
Considerando il dispendio energetico di 0,85 e 1,05 kcal per kg di peso corporeo al chilometro, il fabbisogno proteico medio aggiuntivo per chilo di peso corporeo per correre 10, 20, 30, e 40 chilometri, approssimato alla seconda cifra decimale, è pari a:
10 km: [(4,5 + 5,5) / 2] / 70 = 0,07 g
20 km: [(4,5 + 5,5) / 2] / 70 = 0,14 g
30 km: [(4,5 + 5,5) / 2] / 70 = 0,21 g
40 km: [(4,5 + 5,5) / 2] / 70 = 0,29 g
Infine, considerando anche il fabbisogno proteico giornaliero del soggetto adulto si ottiene il fabbisogno proteico complessivo di un atleta di 70 kg impegnato nelle quattro distanze:
10 km: 0,07 + 0,9 = 0,97 g
20 km: 0,14 + 0,9 = 1,04 g
30 km: 0,21 + 0,9 = 1,11 g
40 km: 0,29 + 0,9 = 1,19 g
Con calcoli analoghi ai precedenti si ottiene il fabbisogno proteico complessivo nel caso di un’ossidazione delle proteine pari al 5%.
10 km: 0,12 + 0,9 = 1,02 g
20 km: 0,24 + 0,9 = 1,14 g
30 km: 0,36 + 0,9 = 1,26 g
40 km: 0,48 + 0,9 = 1,38 g
Se si escludono gli atleti che si allenano tutti i giorni per 30 km o più, i valori ottenuti sono di poco superiori a 0,9 grammi per kg di peso corporeo.
In realtà il fabbisogno proteico giornaliero è leggermente superiore a quello calcolato in quanto una certa quantità di azoto, dunque di proteine si perde, oltre che con le urine, anche attraverso la sudorazione.
Perdita di acqua e sali minerali nella corsa
Le perdite di acqua dipendono dalla quantità di sudore che l’atleta produce, che a sua volta dipendente da:
temperatura e umidità dell’aria;
irraggiamento solare.
La perdita sarà tanto maggiore quanto più alti sono questi valori.
Va comunque sottolineato che il sudore è prodotto in quantità diverse da soggetto a soggetto.
Con la sudorazione i principali sali minerali persi sono:
il sodio e il cloro, circa 1 grammo per litro di sudore nell’atleta abituato ad allenarsi in condizioni ambientali che provocano una intensa sudorazione;
il potassio, in quantità corrispondente a circa il 15% del sodio perso;
il magnesio in quantità corrispondente a circa l’1% del sodio perso.
La quantità di sali persi è funzione del volume di sudore prodotto e aumenta negli atleti non acclimatati al caldo.
Nella tabella sono mostrati i valori, espressi in grammi/litro, dei minerali presenti nel sudore di atleti non acclimatati e acclimatati.
Atleti non acclimatati
Atleti acclimatati
Sodio
1,38
0,92
Cloro
1,5
1,00
Potassio
0,20
0,15
Magnesio
0,01
<0,01
Totale
3,09
2,08
Da quanto detto deriva che nel corso dell’attività fisica il minerale più utile da assumere è il sodio.
Dopo l’attività fisica il corridore, o chi suda molto, tende a mangiare più salato. Il fenomeno, scoperto nel corso di studi condotti su operai di fonderie, è conosciuto come fame selettiva. Per il potassio e il magnesio probabilmente non esiste la fame selettiva.
Bibliografia
Sawka M.N., Burke L.M., Eichner E.R., Maughan, R.J., Montain S.J., Stachenfeld N.S. American College of Sports Medicine position stand: exercise and fluid replacement. Med Sci Sport Exercise 2007;39(2):377-390. doi:10.1249/mss.0b013e31802ca597
Shirreffs S., Sawka M.N. Fluid and electrolyte needs for training, competition and recovery. J Sport Sci 2011;29:sup1, S39-S46. doi:10.1080/02640414.2011.614269
L’alfa-amilasi (EC 3.2.1.1) è un enzima che catalizza l’idrolisi di legami glicosidici α-(1→4) presenti all’interno di catene lineari di amilosio e amilopectina, i due principali costituenti dei granuli di amido.[2]
L’enzima è ampiamente diffuso negli animali, piante, funghi, nei phyla Ascomiceti e Basidiomiceti, batteri, nel genere Bacillus, e negli Archea. La sua ampia distribuzione suggerisce un suo ruolo centrale nel metabolismo dei carboidrati.[15]
Nell’uomo l’alfa-amilasi è codificata da due geni differenti, ed è prodotta principalmente delle ghiandole parotidi e dal pancreas esocrino.[9] Nelle piante sei differenti geni codificano per altrettante isoforme dell’enzima.[11]
Negli animali è coinvolta nel primo passaggio della digestione dell’amido, con liberazione del disaccaride maltosio, degli oligosaccaridi maltotriosio e destrine alfa-limite, e di piccole quantità di glucosio.[5]
Poiché l’amido è la principale fonte di alimentare di carboidrati, e quindi di glucosio ed energia, l’attività catalitica dell’alfa-amilasi può influenzare la regolazione della glicemia postprandiale.[8]
L’alfa-amilasi è utilizzata in diversi settori industriali, nonché nella diagnostica clinica.[7]
Nelle piante, e in particolare nelle graminacee quali grano, orzo e riso, è cruciale per la germinazione dei semi e la maturazione dei chicchi.[11][17]
La prima rilevazione di un’attività amilasica risale al 1811, a opera del chimico russo Konstantin Sigizmundovič Kirchhoff, che, lavorando con il grano, scoprì la prima attività enzimatica capace di degradare l’amido del cereale.[16]
A questo lavoro seguirono, nel 1831, gli studi Erhard Friedrich Leuchs che osservò la degradazione dell’amido quando veniva miscelato con la saliva umana. Leuchs chiamò ptialina l’agente responsabile di questa reazione chimica.
Nel 1833, Anselme Payen e Jean-François Persoz, chimici francesi che lavoravano in uno zuccherificio, isolarono dall’orzo un enzima in grado di degradare l’amido, enzima che fu chiamato distasi.[5]
Il nome di alfa-amilasi si deve a Richard Kuhn, che nel 1925 scoprì che i prodotti dell’idrolisi catalizzata dall’enzima erano in configurazione alfa.[13]
L’alfa-amilasi fu anche il primo enzima a essere prodotto industrialmente, nel 1894, estratto da un fungo e utilizzato per il trattamento di disordini digestivi. In seguito, a partire dal 1917, venne estratto anche da Bacillus subtilis e Bacillus mesentericus. Attualmente la maggior parte viene prodotta da organismi geneticamente modificati.[15]
L’alfa-amilasi, e più in generale le amilasi, di cui ne esistono almeno altri due tipi, la beta-amilasi (EC 3.2.1.2) e la gamma-amilasi (EC 3.2.1.3), trovano largo impiego in settori industriali differenti, come quello alimentare, dei mangimi, il settore tessile, la produzione delle birra e del whisky, l’industria farmaceutica, cosmetica e la diagnostica clinica.[7]
Le alfa-amilasi possono essere classificate, sulla base del grado d’idrolisi del substrato, come liquefanti, se idrolizzano il 30-40% dei legami glicosidici dell’amido, e saccarificanti, se l’idrolisi arriva al 50-60%.[15]
Geni dell’alfa-amilasi umana
In molti mammiferi, compreso l’uomo, l’alfa-amilasi è codificata da due geni distinti ma strettamente correlati, presenti sul braccio corto del cromosoma 1, e indicati come:
AMY1, che codifica per l’isoforma presente nella saliva, nella ghiandola mammaria, e in alcuni tipi di tumore, come quello del polmone;[5]
AMY2, che codifica per l’enzima prodotto dal pancreas. AMY2 è presente in due forme, AMY2A e AMY2B, il cui numero di copie per gene differisce nelle diverse popolazioni, sebbene due copie per gene sembra essere un pattern frequente. Sembra anche che il numero di copie di AMY2 sia associato a quello di AMY1.[9]
Azione catalitica
L’alfa-amilasi catalizza il primo passaggio nella digestione dell’amido, vale a dire l’idrolisi dei legami glicosidici α-(1→4) presenti all’interno delle catene lineari di amilosio e amilopectina, agendo su posizioni casuali lungo gli α-glucani. L’enzima è quindi una endoglicosidasi, ha un pH ottimale di circa 7, richiede la presenza di ioni calcio, e porta al rilascio di disaccaridi e oligosaccaridi in configurazione alfa, nello specifico, maltosio e maltotriosio dall’amilosio, e maltosio, destrine alfa-limite e glucosio, quest’ultimo in minima quantità, dall’amilopectina.[2]
Alfa-amilasi
L’alfa-amilasi, al pari di altri enzimi coinvolti nel metabolismo dell’amido, come la pullulanasi (EC 3.2.1.41), la isoamilasi (EC 3.2.1.68), gli enzimi ramificanti l’amido (EC 2.4.1.18) e gli enzimi deramificanti l’amido, appartiene alla famiglia delle glicosil idrolasi 13 o GH-13, enzimi in grado di agire su substrati contenenti legami α-glicosidici.[10]
A questa famiglia appartengono anche due enzimi coinvolti nel metabolismo del trealosio, la trealosio sintasi (EC 5.4.99.16), che catalizza l’interconversione tra maltosio e trealosio, e la trealosio-6-fosfato idrolasi (EC 3.2.1.93), che idrolizza il trealosio a dare una molecola di glucosio e una di glucosio-6-fosfato.
Alfa-amilasi salivare
L’alfa-amilasi salivare o ptialina è sintetizzata principalmente dalle ghiandole parotidi, e in misura minore dalle ghiandole sottomandibolari.
Presente in due forme, una glicosilata e una non glicosilata, è coinvolta nella prima, limitata, digestione dell’amido che avviene nella cavità orale.
L’enzima è una delle proteine salivari più abbondanti, rappresentando il 40-50% di tutte quelle presenti, e, con la lipasi linguale (EC 3.1.1.3), coinvolta nella digestione dei lipidi, quali i trigliceridi e gli esteri del colesterolo, è uno dei più importanti enzimi salivari.[3]
Alfa-amilasi pancreatica
L’alfa-amilasi pancreatica è prodotta dalle cellule acinose del pancreas.
E’ una delle principali proteine del succo pancreatico, e con esso viene secreta nel duodeno dove attacca amilosio e amilopectina, portando a termine la prima parte dell’amilolisi. I prodotti dell’idrolisi sono quindi attaccati dalle alfa-glucosidasi dell’orletto a spazzola degli enterociti, ossia la saccarasi-isomaltasi (EC 3.2.1.48) e la maltasi-glucoamilasi (EC 3.2.1.20), enzimi che idrolizzano anche il legame glicosidico α-(1→2) del saccarosio.[14]
L’alfa-amilasi pancreatica dei mammiferi è in grado di legarsi a N-glicani di glicoproteine dell’orletto a spazzola.[1] Questo legame consente la cooperazione con la saccarasi-isomaltasi, il che determina un notevole aumento del rilascio di glucosio. Tuttavia è stato osservato che l’uptake del glucosio da parte di SGLT1, uno dei trasportatori intestinali coinvolti nell’assorbimento dei monosaccaridi, è inibito da elevate concentrazioni intestinali di alfa-amilasi. E’ stato ipotizzato che questo processo possa rappresentare un meccanismo di regolazione innata per evitare improvvisi aumenti dell’assorbimento del glucosio, e quindi facilitare la regolazione della glicemia postprandiale.[6]
Controllo della glicemia
Il controllo della glicemia postprandiale è ritenuto una valida strategia nella prevenzione dello sviluppo del diabete di tipo II. Tra le metodiche non farmacologiche che è possibile adottare c’è la riduzione della capacità dell’alfa-amilasi di attaccare l’amido attraverso una lavorazione degli alimenti che porti alla retrogradazione dell’amido stesso, o a mezzo di molecole presenti nei cibi che, con diverse modalità d’azione, ne riducano l’attività.[8]
Lavorazione dei cibi
I processi idrotermici cui i cibi ricchi di amido sono sottoposti, ad esempio la cottura in acqua bollente, fanno si che i granuli di amido vadano incontro al fenomeno della gelatinizzazione, ossia assorbano acqua, si rigonfino e perdano buona parte delle loro strutture cristalline, sostituite da strutture amorfe, più facilmente attaccabili dalle alfa-amilasi.[4] Se l’amido gelatinizzato viene fatto raffreddare si può verificare una ricristallizzazione che porta alla formazione di quello che è conosciuto come amido retrogradato, un tipo di amido resistente all’azione dell’alfa-amilasi e che è in grado di raggiungere praticamente intatto il colon, dove viene fermentato dai batteri del microbiota intestinale, al pari della fibra solubile.
Ruolo dei polifenoli
La digestione dell’amido da parte dell’alfa-amilasi è influenzata da molecole presenti negli alimenti, quali i polisaccaridi non amidacei della parete cellulare, proteine, lipidi, e, tra i fitochimici, diversi tipi di polifenoli.[4] Questi composti possono limitare l’azione dell’enzima in almeno quattro modi differenti.[8]
Le proteine e i polisaccaridi non amidacei sono in grado di limitare la perdita della struttura ordinata dell’amido nativo nel corso dei processi idrotermici cui è sottoposto il cibo durante la lavorazione.
I lipidi, i polifenoli, le proteine e i polisaccaridi non amidacei possono formare strutture ordinate con l’amido.
Le proteine, i polisaccaridi non amidacei e i polifenoli possono formare barriere fisiche che riducono l’accesso degli enzimi idrolitici alle catene degli alfa-glucani.
I polifenoli e i polisaccaridi non amidacei sono in grado di ridurre l’attività dell’alfa-amilasi e delle alfa-glucosidasi.
Tra i polifenoli sembrano essere importanti alcuni tipi di flavonoidi come le proantocianidine o tannini condensati.[8] Per essere efficace, l’assunzione di alimenti ricchi di polifenoli, come il tè nero o il te verde, deve essere associata al pasto ricco in carboidrati. Il glutine sembra in grado di ostacolare l’interazione tra polifenoli e granuli di amido, e dunque ridurne l’effetto inibitorio.
Dosaggio dell’amilasi sierica
Il dosaggio dell’attività sierica dell’alfa-amilasi viene utilizzato come test diagnostico di laboratorio.[7]
L’aumento dei livelli sierici dell’attività dell’enzima può derivare da:
una pancreatite acuta o una riacutizzazione di una pancreatite cronica;
un’infiammazione a carico delle ghiandole parotidi;
un’ostruzione dei dotti secretori delle ghiandole parotidi o del dotto pancreatico.
I test attualmente in uso non distinguono tra le isoforme salivare e pancreatica. Tuttavia, se l’aumento dell’attività amilasica è associato a quello dell’attività lipasica, la patologia sottostante è probabilmente di origine pancreatica.
Alfa-amilasi del grano
Nelle piante, e in particolare nel grano, l’alfa-amilasi ha un ruolo nella germinazione e nel processo di maltazione, ossia una germinazione controllata che porta alla conversione del grano in malto che viene utilizzato principalmente per la produzione di whisky e birra.[11]
Nei chicchi di grano l’enzima viene sintetizzato principalmente nelle prime fasi della germinazione, che sono eterotrofiche in quanto dipendono fortemente dalle risorse immagazzinate, e quindi anche sull’amido presente nell’endosperma.[17] La sintesi dell’enzima avviene nello strato aleuronico, e da inizio alla mobilizzazione delle riserve di amido.
L’alfa-amilasi è coinvolta anche in uno dei principali difetti del grano, la germinazione pre-raccolta.
Bibliografia
^ Asanuma-Date K., Hirano Y., Le N., Sano K., Kawasaki N., Hashii N., Hiruta Y., Nakayama K., Umemura M., Ishikawa K., Sakagami H., and Ogawa H. Functional regulation of sugar assimilation by N-glycan-specific interaction of pancreatic α-amylase with glycoproteins of duodenal brush border membrane. J Biol Chem 2012;287(27):23104-18. doi:10.1074/jbc.m111.314658
^ ab Benjamin S., Smitha R.B., Jisha V.N., Pradeep S., Sajith S., Sreedevi S., Priji P., Unni K.N., Sarath Josh M.K. A monograph on amylases from Bacillus spp. Adv biosci biotechnol 2013;4:No.2. doi:10.4236/abb.2013.42032
^ Boehlke C., Zierau O., Hannig C. Salivary amylase – The enzyme of unspecialized euryphagous animals. Arch Oral Biol 2015;60(8):1162-76. doi:10.1016/j.archoralbio.2015.05.008
^ ab Butterworth P.J., Bajka B.H., Edwards C.H., Warren F.J., Ellis P.R. Enzyme kinetic approach for mechanistic insight and predictions of in vivo starch digestibility and the glycaemic index of foods. Trends Food Sci Technol 2022;120:254-264. doi:10.1016/j.tifs.2021.11.015
^ abc Butterworth P.J., Warren F.J. and Ellis P.R. Human α-amylase and starch digestion: an interesting marriage. Starch/Stärke 2011;63:395-405. doi:10.1002/star.201000150
^ Date K., Satoh A., Lida K., Ogawa H. Pancreatic α-amylase controls glucose assimilation by duodenal retrieval through N-glycan-specific binding, endocytosis, and degradation. J Biol Chem 2015;290(28):17439-50. doi:10.1074/jbc.M114.594937
abc de Souza P.M., de Oliveira Magalhães P. Application of microbial α-amylase in industry – A review. Braz J Microbiol 2010;41(4):850-61. doi:10.1590/S1517-83822010000400004
^ abcd Gong L., Feng D., Wang T., Ren Y., Liu Y., Wang J. Inhibitors of α-amylase and α-glucosidase: potential linkage for whole cereal foods on prevention of hyperglycemia. Food Sci Nutr 2020;8(12):6320-6337. doi:10.1002/fsn3.1987
^ ab Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W, Lee C. Detection of large-scale variation in the human genome. Nature Genetics. 2004;36:949–951. doi:10.1038/ng1416
^ Janeček Š., Svensson B., MacGregor E.A. α-Amylase: an enzyme specificity found in various families of glycoside hydrolases. Cell Mol Life Sci 2014;71(7):1149-70. doi:10.1007/s00018-013-1388-z
^ abc Ju L., Pan Z., Zhang H., Li Q., Liang J., Deng G., Yu M., Long H. New insights into the origin and evolution of α-amylase genes in green plants. Sci Rep 2019;9(1):4929. doi:10.1038/s41598-019-41420-w
^ Keller P.J., Allan B.J. The protein composition of human pancreatic juice. J Biol Chem 196725;242(2):281-7. doi:10.1016/S0021-9258(19)81461-8
^ Kuhn R. Mechanism of the action of amylases. Constitution of starch. Justus Liebigs Annalen der Chemie 1925;443:1-71
^ Nichols B.L., Avery S., Sen P., Swallow D.M., Hahn D., Sterchi E. The maltase-glucoamylase gene: common ancestry to sucrase-isomaltase with complementary starch digestion activities. Proc Natl Acad Sci U S A. 2003 Feb 4;100(3):1432-7. doi:10.1073/pnas.0237170100
^ abc Tiwari S.P., Srivastava R., Singh C.S., Shukla K., Singh R.K., Singh P., Singh R., Singh N.L., and Sharma R. Amylases: an overview with special reference to alpha amylase. Journal of Global Biosciences 2015;4(SI1):1886-1901.
^ Zakowski J.J., Bruns D.E. Biochemistry of human alpha amylase isoenzymes. Crit Rev Clin Lab Sci 1985;21(4):283-322. doi:10.3109/10408368509165786
^ ab Zhang Q., Pritchard J., Mieog J., Byrne K., Colgrave M.L., Wang J.R., Ral J.F. Over-expression of a wheat late maturity alpha-amylase type 1 impact on starch properties during grain development and germination. Front Plant Sci 2022;13:811728. doi:10.3389/fpls.2022.811728
L’acido gamma-linolenico o GLA, un acido grasso essenziale appartenente al gruppo degli acidi grassi polinsaturi omega-6, come il suo precursore acido linoleico (il più abbondante acido grasso polinsaturo nell’epidermide, dove è coinvolto nel mantenimento della barriera epidermica all’acqua) svolge un ruolo importante nella fisiologia e fisiopatologia della pelle.
Studi condotti sugli esseri umani hanno rivelato che l’acido gamma-linolenico:
migliora l’umidità della pelle;
migliora la fermezza e la rugosità della pelle;
diminuisce la perdita d’acqua transepidermica (una delle anomalie della pelle negli animali sottoposti a carenza di acidi grassi essenziali).
PGE1
Utilizzando epidermide di cavia come un modello di epidermide umana (sono funzionalmente simili) è stato dimostrato che la supplementazione di animali con cibi ricchi di acido gamma-linolenico si traduce in una grande produzione nella pelle di PGE1 e 15-HETrE (come già dimostrato in a esperimenti in vitro).
Poiché queste molecole hanno entrambe proprietà anti-inflammatorie/anti-proliferative, la supplementazione con alimenti ricchi di acido gamma-linolenico può essere un’aggiunta alla terapia standard per i disturbi infiammatori/proliferativi della pelle.
Fonti principali di acido gamma-linolenico
Le principali fonti di acido gamma-linolenico sono gli oli di semi di:
ribes nero (da 15% al 19% degli acidi grassi totali);
enotera (dal 7% al 14% degli acidi grassi totali);
Pressione arteriosa e acido gamma-linolenico
La relazione tra assunzione di acidi grassi con la dieta e la pressione arteriosa provengono principalmente da studi condotti su ratti geneticamente modificati che spontaneamente sviluppano ipertensione (un modello animale comunemente usato per l’ipertensione umana.
In questi studi sono state osservate molte anomalie della membrana. Pertanto l’ipertensione nel “modello ratto” potrebbe essere correlata ai cambiamenti nel metabolismo degli acidi grassi polinsaturi a livello della membrana cellulare.
Riguardo agli acidi grassi polinsaturi, vari gruppi di ricerca hanno riportato che l’acido gamma-linolenico riduce la pressione arteriosa in ratti normali e geneticamente modificati (effetto maggiore) ed è stato ipotizzato che si verifichi attraverso l’interferenza con il sistema renina-angiotensina (che promuove la resistenza vascolare e la ritenzione renale), alterando le proprietà della membrana cellulare delle cellule muscolari lisce vascolari e quindi interferendo con l’azione dell’angiotensina II.
Un altro possibile meccanismo d’azione dell’acido gamma-linolenico per ridurre la pressione arteriosa potrebbe essere attraverso il suo metabolita acido diomo-gamma-linolenico, il quale può essere incorporato nei fosfolipidi della membrana delle cellule muscolari lisce vascolari, poi rilasciato dall’azione della fosfolipasi A2, e trasformato dalla COX-1 in PGE1 che induce il rilassamento della muscolatura liscia vascolare.
Artrite reumatoide e acido gamma-linolenico
In uno studio condotto da Leventhal e colleghi nel 1993 è stato dimostrato che l’assunzione di una elevata concentrazione di olio di borragine (circa 1400 mg/die di acido gamma-linolenico) per 24 settimane, ha portato a una riduzione clinicamente significativa dei segni e sintomi di attività dell’artrite reumatoide.
In uno studio successivo condotta da Zurier e colleghi nel 1996, l’assunzione di una dose più alta (circa 2,8 g/die di acido gamma-linolenico) per 6 mesi ha ridotto, in modo clinicamente rilevante, segni e sintomi dell’attività della malattia. I pazienti che sono rimasti per un anno sulla dose di 2,8 g/die nella dieta hanno mostrato un miglioramento continuato dei sintomi (l’uso di acido gamma-linolenico anche alla dose più alta, è ben tollerato, con minimi effetti collaterali).
Questi dati sottolineano che la quantità giornaliera e la durata dell’assunzione di acido gamma-linolenico con la dieta si correlano con l’efficacia clinica.
Bibliografia
Akoh C.C. and Min D.B. “Food lipids: chemistry, nutrition, and biotechnology” 3th ed. 2008
Chow Ching K. “Fatty acids in foods and their health implication” 3th ed. 2008
Fan Y.Y. and Chapkin R.S. Importance of dietary γ-linolenic acid in human health and nutrition. J Nutr 1998;128:1411-1414. doi:10.1093/jn/128.9.1411
Leventhal L.J., Boyce E.G. and Zurier R.B. Treatment of rheumatoid arthritis with gammalinolenic acid. Ann Intern Med 1993;119:867-873. doi:10.7326/0003-4819-119-9-199311010-00001
Miller C.C. and Ziboh V.A. Gammalinolenic acid-enriched diet alters cutaneous eicosanoids. Biochem Biophys Res Commun 1988;154:967-974. doi:10.1016/0006-291X(88)90234-3
Zurier R.B., Rossetti R.G., Jacobson E.W., DeMarco D.M., Liu N.Y., Temming J.E., White B.M. and Laposata M. Gamma-linolenic acid treatment of rheumatoid arthritis. A randomized, placebocontrolled trial. Arthritis Rheum 1996;39:1808-1817. doi:10.1002/art.1780391106
Molti studi hanno dimostrato una relazione diretta, dose-dipendente tra l’assunzione di alcool e la pressione sanguigna, in particolare per assunzioni superiori a due drink/die.
Tale relazione è indipendente da:
età;
apporto di sale;
obesità;
infine, persiste a prescindere dal tipo di bevanda.
Inoltre il forte consumo di bevande alcoliche per lunghi periodi di tempo è uno dei fattori predisponenti all’ipertensione; dal 5 al 7% dei casi della malattia sono dovuti a un eccessivo consumo di alcool.
Una meta-analisi di 15 studi clinici controllati randomizzati ha mostrato che la riduzione dell’assunzione di bevande alcoliche porta benefici terapeutici sia per gli ipertesi che i normotesi con riduzioni simili della pressione sistolica e diastolica (negli ipertesi la riduzione si verifica in poche settimane).
Assunzione di alcool e prevenzione dell’ipertensione
Le linee guida sulla prevenzione primaria dell’ipertensione raccomandano che il consumo di alcool (etanolo) nella maggior parte degli uomini, in assenza di controindicazioni, dovrebbe essere inferiore ai 28 g/die, limite entro il quale può ridurre il rischio di malattia cardiaca coronarica. Il consumo limitato a questi quantitativi deve essere ottenuto con l’assunzione di bevande a basso contenuto alcolico, preferibilmente ai pasti (bere quantità anche piccole o moderate al di fuori dei pasti aumenta la probabilità di sviluppare ipertensione): ciò significa non più di 680 ml di una birra normale o 280 ml di vino (12% vol), soprattutto in caso di ipertensione; per le donne e i soggetti esili il consumo dovrebbe essere dimezzato1.
Da evitare il consumo di bevande ad alta gradazione alcolica anche se il contenuto totale di etanolo non supera i 28 g/die.
Effetti dell’etanolo sulla pressione arteriosa
In ogni caso resta incertezza riguardo i benefici o i rischi attribuibili a un consumo di etanolo leggero/moderato sul rischio di ipertensione.
In uno studio pubblicato nell’aprile 2008 gli autori hanno esaminato l’associazione tra l’assunzione di etanolo e il rischio di sviluppare ipertensione in 28848 donne arruolate nel “Women’s Health Study” e 13455 uomini dal “Physicians’ Health Study”.
Lo studio conferma che un forte consumo di bevande alcoliche, superiore a 2 drink/die, aumenta il rischio di ipertensione sia negli uomini che nelle donne ma, sorprendentemente, ha rilevato che l’associazione tra un consumo leggero/moderato, fino a due drink/die, e il rischio di sviluppare l’ipertensione è diverso nei due sessi. Le donne hanno un rischio potenziale ridotto di ipertensione da un consumo di alcool leggero/moderato con un’associazione a forma di J2; gli uomini non hanno benefici da tale consumo ma anzi un aumento del rischio.
Tuttavia, le linee guida per la prevenzione primaria dell’ipertensione limitano il consumo di bevande alcoliche a meno di due drink/die negli uomini e meno di uno nei soggetti più esili e nelle donne.
1. Un drink standard contiene circa 14 g di etanolo, vale a dire una birra normale da 340 ml, 140 ml di vino (12% vol), o 42 ml di bevande alcoliche distillate, che sono comunque sconsigliate.
2. Molti studi hanno mostrato una relazione a forma di J tra l’assunzione di etanolo e la pressione sanguigna. I bevitori leggeri (non più di 28 g di etanolo/die) hanno una pressione più bassa rispetto agli astemi; invece chi consuma più di 28 g di etanolo/die ha una pressione più alta dei non bevitori. Quindi l’alcool potrebbe essere un vasodilatatore a basse dosi e un vasocostrittore a dosi maggiori.
Sesso H.D., Cook N.R., Buring J.E., Manson J.E. and Gaziano J.M. Alcohol consumption and the risk of hypertension in women and men. Hypertension 2008;51:1080-1087. doi:10.1161/HYPERTENSIONAHA.107.104968
Writing Group of the PREMIER Collaborative Research Group. Effects of comprehensive lifestyle modification on blood pressure control: main results of the PREMIER Clinical Trial. JAMA 2003;289:2083-2093. doi:10.1001/jama.289.16.2083
World Health Organization, International Society of Hypertension Writing Group. 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. Guidelines and recommendations. J Hyperten 2003;21:1983-1992.
Gli acidi grassi trans o grassi trans o TFA, acronimo dell’inglese trans fatty acids, sono acidi grassi insaturi, una sottoclasse di lipidi, con almeno un doppio legame in configurazione trans.
I doppi legami carbonio-carbonio hanno una conformazione planare e quindi possono essere considerati come dei piani ai cui lati opposti si attacca e prosegue la catena di atomi di carbonio. “L’ingresso” e “l’uscita” della catena può avvenire sullo stesso lato del piano, e in questo caso il doppio legame è detto in configurazione cis, o su lati opposti, e in quel caso si parla di configurazione trans. Questo è un esempio di isomeria geometrica o isomeria cis-trans.
Acidi Grassi trans, cis e saturi
Gli acidi grassi insaturi più comunemente hanno i loro doppi legami in configurazione cis; l’altra configurazione meno comune è appunto la trans.
Il legame cis determina una curvatura nella catena carboniosa dell’acido grasso, mentre la geometria del legame trans raddrizza la catena, impartendo alla molecola una struttura più simile a quella degli acidi grassi saturi.
Di seguito alcune caratteristiche distintive dei grassi a elevato tenore di acidi grassi trans, particolarmente vantaggiose per produrre margarine e grassi da pasticceria, ristorazione commerciale e per i processi di lavorazione industriale.
Le molecole curve non possono impacchettarsi facilmente mentre quelle lineari possono farlo. Ciò significa che i grassi trans concorrono, assieme ai geometricamente simili acidi grassi saturi, alla compattezza del grasso in cui si trovano conferendogli un punto di fusione più alto.
Innalzare il punto di fusione dei grassi significa che è possibile convertirli dalla forma liquida a quella semisolida o solida a temperatura ambiente.
Nota: gli acidi grassi trans tendono a essere meno solidi rispetto agli acidi grassi saturi.
Hanno, oltre a un punto di fusione, anche consistenza e gusto simili a quelli del burro.
Si conservano a lungo a temperatura ambiente.
Non vanno incontro a variazioni di sapore.
Sono stabili durante la frittura.
Origine
Gli acidi grassi trans presenti nella dieta possono essere di origine industriale, casalinga, vegetale o animale.
Nei paesi industrializzati la maggior parte dei grassi trans consumati nella dieta, negli Usa circa l’80% del totale, sono prodotti industrialmente, in quantità variabili, nel corso della parziale idrogenazione di oli alimentari ricchi di acidi grassi insaturi.
Sono prodotti a livello casalingo durante la frittura utilizzando oli vegetali contenenti acidi grassi insaturi.
Derivano dalla trasformazione batterica nel rumine dei ruminanti degli acidi grassi insaturi ingeriti dagli animali.
Un’altra fonte naturale è rappresentata da alcune piante come porri, piselli, lattuga e spinaci che contengono l’acido trans-3-esadecenoico, e l’olio di colza dove sono presenti l’acido brassidico (22:1∆13t) e l’acido gondoico (C20:1∆11t). In queste alimenti i grassi trans sono presenti in piccole quantità.
Piccolissime quantità, inferiori al 2%, si formano nel corso della deodorizzazione degli oli vegetali. Si tratta di un processo necessario nella raffinazione degli oli commestibili nel corso del quale si formano piccole quantità di acidi grassi trans con più di due doppi legami. Questi isomeri sono presenti anche nei cibi fritti e in quantità elevate in alcuni oli vegetali parzialmente idrogenati.
Acidi grassi trans industriali
L’idrogenazione è una reazione chimica nella quale l’idrogeno gassoso reagisce, in presenza di un catalizzatore, con una molecola.
L’idrogenazione degli acidi grassi insaturi consiste nell’addizione di atomi di idrogeno ai doppi legami presenti lungo la catena carboniosa degli acidi grassi. La reazione avviene in presenza di catalizzatori metallici ed è favorita dal riscaldamento degli oli vegetali in cui sono presenti i suddetti acidi grassi.
La parziale idrogenazione degli oli vegetali
Il processo di idrogenazione venne scoperto nel 1897 dal francese Paul Sabatier, premio Nobel per la chimica, insieme con il collega francese Victor Grignard, utilizzando come catalizzatore il nickel.
La parziale idrogenazione degli oli vegetali venne sviluppata nel 1903 da un chimico tedesco, Wilhelm Normann, che depositò un brevetto britannico intitolato “Process for converting unsaturated fatty acids or their glycerides into saturated compounds”. Il termine acidi grassi trans comparve per la prima volta nella Remark column della quinta edizione di “Standard Tables of Food Composition” in Giappone.
Nella parziale idrogenazione si verifica una saturazione incompleta dei doppi legami lungo la catena carboniosa dell’acido grasso insaturo. Ad esempio, per quanto riguarda l’olio di pesce il contenuto in acidi grassi trans in assenza di idrogenazione e in caso di forte idrogenazione è rispettivamente dello 0,5 e 3,6% mentre nel parziale idrogenazione è e del 30%.
Da Acido Oleico ad Acido Vaccenico
Ma, cosa più importante, alcuni dei doppi legami che restano possono cambiare posizione lungo la catena e produrre isomeri geometrici e di posizione, ossia i doppi legami possono essere modificati sia nella posizione che nella conformazione, con formazione appunto dei legami trans.
Inoltre si verificano anche le seguenti modificazioni.
Si formano monomeri ciclici e dimeri intramolecolari lineari.
Gli oli vegetali parzialmente idrogenati furono sviluppati per la produzione di grassi vegetali, un’alternativa più economica rispetto ai grassi animali. Infatti tramite questo processo, oli come quello di semi di lino, semi di cotone, germe di grano, soia, mais, girasole, sesamo, cartamo e vinacciolo, ricchi in acidi grassi insaturi, sono convertiti in grassi semisolidi.
Il primo olio idrogenato fu l’olio di semi di cotone negli USA nel 1911 per la produzione di grasso vegetale per pasticceria.
Negli anni ’30 la parziale idrogenazione divenne popolare con lo sviluppo della margarina.
Attualmente negli USA sono prodotte 2,7-3,6 milioni di tonnellate di oli idrogenati all’anno.
Acidi grassi trans dai ruminanti
Gli acidi grassi trans di origine animale sono prodotti dai batteri presenti nel rumine dei ruminanti, ad esempio mucche, pecore e capre, utilizzando come substrato una parte della relativamente piccola quantità degli acidi grassi insaturi contenuti negli alimenti di cui si nutrono, quindi mangimi, piante ed erbe.
E, considerando un animale che viva almeno un anno e abbia la possibilità di pascolare e/o mangiare fieno, esiste una variabilità stagionale che interessa gli acidi grassi insaturi assunti e i grassi trans prodotti. Infatti, in estate e primavera i pascoli possono contenere più acidi grassi insaturi rispetto ai mangimi che gli animali consumano durante l’inverno.
Si ritrovano di seguito in piccole ma significative quantità nei trigliceridi della carne e dei prodotti lattiero-caseari non magri, dove costituiscono tipicamente meno del 5% del totale degli acidi grassi. Nei trigliceridi, tali acidi grassi sono localizzati in posizione sn-1 e sn-3 della molecola, mentre nelle margarine e negli altri prodotti idrogenati di origine industriale sembrano essere concentrati in posizione sn-2.
Gli acidi grassi trans prodotti dai ruminanti sono principalmente acidi grassi monoinsaturi, con un numero di atomi di carbonio compreso tra 16 e 18, e costituiscono una piccola percentuale degli acidi grassi trans assunti con l’alimentazione (vedi sotto).
Isomeri
Sia per gli acidi grassi trans industriali che di origine animale, il gruppo più importante è rappresentato dagli isomeri trans del C18:1, ossia molecole contenenti 18 atomi di carbonio e un doppio legame trans la cui posizione varia tra il carbonio Δ6 e quello Δ16, con gli isomeri più comuni per entrambe le fonti con il doppio legame in posizione tra Δ9 e Δ11.
Tuttavia, sebbene una stessa molecola possa essere presente sia nel gruppo di derivazione industriale che animale, esiste una considerevole differenza quantitativa. Ad esempio, nel gruppo prodotto nei ruminanti, l’acido vaccenico (C18:1Δ11t) costituisce oltre il 60% degli isomeri trans-C18:1, mentre in quelli industriali l’acido elaidico (C18:1Δ9t) rappresenta il 15-20%, e il C18:1Δ10t e l’acido vaccenico oltre il 20% ciascuno.
Isomeri trans del C18:1
Effetti sulla salute
Gli acidi grassi trans che derivano dai ruminanti, nelle quantità effettivamente consumate nella dieta, non sono pericolosi per la salute umana.
Al contrario, il consumo dei grassi trans industriali non ha effetti benefici evidenti ne alcun valore intrinseco, oltre al loro contributo calorico, e, dal punto di vista della salute è solo dannoso, avendo effetti nocivi su:
livelli dei lipidi sierici;
cellule endoteliali;
processo infiammatorio;
altri fattori di rischio per le malattie cardiovascolari.
Inoltre sono associati positivamente con il rischio di cardiopatia coronarica, e di morte improvvisa per cause cardiache e diabete.
Nota: nel proseguo dell’articolo, se non diversamente specificato, ci si riferirà agli acidi grassi trans industriali, che verranno indicati semplicemente come acidi grassi trans o grassi trans.
Effetti a livello plasmatico
I livelli plasmatici del colesterolo LDL e del colesterolo HDL sono marcatori di rischio ben documentati per lo sviluppo di malattia coronarica o CHD, acronimo dell’inglese coronary heart disease:
elevati livelli di colesterolo LDL sono associati con un’aumentata incidenza di malattia cardiaca ischemica;
elevati livelli di colesterolo HDL si associano a una ridotta incidenza del rischio.
Per questo motivo il rapporto tra il livello del colesterolo totale e del colesterolo HDL è spesso usato come indicatore di rischio combinato per questi due componenti in relazione allo sviluppo della malattia cardiaca: maggiore è il rapporto, maggiore è il rischio.
I grassi trans hanno effetti nocivi sui lipidi sierici. Questi effetti sono stati valutati in numerosi studi di alimentazione controllata nei quali veniva effettuata una sostituzione isocalorica di acidi grassi saturi o cis-insaturi con grassi trans. E’ stato dimostrato che tale sostituzione:
aumenta livelli del colesterolo LDL;
riduce quelli del colesterolo HDL, a differenza degli acidi grassi saturi che invece aumentano i livelli di colesterolo HDL se utilizzati come sostituti in studi analoghi;
aumenta il rapporto tra colesterolo totale e colesterolo HDL, fino a valori pari a circa il doppio di quelli ottenuti con acidi grassi saturi, e, sulla base di questo solo effetto, è stato stimato che gli acidi grassi trans provochino circa il 6% degli eventi coronarici negli Stati Uniti.
Inoltre gli acidi grassi trans:
causano un pericoloso aumento delle LDL piccole e dense, che sono associate a un notevole aumento del rischio di CHD, anche in presenza di colesterolo LDL relativamente normale;
aumentano i livelli ematici dei trigliceridi, e questo è un fattore di rischio indipendente per la CHD;
aumentano i livelli della lipoproteina(a) o Lp(a), un altro importante fattore di rischio coronarico.
E tuttavia nel 2004 studi prospettici hanno dimostrato che la relazione tra l’assunzione di grassi trans e l’incidenza di CHD è maggiore di quella prevista da cambiamenti nei livelli dei lipidi sierici da soli. Ciò suggerisce che tali acido grassi influenzino altri fattori di rischio per la CHD, come l’infiammazione e la disfunzione delle cellule endoteliali.
Infiammazione e disfunzione delle cellule endoteliali
Il ruolo dell’infiammazione nell’aterosclerosi, e di conseguenza nella CHD, è cresciuto nell’ultimo decennio.
L’interleuchina-6, la proteina C-reattiva o CRP, acronimo dell’inglese C-reactive protein, e un’aumentata attività del fattore di necrosi tumorale o TNF, acronimo dell’inglese tumor necrosis factor, sono marcatori dell’infiammazione.
Nelle donne una maggiore assunzione di grassi trans è associata a un’aumentata attività del sistema TNF, e in quelle con un elevato indice di massa corporea con aumentati livelli di interleuchina-6 e CRP. Ad esempio, la differenza nei livelli di proteina C-reattiva osservata con un’assunzione media di grassi trans pari al 2,1% dell’assunzione di energia giornaliera totale, rispetto allo 0,9%, corrisponde a un aumento del rischio di malattia cardiovascolare del 30%. Risultati simili sono stati riportati in pazienti con malattia cardiaca, in studi randomizzati e controllati, in studi in vitro e in studi in cui sono stati analizzati i livelli di membrana degli acidi grassi trans, un biomarker della loro assunzione dietetica.
Quindi, i grassi trans promuovono l’infiammazione, e la loro azione pro-infiammatoria può spiegare parte dei loro effetti sulla CHD che, come visto, sono maggiori di quanto ci si aspetterebbe a seguito dei soli effetti sulle lipoproteine sieriche.
Attenzione: la presenza di infiammazione è un fattore di rischio indipendente non solo per la CHD ma anche per la resistenza all’insulina, il diabete, le dislipidemie e l’insufficienza cardiaca.
Un altro sito di azione degli acidi grassi trans può essere la funzione endoteliale. Diversi studi hanno suggerito l’associazione tra una loro maggiore assunzione e un aumento dei livelli dei biomarcatori circolanti della disfunzione endoteliale, come E-selectina, sICAM-1 e sVCAM-1.
Altri effetti
Studi in vitro hanno dimostrato che i grassi trans influenzano il metabolismo lipidico attraverso diversi percorsi.
Alterano la secrezione, la composizione lipidica e le dimensioni dell’apolipoproteina B-100 o Apo B-100.
Aumentano l’accumulo cellulare e la secrezione di colesterolo libero e di esteri del colesterolo da parte degli epatociti.
Alterano, negli adipociti, l’espressione dei geni per il PPAR-gamma, acronimo dell’inglese peroxisome proliferator-activated receptor gamma, la lipoprotein lipasi e la resistina, proteine con un ruolo centrale nel metabolismo degli acidi grassi e del glucosio.
Cardiopatia coronarica
Gli acidi grassi trans sono un fattore di rischio cardiovascolare indipendente.
Dai primi anni ’90 del secolo scorso l’attenzione si è focalizzata sui loro effetti sui lipidi plasmatici e sulla concentrazione delle lipoproteine.
Dal punto di vista epidemiologico, quattro importanti studi prospettici, che hanno coinvolto circa 140.000 soggetti monitorati per 6-14 anni, hanno tutti trovato riscontri positivi tra i livelli di questi acidi grassi nella dieta, valutati con l’aiuto di un questionario dettagliato sulla composizione della dieta, e il rischio di CHD.
I quattro studi sono:
The Health Professionals Follow-up study;
The Alpha-Tocopherol Beta-Carotene Cancer Prevention Study;
The Nurses’ Health Study;
The Zutphen Elderly Study.
Questi studi riguardano popolazioni talmente diverse che molto probabilmente i risultati sono applicabili all’intera popolazione mondiale.
Una meta-analisi degli studi suddetti ha mostrato che un 2% di aumento nell’apporto energetico dai grassi trans era associato con un incremento del 23% nell’incidenza di cardiopatia coronarica. Il rischio relativo di malattia cardiaca era di 1,36 nel “The Health Professionals Follow-up Study”, 1,14 nel “The Alpha-Tocopherol Beta-Carotene Cancer Prevention Study”; 1,93 (1,43-2,61) nel “The Nurses’ Health Study” e 1,28 (1,01-1,61) nel “The Zutphen Elderly Study”.
Quindi, anche un piccolo incremento del loro consumo si associa a un sostanziale aumento del rischio: il 2% dell’apporto energetico giornaliero, per una dieta di 2000 Kcal corrisponde a 40 Kcal o 4-5 grammi di grasso cioè l’equivalente di un cucchiaino di grasso!
Inoltre, in tre di questi studi l’associazione tra l’assunzione di grassi trans e il rischio di cardiopatia coronarica era più forte di una corrispondente associazione con l’assunzione di acidi grassi saturi. Nel “The Zutphen Elderly Study” questa associazione non è stata indagata.
A causa degli effetti negativi degli acidi grassi trans, per gli autori della meta-analisi non è etico condurre studi clinici randomizzati a lungo termine per saggiarne gli effetti sull’incidenza della cardiopatia coronarica. Per questi autori “studi osservazionali eseguiti in modo accurato forniscono un approccio ragionevole per valutarne gli effetti sulla salute cardiovascolare”.
Quindi, per evitare i loro effetti nocivi, di gran lunga peggiori in media a quelli dovuti ai contaminanti alimentari o ai residui dei pesticidi, è necessario evitarli o limitarne il consumo a meno dello 0,5% dell’apporto energetico giornaliero.
Ulteriori prove
Uno studio condotto su una popolazione australiana con un primo attacco di cuore e nessuna precedente storia di cardiopatia coronarica o iperlipidemia ha evidenziato un’associazione positiva tra i livelli di acidi grassi trans nel tessuto adiposo e il rischio di un infarto miocardico non fatale.
E’ stato dimostrato che la presenza nel tessuto adiposo di un acido grasso trans 18:1 con il doppio legame in posizione 7 (C18:1Δ7t), che si ritrova sia nei grassi animali che vegetali, era un predittore indipendente di un primo infarto miocardico, ossia il suo livello nel tessuto adiposo rimane un fattore predittivo per la malattia cardiaca dopo aggiustamento per i livelli di colesterolo totale. Di nuovo, sembra che solo una minima parte degli effetti negativi di grassi trans si attui per mezzo delle lipoproteine plasmatiche.
Nel corso di questo studio, a metà del 1996, i grassi trans furono eliminati dalle margarine vendute in Australia. Questa è stata un’opportunità unica per studiare la relazione temporale tra la loro l’assunzione e i loro livelli nel tessuto adiposo. E’ stato dimostrato che scomparivano dal tessuto adiposo sia dei pazienti che dei controlli con un tasso di circa il 15% del totale degli acidi grassi trans/anno.
Un altro studio condotto in Costarica ha trovato un’associazione positiva tra infarto miocardico e acidi grassi trans .
E’ interessante notare che in uno studio più grande, basato su una comunità caso-controllo, i livelli degli acidi grassi trans nelle membrane dei globuli rossi sono stati associati, dopo correzione per gli altri fattori di rischio, con un aumento del rischio di morte cardiaca improvvisa. Inoltre, l’aumento del rischio sembra essere correlato ai livelli dei trans-C18:2, i cui livelli sono stati associati con una triplicazione del rischio di morte improvvisa per cause cardiache, ma non con i livelli nella membrana cellulare di trans-C18:1, la principale classe di isomeri degli acidi grassi trans presenti nei cibi.
Diabete
In uno studio prospettico che ha coinvolto 84.204 infermiere di età compresa tra i 34 e i 59 anni, senza nessuna storia pregressa di diabete, malattia cardiovascolare e cancro, analizzate tra il 1980 e il 1996, e appartenenti al “The Nurses’ Health Study”, l’apporto di acidi grassi trans era correlato in modo significativo con il rischio di sviluppare il diabete di tipo 2. Dopo aggiustamenti per altri fattori di rischio l’apporto di grassi trans era positivamente associato con l’incidenza del diabete, con un incremento del rischio superiore al 39%.
Dati da studi di intervento controllati hanno mostrato che gli acidi grassi trans potrebbero danneggiare la sensibilità all’insulina in soggetti con resistenza all’insulina e diabete di tipo 2 più di quanto facciano gli acidi grassi insaturi, in particolare gli isomeri dell’acido linoleico coniugato o CLA, acronimo dell’inglese conjugated linoleic acid, trans-10,cis-12-CLA.
Attenzione perché alcuni integratori contengono isomeri del CLA e possono essere diabetogeni e proaterogenici in soggetti con resistenza all’insulina.
Nota: anche gli acidi grassi saturi innescano la medesima risposta, con nessuna differenza significativa tra loro e gli acidi grassi trans).
Nessun effetto significativo riguardo alla sensibilità all’insulina è stato invece osservato in soggetti sani e magri.
Acidi grassi trans dai ruminanti e cardiopatia coronarica
Quattro studi prospettici hanno valutato la relazione tra l’assunzione di acidi grassi trans dai ruminanti e il rischio di CHD: non è stata identificata alcuna associazione significativa.
In un altro studio pubblicato nel 2008 sono stati analizzati i dati provenienti da quattro studi di coorte danesi, che coinvolgevano 3686 adulti, arruolati tra il 1974 e il 1993, e seguiti per una mediana di 18 anni. In Danimarca il consumo di prodotti lattiero-caseari è relativamente alto e l’intervallo di assunzione dei grassi trans che provengono dai ruminati piuttosto ampio, sino al 1,1% dell’energia. Negli altri paesi, per la maggior parte delle persone, il consumo di grassi trans dai ruminati è sostanzialmente più basso del 1% dell’energia; negli USA è circa lo 0,5%.
Dopo correzione per altri fattori di rischio, non è stata trovata alcuna significativa associazione tra il loro consumo e l’incidenza di CHD, confermando, in una popolazione con un’assunzione relativamente alta di questo tipo di acidi grassi, le conclusioni dei quattro precedenti studi prospettici.
Quindi gli acidi grassi trans dai ruminanti, nelle quantità effettivamente consumate nella dieta, non aumentano il rischio di cardiopatia coronarica.
L’assenza di rischio rispetto ai grassi trans industriali può essere dovuta alla minore assunzione. Basti considerare che per gli Statunitensi la maggior parte degli acidi grassi trans sono di origine industriale, e che i livelli di grassi trans nel latte e nella carne sono relativamente bassi, dall’1% all’8% del totale dei grassi.
L’assenza di un rischio più elevato di CHD può essere dovuta anche alla presenza di isomeri diversi. Gli acidi grassi trans dai ruminanti e quelli industriali condividono molti isomeri, ma ci sono alcune differenze quantitative:
il livello di acido vaccenico è più alto nei grassi dei ruminanti, 30-50% degli isomeri trans-C18:1;
il gruppo del trans-C18:2, presente negli oli fritti, deodorinizzati e in alcuni oli parzialmente idrogenati, non lo è in quantità apprezzabile nel grasso dei ruminanti.
Infine altri fattori, ancora non noti e potenzialmente protettivi, potrebbero superare gli effetti nocivi dovuti ai grassi trans dai ruminanti.
Acidi grassi trans: esempi di legislazione in diversi paesi
USA Fino al 1985 non erano stati dimostrati effetti avversi dei grassi trans sulla salute umana e, addirittura, nel 1975 uno studio della Procter & Gamble non mostrò alcun effetto avverso sul colesterolo.
Il loro uso nei fast food crebbe dagli anni ’80, quando divenne chiaro il ruolo degli acidi grassi saturi nell’aumento del rischio cardiovascolare. Fu quindi condotta con successo una campagna per indurre la McDonald a passare dall’utilizzo del sego di bue a quello dell’olio vegetale per cucinare le patatine fritte. Nel frattempo diversi studi iniziarono a sollevare dubbi riguardo gli effetti dei grassi trans sulla salute.
La Food and Drug Administration, nel 1985, concluse che tali acidi grassi e l’acido oleico influenzavano il livello di colesterolo in maniera simile ma, dalla seconda metà del 1985 la pericolosità degli acidi grassi trans divenne chiara e la prova finale arrivò da studi di alimentazione controllata e da studi epidemiologici prospettici.
Nel 2003 la FDA ha stabilito che nei valori nutrizionali degli alimenti, sia per i cibi classici che per gli integratori, sia mostrato il loro contenuto a partire dal primo gennaio 2006. In particolare, questa ordinanza rappresenta il primo sostanziale cambiamento alla etichettatura dei prodotti alimentari dall’obbligo, risalente al 1990, di mettere le informazioni per porzione.
Nel 2005 il Dipartimento dell’Agricoltura statunitense ha fatto di un apporto minimo di grassi trans una raccomandazione chiave della nuova piramide alimentare.
Nel 2006 l’American Heart Association raccomanda di limitarne l’apporto all’1% del consumo calorico giornaliero e suggerisce ai produttori di alimenti e ai ristoranti di passare ad altri grassi.
Sempre nel 2006, il Dipartimento della Sanità della città di New York annuncia il divieto del loro utilizzo nei suoi 40.000 ristoranti entro il primo giugno 2008, seguito nel 2010-2011 dallo stato della California.
Australia Dopo il giugno 1996 sono stati eliminati dalla margarina venduta in Australia, che in passato contribuiva per circa il 50% al loro apporto giornaliero in quel paese.
Europa
Dall’11 marzo 2003 il governo danese, dopo un dibattito iniziato nel 1994 e due nuovi rapporti nel 2001 e nel 2003, ha deciso di eliminarne gradualmente l’uso nei cibi prima della fine del 2003.
Due anni più tardi però, la Commissione Europea ha chiesto alla Danimarca di ritirare questa legge che, sfortunatamente, non è stata accettata a livello comunitario.
Tuttavia, nel 2007, la stessa Commissione Europea ha ritirato la procedura d’infrazione nei confronti della Danimarca grazie alle maggiori prove scientifiche sulla pericolosità di questo tipo di acidi grassi.
L’esempio danese è stato seguito nel 2009 dall’Austria e dalla Svizzera, nel 2011 dall’Islanda, dalla Norvegia e Ungheria nel 2014, e più di recente Estonia e Georgia. Quindi circa il 10% della popolazione della Comunità Europea, circa 500 milioni di persone, vive in paesi dove è illegale vendere prodotti con un elevato contenuto di grassi trans. I governi degli altri paesi della Comunità Europea fanno invece affidamento sulla volontà dei produttori di alimenti di ridurne il contenuto nei propri prodotti. Tale strategia si è dimostrata efficace solo nei paesi dell’Europa Occidentale.
Canada
Il Canada sta considerando la legislazione per eliminarli dalle forniture alimentari e nel 2005 ha introdotto l’obbligo di mostrare il loro contenuto negli alimenti preconfezionati.
In conclusione, con l’eccezione di pochi paesi nei quali l’uso a scopi alimentari dei grassi trans è stato vietato, negli altri l’unico modo per ridurne l’apporto è la scelta del consumatore di preferire cibi in cui non siano presenti, leggendo sempre gli ingredienti, anche nel caso in cui il loro contenuto sulla confezione sia dichiarato pari a 0, potendo provenire dalla margarina, dai grassi vegetali per pasticceria, dall’olio vegetale e dalla frittura. Infatti ad esempio negli USA, i produttori di alimenti che ne contengano meno di 0,5 g per porzione possono dichiararne sulla confezione un contenuto pari a 0. Questo contenuto è basso, ma se un consumatore mangia più porzioni, ne consuma quantità sostanziali.
Attenzione: la lista degli ingredienti non è obbligatoria nei ristoranti, forni e in molti altri esercizi alimentari al dettaglio.
Riformulazione degli alimenti per ridurre gli acidi grassi trans
Le organizzazioni di sanità pubblica, compresa l’Organizzazione Mondiale della Sanità a partire dal settembre del 2006, hanno raccomandato di ridurre il consumo di acidi grassi trans. Solo negli USA la loro quasi eliminazione potrebbe evitare tra i 72.000 e i 280.000 casi di malattie cardiovascolari degli 1,2 milioni che si verificano annualmente.
I produttori di alimenti e i ristoranti possono ridurre l’uso dei grassi trans scegliendo alternative agli oli parzialmente idrogenati.
In Danimarca la loro eliminazione dagli oli vegetali non ha aumentato il consumo dei grassi saturi in quanto sono stati per lo più sostituiti con acidi grassi cis-insaturi. Inoltre, non ci sono stati effetti evidenti per i consumatori: ne aumenti nei costi ne riduzioni della disponibilità e della qualità dei prodotti alimentari.
Nel 2009, Stender e colleghi, analizzando ciò che è accaduto negli USA, in Sud Africa e molte nazioni europee, hanno dimostrato che gli acidi grassi trans in alimenti come patatine fritte, biscotti, torte e pop corn per il microonde possono essere sostituiti, a un prezzo simile, con una miscela di acidi grassi saturi, monoinsaturi e polinsaturi. Una tale sostituzione assicura anche maggiori benefici nutrizionali rispetto a quella uno a uno tra i grassi trans e i grassi saturi da soli.
Tuttavia attenzione perché ad esempio solamente nelle patatine fritte con bassi livelli di grassi trans la percentuale di acidi grassi saturi rimane costante, mentre nei biscotti e nelle torte è in media del 33% maggiore e nei pop corn per forno a microonde del 24%: gli acidi grassi saturi sono si meno pericolosi degli grassi trans ma di più rispetto agli acidi grassi mono- e polinsaturi.
Lo stesso gruppo di ricerca, analizzando alcuni cibi di comune consumo in Europa, acquistati in supermercati, anche della stessa catena, e in fast food (McDonald’s e Kentucky Fried Chicken, KFC) dal 2005 al 2014, ha dimostrato come il loro contenuto in grassi trans sia ridotto o addirittura assente in diversi paesi dell’Europa Occidentale mentre rimanga ancora alto in paesi dell’Europa dell’Est e del Sud-Est.
Nel 2010 Mozaffarian e colleghi hanno valutato i livelli degli acidi grassi trans e dei grassi saturi nei cibi dei principali supermercati di marca statunitensi e dei ristoranti dopo la riformulazione per ridurre il contenuto dei primi in due momenti: dal 1993 sino al 2006 e dal 2006 al 2009. E’ stata rilevata una generale riduzione del loro contenuto senza alcun sostanziale od equivalente aumento nel contenuto degli acidi grassi saturi.
Fonti alimentari
Nel mondo sono molti gli alimenti ricchi di grassi trans comunemente consumati.
Negli USA gran parte proviene da oli vegetali parzialmente idrogenati, con un consumo medio da questa fonte che è rimasto costante dagli anni ’60 dello scorso secolo.
Da notare che i valori degli acidi grassi trans che si incontreranno devono essere interpretati con cautela perché molti fast food, ristoranti e industrie possono aver cambiato il tipo di grasso utilizzato per la frittura e la cottura da quando gli articoli analizzati sono stati pubblicati.
I valori riportati, se non diversamente specificato, si riferiscono al contenuto percentuale in acidi grassi trans/100 g di acidi grassi.
Margarina
Tra gli alimenti ricchi di grassi trans la margarina, in bastoncini o dura, ne ha avuto la percentuale più alta. Tuttavia, i loro livelli hanno iniziato a ridursi quando il miglioramento della tecnologia ha permesso la produzione di margarine più morbide, che nel tempo sono divenute popolari. Esistono comunque differenze nel contenuto di grassi trans di margarine provenienti da diversi paesi. Di seguito alcuni esempi.
Il contenuto più alto, 13-16,5%, si ritrova nelle margarine morbide provenienti dall’Islanda, Norvegia e Regno Unito.
Un contenuto minore è presente in quelle dall’Italia, Germania, Finlandia e Grecia, rispettivamente il 5,1%, 4,8%, 3,2%, e 2,9%.
In Portogallo, Paesi Bassi, Belgio, Danimarca, Francia, Spagna e Svezia il contenuto è inferiore al 2%.
USA e Canada sono in ritardo rispetto all’Europa, ma negli USA sono in corso dei cambiamenti a seguito dell’introduzione dei grassi trans nell’etichettatura degli alimenti e della maggior conoscenza dei pericoli associati al loro consumo da parte degli acquirenti. Per questo motivo, al momento, negli USA la margarina è considerata contribuire in misura minore al totale dei grassi trans, mentre le fonti principali sono i prodotti commerciali da forno e dei fast food, come torte, biscotti, wafer, cracker, crocchette di pollo, patatine fritte o pop corn per il forno a microonde (vedi sotto).
Grassi da pasticceria
Il contenuto in grassi trans dei grassi da pasticceria è compreso tra il 6% e il 50%, e varia nei diversi paesi: in Germania, Austria e Nuova Zelanda è più basso che in Francia o negli USA.
Tuttavia, come per la margarina, anche per i grassi da pasticceria il loro contenuto è in diminuzione. In Germania è calato dal 12% nel 1994 al 6% nel 1999, in Danimarca è il 7% (1996), mentre in Nuova Zelanda è di circa il 6% (1997).
Oli vegetali
Al momento gli oli vegetali non idrogenati utilizzati per condire le insalate o per cucinare non contengono acidi grassi trans o ne contengono solo piccole quantità.
La lavorazione di questi oli può comportarne la produzione di piccole quantità, variabili da 0,05 g/100 g di prodotto per l’olio di oliva extra-vergine a 2,42 g/100 g di prodotto per l’olio di colza. Quindi il loro contributo al contenuto di grassi trans dei cibi attualmente consumati è molto piccolo.
Un’eccezione è rappresentata dagli oli vegetali idrogenati pachistani, che ne possono contenere dal 14% al 34%.
Minestre pronte
Tra gli alimenti ricchi di grassi trans le minestre pronte ne contengono quantità significative, che variano dal 10% del brodo di manzo al 35% della crema di cipolle; quindi, se consumate di frequente contribuiscono in grande misura all’apporto di tali acidi grassi nella dieta.
Prodotti alimentari trasformati
Gli acidi grassi trans, grazie alle loro proprietà sono utilizzati nella preparazione di molti alimenti trasformati come biscotti, torte, cornetti, dolci e altri prodotti da forno. E, nella dieta consumata in Nord America, i prodotti da forno ne sono la principale fonte.
Naturalmente, il contenuto in grassi trans dipende dal tipo di grassi utilizzati nella lavorazione.
Salse
La maionese, i condimenti per le insalate e altre salse apportano quantità di grassi trans piccole o nulle.
Latte umano e alimenti per neonati
Il contenuto di acidi grassi trans del latte umano riflette il contenuto in tali grassi della dieta materna nel giorno precedente, è compreso tra 1% e il 7% ed è in diminuzione dal 7,1% del 1998 al 4,6% del biennio 2005-2006.
Gli alimenti per lattanti hanno valori di grassi trans in media di 0,1%-4,5%, con una marca che raggiunge il 15,7%.
Gli alimenti per bambini ne contengono più del 5%.
Fast food e ristoranti
Nei fast food e ristoranti, per la frittura, vengono utilizzati grassi usati anche in pasticceria, quindi con elevate quantità di acidi grassi trans; pertanto i cibi venduti possono contenerne quantità relativamente alte. Fonti sono le torte fritte, le patatine fritte, le crocchette di pollo, gli hamburger, le fritture di pesce e il pollo fritto.
In lavori pubblicati da Stender e colleghi dal 2006 al 2009 viene dimostrato che il contenuto di grassi trans delle patatine fritte e delle crocchette di pollo variava ampiamente da paese a paese ma, anche all’interno della stessa catena di fast food, in uno stesso paese, e addirittura in una stessa città, a causa dell’olio utilizzato per cucinare. Ad esempio, l’olio utilizzato per le patatine fritte nei punti vendita di McDonald’s negli USA e in Perù ne conteneva il 23-24%, mentre l’olio utilizzato nella stessa catena in molti paesi europei ne contiene circa il 10%, con alcuni paesi, come la Danimarca, a un livello più basso del 5% e 1%.
E, considerando un pasto a base di patatine fritte e crocchette di pollo, in porzioni rispettivamente da 171 e 160 g, acquistato da McDonald a New York, lo stesso conteneva oltre 10 g di grassi trans mentre, se veniva acquistato da KFC in Ungheria si arrivava a quasi a 25 g.
Di seguito, sempre dai lavori di Stender e colleghi, si può osservare una comparazione tra diversi paesi per quanto riguarda il contenuto di grassi trans delle crocchette di pollo e delle patatine fritte acquistate da McDonald o da KFC.
Crocchette di pollo e patatine fritte da McDonald:
meno di un grammo solo se i pasti sono stati acquistati in Danimarca;
1-5 g in Portogallo, Paesi Bassi, Russia, Repubblica Ceca o Spagna;
5-10 g negli USA, Perù, Regno Unito, Sud Africa, Polonia, Finlandia, Francia, Italia, Norvegia, Spagna, Svezia, Germania o Ungheria.
Crocchette di pollo e patatine fritte da KFC:
meno di 2 g se i pasti sono stati acquistati nel Regno Unito, Danimarca, Russia o Germania;
2-5 g in Germania, Francia, Regno unito, Spagna o Portogallo;
5-10 g alle Bahamas, Sud Africa o USA;
10-25 g in Ungheria, Polonia, Perù o Repubblica Ceca.
Bibliografia
Akoh C.C. and Min D.B. Food lipids: chemistry, nutrition, and biotechnology. 3rd Edition. CRC Press Taylor & Francis Group, 2008
Ascherio A., Katan M.B., Zock P.L., Stampfer M.J., Willett W.C. Trans fatty acids and coronary heart disease. N Engl J Med 1999;340:1994-1998. doi:10.1056/NEJM199906243402511
Ascherio A., Rimm E.B., Giovannucci E.L., Spiegelman D., Stampfer M., Willett W.C. Dietary fat and risk of coronary heart disease in men: cohort follow up study in the United States. BMJ 1996; 313:84-90. doi:10.1080/17482970601069094
Asp N-G. Fatty acids in focus – the good and the bad ones. Scand J Food Nutr 2006;50:155-160. doi:10.1080/17482970601069094
Baylin A., Kabagambe E.K., Ascherio A., Spiegelman D., Campos H. High 18:2 trans-fatty acids in adipose tissue are associated with increased risk of nonfatal acute myocardial infarction in Costa Rican adults. J Nutr 2003;133:1186-1191. doi:10.1093/jn/133.4.1186
Chow C.K. Fatty acids in foods and their health implication. 3rd Edition. CRC Press Taylor & Francis Group, 2008.
Clifton P.M., Keogh J.B., Noakes M. Trans fatty acids in adipose tissue and the food supply are associated with myocardial infarction. J Nutr 2004;134:874-879. doi:10.1093/jn/134.4.874
Costa N., Cruz R., Graça P., Breda J., and Casal S. Trans fatty acids in the Portuguese food market. Food Control 2016;64:128-134. doi:10.1016/j.foodcont.2015.12.010
Eckel R.H., Borra S., Lichtenstein A.H., Yin-Piazza D.Y. Understanding the Complexity of Trans fatty acid reduction in the American diet. American Heart Association trans fat conference 2006 report of the trans fat conference planning group. Circulation 2007;115:2231-2246. doi:10.1161/CIRCULATIONAHA.106.181947
Hu F.B., Manson J.E., Stampfer M.J., Colditz G., Liu S., Solomon C.G., and Willett W.C. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N Engl J Med 2001;345:790-797. doi:10.1056/NEJMoa010492
Hu F.B., Willett W.C. Optimal diet for prevention of coronary heart disease JAMA 2002;288:2569-2578. doi:10.1001/jama.288.20.2569
Lemaitre R.N., King I.B., Raghunathan T.E., Pearce R.M., Weinmann S., Knopp R.H., Copass M.K., Cobb L.A., Siscovick D.S. Cell membrane trans-fatty acids and the risk of primary cardiac arrest. Circulation 2002;105:697-701. doi:10.1161/hc0602.103583
Lemaitre R.N, King I.B, Mozaffarian D., Sootodehnia N., Siscovick D.S. Trans-fatty acids and sudden cardiac death. Atheroscler Suppl 2006; 7(2):13-5. doi:10.1016/j.atherosclerosissup.2006.04.003
Lichtenstein A.H. Dietary fat, carbohydrate, and protein: effects on plasma lipoprotein patterns J. Lipid Res. 2006;47:1661-1667. doi:10.1194/jlr.R600019-JLR200
Lichtenstein A.H., Ausman L., Jalbert S.M., Schaefer E.J. Effect of different forms of dietary hydrogenated fats on serum lipoprotein cholesterol levels. N Engl J Med 1999;340:1933-1940. doi:10.1056/NEJM199906243402501
Lopez-Garcia E., Schulze M.B., Meigs J.B., Manson J.E, Rifai N., Stampfer M.J., Willett W.C. and Hu F.B. Consumption of trans fatty acids is related to plasma biomarkers of inflammation and endothelial dysfunction. J Nutr 2005;135:562-566. doi:10.1093/jn/135.3.562
Masanori S. Trans Fatty Acids: Properties, Benefits and Risks J Health Sci 2002;48(1):7-13.
Mensink R.P., Katan M.B. Effect of dietary trans fatty acids on high-density and low-density lipoprotein cholesterol levels in healthy subjects. N Engl J Med 1990;323:439-445. doi:10.1056/NEJM199008163230703
Mozaffarian D. Commentary: Ruminant trans fatty acids and coronary heart disease-cause for concern? Int J Epidemiol 2008;37(1):182-184. doi:10.1093/ije/dym263
Mozaffarian D., Jacobson M.F., Greenstein J.S. Food Reformulations to reduce trans fatty acids. N Eng J Med 2010;362:2037-2039. doi:https://doi.org/10.1056/NEJMc1001841
Mozaffarian D., Katan M.B., Ascherio A., Stampfer M.J., Willett W.C. Trans fatty acids and cardiovascular disease. N Engl J Med 2006;354:1601-1613. doi:10.1056/NEJMra054035
Mozaffarian D., Pischon T., Hankinson S.E., Joshipura K., Willett W.C., and Rimm E.B. Dietary intake of trans fatty acids and systemic inflammation in women. Am J Clin Nutr 2004;79:606-612. doi:https://doi.org/10.1093/ajcn/79.4.606
Oh K., Hu F.B., Manson J.E., Stampfer M.J., Willett W.C. Dietary fat intake and risk of coronary heart disease in women: 20 years of follow-up of the Nurses’ Health Study. Am J Epidemiol 2005;161(7):672-679. doi:10.1093/aje/kwi085
Okie S. New York to Trans Fats: You’re Out! N Engl J Med 2007;356:2017-2021. doi:10.1056/NEJMp078058
Oomen C.M., Ocke M.C., Feskens E.J., van Erp-Baart M.A., Kok F.J., Kromhout D. Association between trans fatty acid intake and 10-year risk of coronary heart disease in the Zutphen Elderly Study: a prospective population-based study. Lancet 2001; 357(9258):746-751. doi:10.1016/S0140-6736(00)04166-0
Pietinen P., Ascherio A., Korhonen P., Hartman A.M., Willett W.C., Albanes D., VirtamO J.. Intake of fatty acids and risk of coronary heart disease in a cohort of Finnish men: the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study. Am J Epidemiol 1997;145(10):876-887. doi:10.1093/oxfordjournals.aje.a009047
Risérus U. Trans fatty acids, insulin sensitivity and type 2 diabetes. Scand J Food Nutr 2006;50(4):161-165. doi:10.1080/17482970601133114
Salmerón J., Hu F.B., Manson J.E., Stampfer M.J., Colditz G.A., Rimm E.B., and Willett W.C. Dietary fat intake and risk of type 2 diabetes in women. Am J Clin Nutr 2001;73:1019-1026 doi:10.1093/ajcn/73.6.1019
Stender S., Astrup A., and Dyerberg J. A trans European Union difference in the decline in trans fatty acids in popular foods: a market basket investigation. BMJ Open 2012;2(5):e000859. doi:10.1136/bmjopen-2012-000859
Stender S., Astrup A., and Dyerberg J. Artificial trans fat in popular foods in 2012 and in 2014: a market basket investigation in six European countries. BMJ Open 2016;6(3):e010673. doi:10.1136/bmjopen-2015-010673
Stender S., Astrup A.,and Dyerberg J. Tracing artificial trans fat in popular foods in Europe: a market basket investigation. BMJ Open 2014;4(5):e005218. doi:10.1136/bmjopen-2014-005218
Stender S., Astrup A., Dyerberg J. What went in when trans went out?. N Engl J Med 2009;361:314-316. doi:10.1056/NEJMc0903380
Stender S., Dyerberg J. The influence of trans fatty acids on health. Fourth edition. The Danish Nutrition Council; publ. no. 34, 2003.
Stender S., Dyerberg J., Astrup A. Consumer protection through a legislative ban on industrially produced trans fatty acids in Denmark. Scand J Food Nutr 2006;50(4):155-160. doi:10.1080/17482970601069458
Stender S., Dyerberg J., Astrup A. High levels of trans fat in popular fast foods. N Engl J Med 2006;354:1650-1652. doi:10.1056/NEJMc052959
Willett W., Mozaffarian D. Ruminant or industrial sources of trans fatty acids: public health issue or food label skirmish? Am J Clin Nutr 2008;87(3): 515-516. doi:10.1093/ajcn/87.3.515
Il tè nero, al pari delle altre varietà di tè, è un infuso di foglie lavorate di Camellia sinensis, pianta appartenente alla famiglia delle Theaceae.
A differenza di quanto accade per il tè verde, nel corso della lavorazione delle foglie per la produzione del tè nero si verifica la quasi completa ossidazione delle sostanze in esse contenute, in particolare le catechine, un tipo di polifenoli appartenenti al gruppo dei flavonoidi.
Il colore delle foglie lavorate è scuro e l’infuso, da preparare utilizzando un filtro a persona, o un cucchiaino a persona nel caso del prodotto sfuso, per un tempo di infusione di 3-4 minuti in acqua a 95-100 °C, è di colore rosso bruno.
Il tè è una bevanda che ha origini molto antiche, risalenti a quasi 4000 anni fa. E’ una delle bevande più consumate, soprattutto in Asia, dove prevale, in special modo in Cina e Giappone, il consumo del tè verde. Di contro in occidente il tè nero è la varietà più consumata, e lo è anche a livello globale rappresentando circa il 78% del mercato della bevanda.
L’ossidazione subita dalle foglie nel corso della lavorazione riduce i potenziali effetti benefici ascritti ai polifenoli inizialmente presenti.
Per la produzione di tutti i tipi di tè si utilizzano le foglie fresche e più giovani di Camellia sinensis, ritenute di qualità superiore rispetto a quelle più vecchie.
La lavorazione che porterà alla produzione di foglie essiccate pronte per la preparazione del tè nero si compone di quattro fasi: appassimento o asciugatura, arrotolamento, ossidazione, ed essicazione. Questo tipo di lavorazione porta alla quasi totale ossidazione delle sostanze presenti, in particolare delle catechine.
L’appassimento o asciugatura è il processo attraverso il quale viene rimossa l’acqua presente nelle foglie, che ne costituisce circa il 75% del peso, determinando quindi la concentrazione della linfa. L’appassimento, che migliora anche la lavorazione successiva, può essere ottenuto in tre modi differenti:
esponendo le foglie alla luce solare;
attraverso processi indoor, riscaldando in maniera appropriata le stanze in cui sono riposte le foglie;
ricorrendo all’utilizzo di macchinari che ventilano le foglie.
All’appassimento segue l’arrotolamento che, rompendo il tessuto fogliare, facilita la fuoriuscita della linfa e promuove la successiva ossidazione dei polifenoli.
La fase successiva è quella dell’ossidazione, impropriamente detta anche fermentazione. In questo passaggio si verifica l’ossidazione enzimatica, catalizzata dalla polifenolo ossidasi (EC 1.10.3.1), e non enzimatica, per azione dell’ossigeno atmosferico, di circa il 90-95% dei polifenoli presenti. L’ossidazione è accompagnata anche da altri cambiamenti che renderanno le foglie da verdi a rosse. Fattori cruciali sono anche la temperatura, in genere sui 25°C, il pH, l’umidità, 95% o più, la ventilazione e la durata. Questo passaggio è fondamentale nel determinare la qualità del tè, in quanto gli conferisce le sue caratteristiche organolettiche e la composizione in polifenoli, ben diverse da quelle del tè verde che viene prodotto in modo tale da minimizzare i processi di ossidazione.
Da notare che il contenuto in caffeina non varia significativamente.
L’ultima fase è quella dell’essiccazione. Viene condotta a una temperatura di 80-90 °C per circa 20-25 minuti. L’elevata temperatura inattiva la polifenolo ossidasi arrestando i processi di ossidazione enzimatica.
Tearubigine e teaflavine
I processi ossidativi che si verificano durante la produzione del tè nero interessano le catechine monomeriche e gallate, in misura minore i glicosidi delle catechine, in special modo la miricetina, e composti non flavonoidi, ad esempio la teagallina, e portano alla formazione di polifenoli complessi quali le tearubigine, le teaflavine e gli acidi teaflavici.
Le tearubigine, polimeri di catechine di colore brunastro non ancora ben caratterizzati, sono i principali polifenoli del tè nero, rappresentando circa il 20% del peso dell’estratto. Contribuiscono alla ricchezza di gusto, il cosiddetto “corpo”, oltre che al colore.
Le teaflavine, dimeri di catechine di colore rosso-arancio, costituiscono circa il 3-5% del peso dell’estratto e contribuiscono al gusto vivace e astringente, oltre che al colore.
Le principali teaflavine sono:
teaflavina-3-gallato;
teaflavina-3’-gallato;
teaflavina-3-3’-digallato, la più abbondante.
Benefici per la salute
I benefici per la salute del tè nero sono dovuti in gran parte ai suoi polifenoli complessi, tearubigine e teaflavine, essendo le catechine per la maggior parte ossidate nel corso della lavorazione delle foglie.
Di seguito tre esempi.
Sono stati messi in evidenza effetti antivirali delle teaflavine che, analogamente alle catechine, sembrano essere efficaci in particolare nei confronti dei virus a RNA singolo a filamento positivo. Tra questi virus si annoverano anche SARS-CoV-1 e SARS-CoV-2, virus appartenenti alla famiglia dei Coronaviridae. Al pari delle catechine, anche per l’azione antivirale delle teaflavine sembrano essere importanti i gruppi galloile.
I fitochimici presenti nel tè nero, come quelli del tè verde, sembrano in grado di ridurre l’indice glicemico degli alimenti ricchi di amido. L’effetto sembra dovuto all’inibizione dell’attività della alfa-amilasi pancreatica e di altri enzimi digestivi, e al legame diretto con l’amido, in conseguenza del quale si verrebbe a ridurre la superficie disponibile all’attacco degli enzimi digestivi stessi. L’inibizione è maggiore per gli alimenti privi di glutine, sembra a causa dell’azione del glutine sui polifenoli complessi che quindi non sarebbero in grado di interagire con il polisaccaride. Per approfondimenti si veda l’articolo sui polifenoli del tè.
Tearubigine e teaflavine sembrano avere un effetto anticariogeno dovuto all’azione inibitoria sulla amilasi batterica e salivare, azione che appare più efficace di quella delle catechine del tè verde. Il tè nero sembra anche in grado di ridurre la produzione di acidi nella cavità orale.
Bibliografia
Asil M.H., Rabiei B., Ansari R.H. Optimal fermentation time and temperature to improve biochemical composition and sensory characteristics of black tea. Aust J Crop Sci 2012;6(3):550-8.
Kan L., Capuano E., Fogliano V., Oliviero T. and Verkerk R. Tea polyphenols as a strategy to control starch digestion in bread: the effects of polyphenol type and gluten. Food Funct 2020;11:5933-5943. doi:10.1039/D0FO01145B
Kuhnert N. Unraveling the structure of the black tea thearubigins. Arch Biochem Biophys 2010;501(1):37-51. doi:10.1016/j.abb.2010.04.013
Li S., Lo C-Y., Pan M-H., Lai C-S. and Ho C-T. Black tea: chemical analysis and stability. Food Funct 2013;4:10-18. doi:10.1039/C2FO30093A
Mhatre S., Srivastava T., Naik S., Patravale V. Antiviral activity of green tea and black tea polyphenols in prophylaxis and treatment of COVID-19: a review. Phytomedicine 2020;153286. doi:10.1016/j.phymed.2020.153286
Menet M-C., Sang S., Yang C.S., Ho C-T., and Rosen R.T. Analysis of theaflavins and thearubigins from black tea extract by MALDI-TOF mass spectrometry. J Agric Food Chem 2004;52:2455-61. doi:10.1021/jf035427e
Sharma V.K., Bhattacharya A., Kumar A. and Sharma H.K. Health benefits of tea consumption. Trop J Pharm Res 2007;6(3):785-792.
Il tè verde, al pari degli altri tipi di tè, è un infuso di foglie lavorate di Camellia sinensis, un membro della famiglia delle Theaceae.
La lavorazione delle foglie che porterà al prodotto pronto per l’infusione è tale da ridurre al minimo i processi ossidativi, sia enzimatici che chimici, a carico delle sostanze in esse contenute, in particolare i fitochimici come le catechine, polifenoli appartenenti alla classe dei flavonoidi e le principali responsabili dei benefici per la salute ascritti al tè verde.
Avendo subito modificazioni chimiche poco significative, le foglie mantengono il loro colore verde, mentre la bevanda, da ottenersi utilizzando un filtro a persona, o nel caso del tè sfuso un cucchiaino a persona, per un tempo di infusione circa tre minuti in acqua a 75 °C, è di colore giallo oro.
Ed è al tipo di lavorazione delle foglie che si deve parte delle peculiari proprietà organolettiche della bevanda, come il sapore, che risulta più delicato e leggero rispetto a quello del tè nero, e le sue proprietà salutari, da sempre riconosciute nella cultura orientale.
Solo di recente la comunità scientifica ha iniziato a studiarne i benefici per la salute, riconoscendone il valore preventivo riguardo lo viluppo di molte malattie, come le malattie cardiovascolari e alcuni tipi di tumore.
E’ stato dimostrato che i polifenoli del tè, in particolare le catechine, sono in grado di attivare vie di segnalazione intracellulari a seguito del legame a specifici recettori di membrana e/o al passaggio all’interno della cellula dove poi si legano a specifici recettori citoplasmatici, mitocondriali o nucleari, in entrambe i casi attivando o inibendo, a seconda del tipo cellulare, determinati processi cellulari.
Dato l’elevato consumo di tè a livello mondiale, anche piccoli effetti sulla salute delle persone potrebbero avere grandi effetti sulla salute pubblica.
Camellia sinensis è una pianta sempre verde originaria del Sud, Est, e Sud-Est asiatico, che attualmente viene coltivata in almeno 30 paesi, principalmente con clima sub-tropicale o tropicale, sebbene ci siano varietà coltivate in Cornovaglia e nello stato di Washington.
In natura, se indisturbata, può crescere fino a 15-20 metri, mentre nelle piantagioni è tenuta, per facilitarne la coltivazione e la raccolta delle foglie, a un’altezza inferiore al metro e mezzo.
La sua coltivazione si può spingere fino a 1500-2000 metri di altitudine, e molte delle varietà più pregiate di tè sono ottenute da coltivazioni montane grazie al fatto che la pianta cresce più lentamente permettendo alla foglia di acquisire più aromi.
Le varianti maggiormente coltivate, delle quattro conosciute, sono Camellia sinensis var. sinensis, originaria della Cina, e Camellia sinensis var. assamica, originaria dell’India.
Per la produzione delle differenti varietà di tè sono utilizzate le foglie fresche, tra le quali sono scelte le più giovani poiché quelle vecchie sono ritenute di qualità inferiore.
Le foglie fresche sono ricche di polifenoli solubili in acqua, in particolare catechine e catechine glicosilate. Le principali catechine presenti sono l’epigallocatechina-3-gallato o EGCG, la più attiva, l’epigallocatechina, la epicatechina-3-gallato, l’epicatechina. Sono presenti, anche se in quantità minore, la catechina, la gallocatechina, la catechina gallato e la gallocatechina gallato.
Gallocatechina gallato
Questi polifenoli costituiscono il 30-42% del peso secco della foglia. La caffeina costituisce circa il 3% del peso secco della foglia, con variazioni dall’1,4 al 4,5%.
Il cultivar, le caratteristiche del suolo, i metodi di coltivazione, l’altitudine, il clima, il periodo dell’anno in cui avviene la raccolta delle foglie, ovviamente assieme alla lavorazione delle foglie, sono tutti fattori che influenzeranno le caratteristiche organolettiche della bevanda.
Come si produce il tè verde?
Le differenze nella lavorazione delle foglie, che portano alla produzione dei diversi tipi di tè, causano differenti gradi di ossidazione delle sostanze in esse presenti, soprattutto delle catechine.
La lavorazione che porta alla produzione del tè verde è tale da ridurre al minimo i processi ossidativi, sia enzimatici che non enzimatici.
Dopo la raccolta, le foglie sono esposte al sole per 2-3 ore e fatte appassire/asciugare. Di seguito, la lavorazione vera e propria procede attraverso tre fasi principali:
trattamento termico;
arrotolamento;
essicazione.
Il trattamento termico, breve e delicato, è il passaggio cruciale per la qualità e le proprietà finali della bevanda. Può essere effettuato sia con il vapore, che è il metodo tradizionale giapponese, che mediante cottura a secco in pentole calde, dei grandi wok, una sorta di torrefazione, che è il metodo tradizionale cinese. Il trattamento termico permette di inattivare gli enzimi presenti nei tessuti fogliari bloccando i processi di ossidazione enzimatica, in particolare quelli a carico dei polifenoli. Inoltre elimina l’odore d’erba, facendo così risaltare quello del tè, e fa evaporare, nel caso del metodo tradizionale cinese, parte dell’acqua della foglia, di cui ne costituisce circa il 75% del peso, rendendola più morbida, così da facilitare il passaggio successivo.
Al trattamento termico segue una fase di arrotolamento che ha lo scopo di facilitare la fase successiva di essiccamento e di distruggere il tessuto fogliare al fine di favorire il rilascio degli aromi della foglia, migliorando la qualità del prodotto.
L’ultima fase è quella dell’essiccazione, che comporta anche la formazione di nuovi composti e migliora l’aspetto del prodotto.
Benefici per la salute
L’assunzione di tè è da sempre associata, almeno nella cultura orientale, Cina e Giappone in primis, a benefici per la salute. Di seguito una breve rassegna di risultati di studi epidemiologici e di laboratorio che hanno indagato il ruolo preventivo che l’assunzione di tè verde può avere nei confronti dello sviluppo di molte patologie. La EGCG, che è la catechina più abbondante presente nel tè verde rappresentando quasi il 60% dei polifenoli presenti nelle foglie secche, sembra svolgere il ruolo principale.
A livello molecolare, per molti degli effetti esercitati dalle catechine sembrano essere essenziali i gruppi galloile presenti nelle posizione 3 e/o 3’.
Malattie cardiovascolari
Le malattie cardiovascolari sono la principale causa di morte nel mondo, in particolare nei paesi a basso e medio reddito, con una stima di 17,3 milioni di decessi nel 2008, valore che potrebbe aumentare fino a 23,3 milioni entro il 2030.
Un consumo giornaliero di tè, soprattutto tè verde, sembra essere associato a una riduzione del rischio della loro comparsa, in particolare ipertensione e ictus.
Tra i meccanismi proposti sembra essere importante l’accresciuta bioattività del monossido di azoto endoteliale, un potente vasodilatatore, dovuta all’azione dei polifenoli che ne aumenterebbero la sintesi e/o ne ridurrebbero la degradazione mediata dall’anione superossido.
Neoplasie
Molti studi epidemiologici e di laboratorio hanno dato risultati incoraggianti riguardo al possibile ruolo preventivo del tè, in particolare il tè verde, nei confronti dello sviluppo di alcune neoplasie quali i tumori del cavo orale, del tratto digestivo, e del polmone tra coloro che non hanno mai fumato.
I polifenoli del tè sembrano agire non solo come antiossidanti, ma anche come molecole che, direttamente, possono influenzare l’espressione genica e l’attività di diverse vie metaboliche.
Attività antivirale
Recenti studi hanno messo in evidenza gli effetti antivirali delle catechine, in particolare della EGCG del tè verde, e delle teaflavine del tè nero, soprattutto nei confronti dei virus a RNA a singolo filamento positivo. A questo tipo di virus appartiene anche la famiglia dei Coronaviridae, che a sua volta comprende sia il SARS-CoV-1 che il SARS-CoV-2.
Le proprietà antivirali della EGCG sembrano essere dovute alle sue caratteristiche strutturali, ossia alla presenza di gruppi pirogallici e galloilici.
Digestione dell’amido
Studi in vitro hanno evidenziato come i polifenoli del tè verde e del tè nero siano in grado di ridurre l’indice glicemico di alimenti ricchi di amido. Di conseguenza potrebbero essere utili nel controllo del loro indice glicemico. L’effetto sembra essere dovuto all’inibizione della alfa-amilasi pancreatica e di altri enzimi digestivi, e a un’interazione diretta tra l’amido e i fitochimici che ridurrebbe la superficie dei granuli di amido disponibile per l’attacco degli enzimi digestivi stessi. Il tè verde appare essere ugualmente efficace sia nei confronti dei cibi contenenti glutine, nei confronti dei quali il tè nero appare meno efficace, che di quelli privi di glutine.
Perdita di peso
Nella fase di perdita di peso, come durante la fase di mantenimento, è importante mantenere il più costante possibile il dispendio energetico giornaliero.
A partire dagli anni ‘90 dello scorso secolo è stato proposto che il tè verde, grazie al suo contenuto in caffeina e catechine fosse di aiuto per:
mantenere o addirittura aumentare la spesa energetica giornaliera;
incrementare l’ossidazione dei grassi.
Oltre a questi potenziali effetti termogenici e lipolitici, catechine e caffeina potrebbero essere utili agendo su altri bersagli quali l’assorbimento dei lipidi e l’apporto energetico, forse attraverso il loro impatto sul microbiota intestinale, che è parte del più ampio microbiota umano, e sull’espressione genica.
Sono stati quindi commercializzati prodotti per la perdita di peso e il mantenimento del peso perso a base di estratti di tè verde, contenenti concentrazioni di catechine e caffeina molto maggiori rispetto alla bevanda.
Ma quanto c’è di scientificamente comprovato nelle proprietà “brucia grassi” del tè verde?
La questione sembra essere stata risolta da una accurata meta-analisi di 15 studi che ha analizzato la relazione tra perdita di peso e assunzione dei suddetti prodotti. Lo studio ha evidenziato che i prodotti a base di tè verde inducono, in adulti obesi e in sovrappeso, una diminuzione di peso che:
non è statisticamente significativa;
è molto piccola;
probabilmente non è clinicamente importante.
Quindi, sulla base delle ricerche scientifiche, il tè verde non sembra essere di aiuto nella perdita di peso ne nel mantenimento del peso perso.
Attività anticariogena
Studi in vitro e su animali hanno messo in evidenza che il tè, e nello specifico i suoi polifenoli, sembrano possedere:
proprietà antibatteriche nei confronti di patogeni ad azione cariogena, come Streptococcus mutans, come nel caso della EGCG del tè verde;
azione inibitoria sulla amilasi salivare e batterica, ruolo in cui le tearubigine e teaflavine del tè nero sembrano più efficaci delle catechine del tè verde;
azione inibitoria sulla produzione di acidi nella cavità orale.
Tutte queste caratteristiche rendono il tè verde e il tè nero bevande con una potenziale attività anticariogena.
Bibliografia
Arab L., Khan F., and Lam H. Tea consumption and cardiovascular disease risk. Am J Clin Nutr 2013;98:1651S-1659S. doi:10.3945/ajcn.113.059345
Clifford M.N., van der Hooft J.J.J., and Crozier A. Human studies on the absorption, distribution, metabolism, and excretion of tea polyphenols. Am J Clin Nutr 2013;98:1619S-1630S. doi:10.3945/ajcn.113.058958
Dwyer J.T. and Peterson J. Tea and flavonoids: where we are, where to go next. Am J Clin Nutr 2013;98:1611S-1618S. doi:10.3945/ajcn.113.059584
Goenka P., Sarawgi A., Karun V., Nigam A.G., Dutta S., Marwah N. Camellia sinensis (Tea): implications and role in preventing dental decay. Phcog Rev 2013;7:152-156. doi:10.4103/0973-7847.120515
Green R.J., Murphy A.S., Schulz B., Watkins B.A. and Ferruzzi M.G. Common tea formulations modulate in vitro digestive recovery of green tea catechins. Mol Nutr Food Res 2007;51(9):1152-1162. doi:10.1002/mnfr.200700086
Grassi D., Desideri G., Di Giosia P., De Feo M., Fellini E., Cheli P., Ferri L., and Ferri C. Tea, flavonoids, and cardiovascular health: endothelial protection. Am J Clin Nutr 2013;98:1660S-1666S. doi:10.3945/ajcn.113.058313
Huang W-Y., Lin Y-R., Ho R-F., Liu H-Y., and Lin Y-S. Effects of water solutions on extracting green tea leaves. Sci World J 2013;Article ID 368350. doi:10.1155/2013/368350
Hursel R. and Westerterp-Plantenga M.S. Catechin- and caffeine-rich teas for control of body weight in humans. Am J Clin Nutr 2013;98:1682S-1693S. doi:10.3945/ajcn.113.058396
Hursel R., Viechtbauer W. and Westerterp-Plantenga M.S. The effects of green tea on weight loss and weight maintenance: a meta-analysis. Int J Obesity 2009;33:956-961. doi:10.1038/ijo.2009.135
Jurgens T.M., Whelan A.M., Killian L., Doucette S., Kirk S., Foy E. Green tea for weight loss and weight maintenance in overweight or obese adults. Editorial group: Cochrane Metabolic and Endocrine Disorders Group. 2012:12 Art. No.: CD008650. doi:10.1002/14651858.CD008650.pub2
Lambert J.D. Does tea prevent cancer? Evidence from laboratory and human intervention studies. Am J Clin Nutr 2013;98:1667S-1675S. doi:10.3945/ajcn.113.059352
Lorenz M. Cellular targets for the beneficial actions of tea polyphenols. Am J Clin Nutr 2013;98:1642S-1650S. doi:10.3945/ajcn.113.058230
Mathur A., Gopalakrishnan D., Mehta V., Rizwan S.A., Shetiya S.H., Bagwe S. Efficacy of green tea-based mouthwashes on dental plaque and gingival inflammation: a systematic review and meta-analysis. Indian J Dent Res 2018;29(2):225-232. doi:10.4103/ijdr.IJDR_493_17
Mhatre S., Srivastava T., Naik S., Patravale V. Antiviral activity of green tea and black tea polyphenols in prophylaxis and treatment of COVID-19: a review. Phytomedicine 2020;153286. doi:10.1016/j.phymed.2020.153286
Sharma V.K., Bhattacharya A., Kumar A. and Sharma H.K. Health benefits of tea consumption. Trop J Pharm Res 2007;6(3):785-792.
Yang Y-C., Lu F-H., Wu J-S., Wu C-H., Chang C-J. The protective effect of habitual tea consumption on hypertension. Arch Intern Med 2004;164:1534-1540. doi:10.1001/archinte.164.14.1534
Yuan J-M. Cancer prevention by green tea: evidence from epidemiologic studies. Am J Clin Nutr 2013;98:1676S-1681S. doi:10.3945/ajcn.113.058271
Xu J., Xu Z., Zheng W. A review of the antiviral role of green tea catechins. Molecules 2017;22(8):1337. doi:10.3390/molecules22081337
Insieme con le catechine e le proantocianidine, le antocianine o antociani e i loro prodotti di ossidazione sono i flavonoidi più abbondanti nella dieta umana. Le antocianine si ritrovano:
in certe varietà di cereali pigmentati, come il riso nero o il mais viola;
in alcune verdure a foglia e a radice come melanzane, cavoli rossi, cipolle rosse e ravanelli, nei fagioli;
ma soprattutto nella frutta rossa.
Anche nel vino rosso sono presenti antociani (200-350 mg/L) che, nel corso dell’invecchiamento del vino stesso, sono trasformate in varie molecole complesse.
Il contenuto nei cibi è generalmente proporzionale all’intensità del colore del frutto o verdura: aumenta nel corso della maturazione e raggiunge valori fino a 2-4 g/kg di peso fresco nel ribes nero e mirtilli rossi americani (cranberries).
Questi polifenoli si trovano principalmente nella buccia, tranne che in certi tipi di frutta rossa, come ciliegie e frutti di bosco rossi, ad esempio le fragole, dove sono presenti sia nella buccia che nella polpa.
Gli antociani più comuni nei cibi sono i glicosidi della cianidina.
I frutti di bosco sono la principale fonte di antocianine, con valori variabili tra 66,8 e 947,5 mg/100 g di peso fresco.
Altri frutti, come l’uva rossa, le ciliegie e le prugne hanno contenuti variabili tra 2 e 150 mg/100 g di peso fresco.
Infine in frutti come pesche, nettarine e alcuni tipi di pere e mele sono scarsamente presenti, con un contenuto inferiori a 10 mg/100 g peso fresco.
Il mirtillo rosso americano, oltre ad avere un contenuto notevolmente elevato di antociani, è uno dei rari alimenti che contiene glicosidi delle sei antocianidine più comunemente trovate nei cibi: cianidina, peonidina, malvidina, pelargonidina, delfinidina, e petunidina. Gli antociani predominanti sono i 3-O-galattosidi e 3-O-arabinosidi della cianidina e peonidina; sono stati rilevati un totale di 13 antociani, principalmente in forma di 3-O-monoglicosi.
Assorbimento intestinale
Fino a poco tempo fa si riteneva che gli antociani, insieme alle proantocianidine e ai derivati dell’acido gallico delle catechine, fossero i polifenoli meno ben assorbiti dall’intestino, con un tempo di comparsa nel plasma coerente con l’assorbimento sia nello stomaco che nell’intestino tenue. In realtà, alcuni studi hanno rivelato che la loro biodisponibilità è stata sottovalutata dal momento che tutti i loro metaboliti potrebbero non essere ancora stati identificati.
A questo riguardo va sottolineato che solo una piccola parte delle antocianine presenti negli alimenti è assorbita come tale o come prodotti di idrolisi in cui lo zucchero è stato rimosso.
Quindi, una grande quantità di questi polifenoli ingeriti entra nel colon, dove possono anche subire reazioni di glucuronidazione, solfatazione, metilazione, e ossidazione.
Antocianine e microbiota del colon
Sono pochi gli studi che hanno esaminato il metabolismo degli antociani da parte del microbiota intestinale del colon, che è parte del più ampio microbiota umano.
Entro due ore sembra che tutti siano privati della loro componente zuccherina, liberando quindi antocianidine. Le antocianidine sono molecole chimicamente instabili nel pH neutro del colon che possono essere metabolizzate dalla microflora del colon o semplicemente essere degradate chimicamente con produzione di una serie di nuove molecole non ancora completamente identificate ma che comprendono acidi fenolici come:
acido gallico;
acido protocatecuico;
acido siringico;
acido vanillico;
floroglucinolo (1,3,5-triidrossibenzene).
Queste molecole, grazie alla loro maggiore stabilità sia chimica che microbica, potrebbero essere le principali responsabili delle attività antiossidanti e degli altri effetti fisiologici osservati in vivo e attribuiti agli antociani.
Bibliografia
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
de Pascual-Teresa S., Moreno D.A. and García-Viguera C. Flavanols and anthocyanins in cardiovascular health: a review of current evidence. Int J Mol Sci 2010;11:1679-1703. doi:10.3390/ijms11041679
Escribano-Bailòn M.T., Santos-Buelga C., Rivas-Gonzalo J.C. Anthocyanins in cereals. J Chromatogr A 2004:1054;129-141. doi:10.1016/j.chroma.2004.08.152
Han X., Shen T. and Lou H. Dietary polyphenols and their biological significance. Int J Mol Sci 2007;9:950-988. doi:10.3390/i8090950
Manach C., Scalbert A., Morand C., Rémésy C., and Jime´nez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr 2004;79(5):727-747. doi:10.1093/ajcn/79.5.727
Tsao R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010;2:1231-1246. doi:10.3390/nu2121231
Le foglie della pianta del tè, Camellia sinensis, sono ricche di composti dotati di molte attività biologiche, che spaziano dalla prevenzione dello sviluppo di patologie croniche alla riduzione dell’indice glicemico degli alimenti ricchi di amido.
Nell’infuso sono state individuate più di 4000 molecole differenti, di cui circa un terzo sono polifenoli, i fitochimici più importanti nel determinare il valore nutrizionale della tè.
La maggior parte dei polifenoli del tè appartengono al gruppo dei flavonoidi. Esempi sono le catechine del tè verde, tra cui la più importante e abbondante è la epigallocatechina-3-gallato o EGCG, e le tearubigine e teaflavine del tè nero, per le cui azioni a livello molecolare sembrano essere importanti i gruppi galloile presenti nelle posizioni 3 e/o 3’.
Epigallocatechina Gallato
Altri composti naturalmente presenti nelle foglie di Camellia sinensis sono:
alcaloidi, come caffeina, teofillina e teobromina;
aminoacidi, tra i quali uno dei più importanti è la teanina o R-glutamiletilamide, che è anche un neurotrasmettitore cerebrale e uno dei più importanti aminoacidi presenti nel tè verde;
I polifenoli, sia in vitro che in vivo, hanno un ampio spettro di attività biologiche che includono:
proprietà antiossidanti/pro-ossidanti;
un ruolo protettivo nei confronti dello sviluppo del diabete, delle iperlipidemie e di vari tipi di tumori;
inibizione dell’infiammazione;
attività antivirali;
un’attività inibitoria sulla digestione dell’amido;
attività anticariogene.
Data la loro abbondante presenza nel tè, negli ultimi anni è cresciuto l’interesse riguardo ai possibili effetti preventivi della bevanda nei confronti di diverse malattie, in particolare delle malattie cardiovascolari, ad esempio nello sviluppo e progressione dell’aterosclerosi.
Meccanismi d’azione
Attualmente si stanno accumulando molte informazioni sui meccanismi cellulari e molecolari attraverso cui i polifenoli del tè esercitano i loro effetti.
Sembra, almeno in vitro, che siano le catechine per il tè verde e le teaflavine e tearubigine per il tè nero, le molecole responsabili degli effetti fisiologici e dei benefici per la salute esercitati dalla bevanda.
Tra gli meccanismi molecolari con cui i polifenoli del tè sembrano agire, sono state osservate modifiche nell’attività di varie protein chinasi conseguenti al loro legame a specifici recettori presenti sulla membrana plasmatica. Le chinasi a loro volta andranno a fosforilare proteine bersaglio, quali ad esempio fattori di trascrizione che potranno così traslocare nel nucleo modificandone l’espressione genica. Questo sembra essere il meccanismo d’azione di EGCG e il meccanismo proposto per le tearubigine, molecole di grandi dimensioni che potrebbero non essere in grado di attraversare la membrana plasmatica.
In aggiunta al legame polifenolo-recettore di membrana, alcuni polifenoli potrebbero essi stessi passare all’interno della cellula legandosi poi a specifici bersagli citoplasmatici, mitocondriali o nucleari.
E, a seconda del tipo cellulare e della loro quantità, i polifenoli del tè potranno attivare o inibire determinati processi cellulari.
Digestione dell’amido
I polifenoli del tè esercitano un effetto inibitorio sulla digestione dell’amido.
Studi in vitro hanno evidenziato che estratti di tè verde, che contengono polifenoli monomerici, hanno una pari efficacia inibitoria sulla digestione dell’amido del pane con e senza glutine, mentre estratti di tè nero, ricchi di tannini, ossia polifenoli polimerici, risultano meno efficaci nel caso del pane con glutine.
Sembra quindi che l’azione inibitoria operata dai tannini sia influenzata negativamente dal glutine, mentre il glutine ha uno scarso effetto inibitorio nei confronti dell’azione dei polifenoli monometrici.
L’effetto inibitorio dei fitochimici è stato attribuito a diversi meccanismi, di seguito brevemente descritti.
Un’inibizione competitiva sulla alfa-amilasi pancreatica, azione in cui sembrano essere importante i gruppi galloile.
L’inibizione di altri enzimi digestivi presenti nel tratto gastrointestinale.
L’interazione con l’amido. I polifenoli potrebbero interagire direttamente con i granuli di amido, attraverso legami idrogeno e forze idrofobiche, riducendo in questo modo la superficie disponibile all’attacco da parte degli enzimi digestivi.
Di contro il glutine potrebbe ridurre la quantità di polifenoli in grado di interagire con l’amido e quindi in grado di inibirne la digestione.
I polifenoli del tè potrebbero rappresentare un mezzo per il controllo dell’indice glicemico dei cibi ricchi di amido. Tuttavia va sottolineato che, ad esempio nel caso del pane, per raggiungere un effetto inibitorio reale, l’ingestione di 100 g di pane deve essere accompagnata dalla coingestione di due tazze e mezzo di tè verde o due di tè nero.
Bibliografia
Arab L., Khan F., and Lam H. Tea consumption and cardiovascular disease risk. Am J Clin Nutr 2013;98:1651S-1659S. doi:10.3945/ajcn.113.059345
Dwyer J.T. and Peterson J. Tea and flavonoids: where we are, where to go next. Am J Clin Nutr 2013;98:1611S-1618S. doi:10.3945/ajcn.113.059584
Grassi D., Desideri G., Di Giosia P., De Feo M., Fellini E., Cheli P., Ferri L., and Ferri C. Tea, flavonoids, and cardiovascular health: endothelial protection. Am J Clin Nutr 2013;98:1660S-1666S. doi:10.3945/ajcn.113.058313
Kan L., Capuano E., Fogliano V., Oliviero T. and Verkerk R. Tea polyphenols as a strategy to control starch digestion in bread: the effects of polyphenol type and gluten. Food Funct 2020;11:5933-5943. doi: 10.1039/D0FO01145B
Lambert J.D. Does tea prevent cancer? Evidence from laboratory and human intervention studies. Am J Clin Nutr 2013;98:1667S-1675S. doi:10.3945/ajcn.113.059352
Lenore Arab L., Khan F., and Lam H. Tea consumption and cardiovascular disease risk. Am J Clin Nutr 2013;98:1651S-1659S. doi:10.3945/ajcn.113.059345
Lorenz M. Cellular targets for the beneficial actions of tea polyphenols. Am J Clin Nutr 2013;98:1642S-1650S. doi:10.3945/ajcn.113.058230
Mathur A., Gopalakrishnan D., Mehta V., Rizwan S.A., Shetiya S.H., Bagwe S. Efficacy of green tea-based mouthwashes on dental plaque and gingival inflammation: a systematic review and meta-analysis. Indian J Dent Res 2018;29(2):225-232. doi:10.4103/ijdr.IJDR_493_17
Mhatre S., Srivastava T., Naik S., Patravale V. Antiviral activity of green tea and black tea polyphenols in prophylaxis and treatment of COVID-19: a review. Phytomedicine 2020;153286. doi:10.1016/j.phymed.2020.153286
Sharma V.K., Bhattacharya A., Kumar A. and Sharma H.K. Health benefits of tea consumption. Trop J Pharm Res 2007;6(3):785-792.
Yuan J-M. Cancer prevention by green tea: evidence from epidemiologic studies. Am J Clin Nutr 2013;98:1676S-1681S. doi:10.3945/ajcn.113.058271
Xu J., Xu Z., Zheng W. A review of the antiviral role of green tea catechins. Molecules 2017;22(8):1337. doi:10.3390/molecules22081337
Gli isoflavoni sono polifenoli privi di colore appartenenti alla famiglia dei flavonoidi.
A differenza della maggior parte degli altri flavonoidi, hanno una distribuzione tassonomica limitata, trovandosi quasi esclusivamente nelle piante appartenenti alla famiglia delle Leguminose o Fabacee, in particolare nella soia.
Poiché i legumi, la soia in primis, sono una parte importante della dieta in molte culture, questi flavonoidi potrebbero avere un grande impatto sulla salute umana.
Si trovano anche nei fagioli e nelle fave, ma in concentrazioni molto minori rispetto a quelle presenti nella soia e nei prodotti derivati.
Un’altra buona fonte di tali molecole è il trifoglio rosso o trifoglio dei prati (Trifolium pratense), anch’esso appartenente alla famiglia delle Leguminose.
Nella frutta e verdura, al momento, non ne sono stati trovati.
Insieme agli acidi fenolici, quali l’acido caffeico e l’acido gallico, e ai glicosidi della quercetina, sono i polifenoli meglio assorbiti, seguiti dalle catechine (ma non le gallocatechine) e dai flavanoni.
Nelle piante alcuni isoflavoni sono dotati di attività antimicrobica e sono sintetizzati in risposta ad attacchi da parte di batteri o funghi; agiscono quindi come fitoalesine.
Mentre nella maggior parte dei flavonoidi l’anello B si lega all’anello C in posizione 2, negli isoflavoni l’anello B si lega all’anello C in posizione 3.
Scheletro di Base degli Isoflavoni
Anche se non sono steroidi, sono strutturalmente simili agli estrogeni, in particolare all’estradiolo. Questo conferisce loro proprietà pseudormonali, compresa la capacità di legarsi ai recettori per gli estrogeni, e sono per questo considerati fitoestrogeni o estrogeni vegetali. I benefici spesso ascritti alla soia e ai cibi a base di soia, ad esempio il tofu, si ritiene derivino dalla capacità degli isoflavoni presenti di agire come fitoestrogeni.
Va però sottolineato che il legame ai recettori per gli estrogeni sembra perdere forza con il tempo, per cui la loro efficacia non andrebbe sopravvalutata.
Negli alimenti sono presenti in quattro forme:
aglicone;
7-O-glucoside;
6’-O-acetil-7-O-glucoside;
6’-O-malonil-7-O-glucoside.
Isoflavoni della soia: genisteina, daidzeina e gliciteina
La soia e i suoi derivati, come il latte, il tofu, il tempeh e il miso, sono la principale fonte di isoflavoni nella dieta umana.
Il contenuto in isoflavoni della soia e dei prodotti derivati varia in modo considerevole in funzione della zona geografica e delle condizioni di crescita e lavorazione; ad es. la soia ne contiene tra 580 e 3800 mg/kg di peso fresco mentre il latte di soia tra i 30 e i 175 mg/L.
I più abbondanti in questi alimenti sono la genisteina, la daidzeina e la gliciteina, in genere presenti in rapporto di concentrazione 1:1:0,2; altri isoflavoni presenti sono la biocanina A e la formononetina.
Esempi di Isoflavoni
I 6’-O-malonil derivati hanno un gusto sgradevole, amaro e astringente, e quindi conferiscono un cattivo sapore ai cibi in cui sono contenuti. Tuttavia, essendo sensibili alla temperatura, sono spesso idrolizzati a glicosidi nel corso dei processi industriali, come la produzione del latte di soia.
I processi di fermentazione che sono necessari nella preparazione di cibi come il tempeh e il miso determinano a loro volta l’idrolisi dei glicosidi ad agliconi, ossia la molecola priva di zucchero.
I glicosidi degli isoflavoni della soia e dei prodotti della soia possono essere deglicosilati anche per azione delle β-glicosidasi dell’intestino tenue umano.
Gli agliconi sono molto resistenti al calore.
Sebbene molti composti presenti nella dieta siano convertiti dai batteri intestinali in molecole meno attive, in altri casi si verifica la conversione in molecole dotate di maggiore attività biologica. Questo è il caso degli isoflavoni, ma anche dei prenilflavonoidi del luppolo (Humulus lupulus), e dei lignani, anch’essi fitoestrogeni.
Fitoestrogeni e menopausa
Nelle donne in perimenopausa, anche detta transizione menopausale, e in menopausa vera e propria, i sintomi vasomotori, come le vampate di calore e le sudorazioni notturne, e la perdita di massa ossea sono molto comuni. La terapia sostitutiva ormonale (TOS) si è dimostrata un trattamento molto efficace per queste problematiche.
Il ricorso a terapie alternative a base di fitoestrogeni è aumentato a seguito della pubblicazione dei risultati del “Women’s Health Initiative” (WHI), i quali suggeriscono che la terapia sostitutiva ormonale potrebbero portare più rischi, in particolare un aumento della probabilità di sviluppare di alcune malattie croniche, che benefici.
Tra i fitoestrogeni più utilizzati dalle donne in menopausa ci sono gli isoflavoni della soia, spesso assunti in forma di alimenti fortificati o compresse. Molti studi hanno però messo in evidenza la mancanza di efficacia degli isoflavoni di soia, e del trifoglio rosso, anche in grandi dosi, nella prevenzione dei sintomi vasomotori, quali le vampate di calore e sudorazioni notturne, e della perdita di massa ossea durante la menopausa.
Bibliografia
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
Han X., Shen T. and Lou H. Dietary polyphenols and their biological significance. Int J Mol Sci 2007;9:950-988. doi:10.3390/i8090950
Lagari V.S., Levis S. Phytoestrogens for menopausal bone loss and climacteric symptoms. J Steroid Biochem Mol Biol 2014;139:294-301 doi:10.1016/j.jsbmb.2012.12.002
Lethaby A., Marjoribanks J., Kronenberg F., Roberts H., Eden J., Brown J. Phytoestrogens for menopausal vasomotor symptom. Cochrane Database of Systematic Reviews 2013, Issue 12. Art. No.: CD001395. doi:10.1002/14651858.CD001395.pub4
Levis S., Strickman-Stein N., Ganjei-Azar P., Xu P., Doerge D.R., Krischer J. Soy isoflavones in the prevention of menopausal bone loss and menopausal symptoms: a randomized, double-blind trial. Arch Intern Med 2011:8;171(15):1363-1369 doi:10.1001/archinternmed.2011.330
Manach C., Scalbert A., Morand C., Rémésy C., and Jime´nez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr 2004;79(5):727-747. doi:10.1093/ajcn/79.5.727
Tsao R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010;2:1231-1246. doi:10.3390/nu2121231
L’interesse sulle proantocianidine, in particolare sulle procianidine, loro forme dimeriche, è cresciuto a seguito della scoperta, conseguente a studi clinici e di laboratorio, delle loro proprietà antiinfiammatorie, anti-infettive, anti-carcinogeniche e cardioprotettive. Questi effetti protettivi sono stati attribuiti alla:
loro capacità di agire come scavenger nei confronti dei radicali liberi e di inibire la perossidazione dei lipidi di membrana;
sono inoltre in grado di agire su vari bersagli molecolari proteici all’interno della cellula, modulandone l’attività.
Le proantocianidine in alimenti differenti variano notevolmente in termini di:
In alcuni alimenti, come gli anacardi e i fagioli neri, sono presenti solamente dimeri, le procianidine di tipo A e B, mentre nella maggior parte degli altri si ritrovano in una vasta gamma di polimerizzazioni, da 2 a 10 unità e oltre.
Gli alimenti più ricchi di proantocianidine sono la cannella e il sorgo, che ne contengono rispettivamente circa 8000 e fino a 4000 mg/100 g di prodotto fresco.
Un’altra ricca fonte sono i semi d’uva (Vitis vinifera), con un contenuto di circa 3500 mg/100 g di peso secco.
Altre fonti importanti sono la frutta e i frutti di bosco, alcuni legumi, il vino rosso e in misura minore la birra, nocciole, pistacchi, mandorle, noci, e cacao.
Il caffè non è una buona fonte.
Nella maggior parte delle verdure le proantocianidine non sono rilevabili; in piccole concentrazioni sono state trovate nella zucca indiana.
Anche nel mais, riso e grano non sono rilevabili, mentre sono presenti nell’orzo.
Sebbene molti alimenti vegetali e prodotti derivati contengano elevate quantità di proantocianidine, solo pochi, come le prugne, l’avocado, le arachidi o la cannella, contengono procianidine di tipo A, ma nessuno in quantità pari ai mirtilli rossi americani (Cranberries, Vacciniun macrocarpon).
Le procianidine di tipo A mostrano, in vitro, una capacità di inibizione dell’adesione di Escherichia coli P-fimbriata alle cellule uroepiteliali, adesione che rappresenta la fase iniziale delle infezioni urogenitali, maggiore rispetto alle procianidine di tipo B.
Fonti alimentari di procianidine di tipo B
Le procianidine di tipo B, formate da catechina e/o epicatechina come unità costitutive, sono state rilevate come proantocianidine esclusive in 20 tipi di alimenti tra cui mirtilli neri (Vaccinium myrtillus), more, bacche dell’Aronia, semi d’uva, mele, pesche, pere, nettarine, kiwi, mango, datteri, banane, zucca indiana, sorgo, orzo, piselli occhio nero, fagioli neri, noci e anacardi.
Proantocianidine nella frutta
Nella dieta occidentale la frutta rappresenta la fonte più importante di proantocianidine.
Le principali fonti sono rappresentate dai:
frutti di bosco, come i mirtilli neri, mirtilli rossi americani e ribes nero, e susine o prugne, con un contenuto di circa 200 mg/100 g di prodotto fresco;
fonti intermedie sono mele e uva (60-90 mg/100 g);
negli altri frutti il contenuto è inferiore a 40 mg/100 g.
Nella frutta, le più comuni proantocianidine sono tetrameri, esameri e polimeri.
Buone fonti di proantocianidine sono anche alcuni succhi di frutta.
Proantocianidine nei semi d’uva
Come detto, una fonte particolarmente ricca di proantocianidine è rappresentata dai semi dell’uva.
Le proantocianidine presenti nei semi dell’uva sono solo procianidine di tipo B, per la maggior parte presenti come dimeri, trimeri e oligomeri altamente polimerizzati.
Le proantocianidine da questa fonte si sono dimostrate potenti antiossidanti e scavenger di radicali liberi, essendo più efficaci della vitamina C o della vitamina E.
Esperimenti condotti sia in vivo che in vitro supportano l’idea che le proantocianidine, in particolare quelle dei semi dell’uva, possano agire come agenti anti-carcinogenici; sembra che, nelle cellule cancerose, siano coinvolte nella:
riduzione della proliferazione cellulare;
nell’aumento dell’apoptosi;
nell’arresto del ciclo cellulare;
nella modulazione dell’espressione e dell’attività di NF-kB e dei geni bersaglio di NF-kB.
Bibliografia
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
Gu L., Kelm M.A., Hammerstone J.F., Beecher G., Holden J., Haytowitz D., Gebhardt S., and Prior R.L. Concentrations of proanthocyanidins in common foods and estimations of normal consumption. J Nutr 2004;134(3):613-617. doi:10.1093/jn/134.3.613
Han X., Shen T. and Lou H. Dietary polyphenols and their biological significance. Int J Mol Sci 2007;9:950-988. doi:10.3390/i8090950
Manach C., Scalbert A., Morand C., Rémésy C., and Jime´nez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr 2004;79(5):727-747. doi:10.1093/ajcn/79.5.727
Nandakumar V., Singh T., and Katiyar S.K. Multi-targeted prevention and therapy of cancer by proanthocyanidins. Cancer Lett 2008;269(2):378-387. doi:10.1016/j.canlet.2008.03.049
Ottaviani J.I., Kwik-Uribe C., Keen C.L., and Schroeter H. Intake of dietary procyanidins does not contribute to the pool of circulating flavanols in humans. Am J Clin Nutr 2012;95:851-858. doi:10.3945/ajcn.111.028340
Tsao R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010;2:1231-1246. doi:10.3390/nu2121231
Wang Y.,Chung S., Song W.O., and Chun O.K. Estimation of daily proanthocyanidin intake and major food sources in the U.S. diet. J Nutr 2011;141(3):447-452. doi:10.3945/jn.110.133900
Sono molecole prive di colore che si accumulano principalmente nei tessuti esterni e aerei, quindi pelle e foglie, di frutta e verdura, poiché la loro biosintesi è stimolata dalla luce solare, mentre sono praticamente assenti nella polpa.
Sono i flavonoidi più diffusi nella frutta e verdura, dove sono presenti generalmente in concentrazioni relativamente basse.
Data la loro diffusione in natura e nei cibi consumati dall’uomo, tali molecole devono essere tenute in considerazione quando si va ad analizzare l’effetto positivo sulla salute associato al consumo di frutta e verdura.
Il loro effetto è probabilmente legato alla loro capacità di:
agire come antiossidanti;
agire come agenti ad azione antiinfiammatoria;
agire come fattori antitumorali;
modulare diverse vie di segnalazione cellulare; un esempio è l’azione della quercetina, il flavonolo più diffuso, sulla attività ossidante delle MAPK indotta dallo stress.
Chimicamente si distinguono da molti altri flavonoidi in quanto presentano un doppio legame tra le posizioni 2 e 3 e un ossigeno in posizione 4 dell’anello C, al pari dei flavoni da cui però differiscono per la presenza di un gruppo ossidrilico in posizione 3. Dunque si può dire lo scheletro dei flavonoli è un 3-idrossiflavone.
3-Idrossiflavone
Il gruppo ossidrilico in posizione 3 può legare uno zucchero ossia può essere glicosilato.
Al pari di molti altri flavonoidi, la maggior parte di essi si trova nella frutta e verdura, e nei prodotti derivati, in forma glicosilata. Lo zucchero associato ai flavonoli è spesso rappresentato dal glucosio o dal ramnosio, ma possono essere coinvolti anche altri zuccheri, come:
I flavonoli sono rappresentati principalmente dai glicosidi di:
quercetina;
campferolo;
miricetina;
isoramnetina.
Flavonoli
I più diffusi sono i derivati glicosilati di quercetina e campferolo; in natura queste due molecole hanno rispettivamente almeno 279 e 347 diverse combinazioni glicosidiche.
Va infine sottolineato che il residuo di zucchero influenza la biodisponibilità del flavonolo.
Fonti alimentari
Le fonti principali nell’alimentazione umana sono:
frutta;
verdura;
bevande quali tè e vino rosso.
La fonte più ricca è rappresentata dai capperi, che ne contengono fino a 490 mg/100 g di peso fresco, ma si trovano abbondanti anche nelle cipolle, nel cavolo riccio, broccoli, porri, frutti di bosco, come i mirtilli, nell’uva e in alcune erbe e spezie, come l’aneto (Anethum graveolens). In queste fonti il loro contenuto varia da 10 a 100 mg/100 g di peso fresco.
Anche il cacao, il tè verde e il tè nero, ed il vino rosso ne sono fonti. Nel vino, insieme ad altri polifenoli come le catechine, le proantocianidine e polifenoli a basso peso molecolare, concorrono al carattere astringente della bevanda.
Principali flavonoli negli alimenti
I principali flavonoli presenti negli alimenti, in ordine decrescente di abbondanza, sono la quercetina, il kempferolo, la miricetina e la isoramnetina
Quercetina
L’alimento più ricco di quercetina è rappresentato dai capperi, seguiti da cipolle, asparagi, lattuga e frutti di bosco; in molta altra frutta e verdura è presente in quantità minori, attorno a 0,1-5 mg/100 g di peso fresco.
Questo flavonolo è presente anche nel cacao e potrebbe essere uno dei suoi principali fattori di protezione nei confronti dell’ossidazione delle LDL.
Insieme agli isoflavoni, i glicosidi della quercetina sono i polifenoli meglio assorbiti, seguiti dai flavanoni e dalle catechine. Al contrario i derivati dell’acido gallico delle catechine sono tra i polifenoli meno assorbiti, insieme con gli antociani e le proantocianidine.
Campferolo
Fonti caratteristiche di campferolo sono gli ortaggi, come indivia, cavolo e spinaci, con concentrazioni di circa 0,1-26,7 mg/100 g peso fresco, e alcune spezie, come erba cipollina, dragoncello, e finocchio, con concentrazioni di circa 6,5-19 mg/100 g di peso fresco.
I frutti sono una fonte povera della molecola, con un contenuto inferiore a 0,1 mg/100 g di peso fresco.
Miricetina
La miricetina è il terzo flavonolo più abbondante e si trova in alcune spezie, come prezzemolo, origano e finocchio con concentrazioni di circa 2-19,8 mg/100 g di peso fresco, ma anche nel tè, 0,5-1,6 mg/100 ml, e nel vino rosso, 0-9,7 mg/100 ml.
Nella frutta è presente in elevate concentrazioni solo nei frutti di bosco, mentre nella maggior parte dell’altra frutta e nella verdura è presente con un contenuto inferiore a 0,2 mg/100 g di peso fresco.
Isoramnetina
Un quarto flavonolo, meno abbondante rispetto ai precedenti, è la isoramnetina, presente solo in alcuni alimenti come alcune spezie quali: finocchio 9,3 mg/100 g di peso fresco, erba cipollina 5,0-8,5 mg/100 g di peso fresco, dragoncello 5 mg/100 g di peso fresco.
Nella frutta e verdura è presente solo nelle mandorle, dove varia tra 1,2 e 10,3 mg/100 g di peso fresco, nelle pere e cipolle.
Bibliografia
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
Han X., Shen T. and Lou H. Dietary polyphenols and their biological significance. Int J Mol Sci 2007;9:950-988. doi:10.3390/i8090950
Manach C., Scalbert A., Morand C., Rémésy C., and Jime´nez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr 2004;79(5):727-747. doi:10.1093/ajcn/79.5.727
Tsao R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010;2:1231-1246. doi:10.3390/nu2121231
Gli antociani o antocianine sono un sottogruppo di flavonoidi, e dunque di polifenoli, che conferisce alle piante i colori caratteristici.
Sono pigmenti solubili in acqua, si trovano disciolti nella linfa vacuolare dei tessuti epidermici di fiori e frutta, e sono responsabili dei colori della maggior parte dei petali, della frutta e verdura, e di alcune varietà di cereali come il riso nero.
A loro si devono i colori rosso, rosa e dal viola al blu dei frutti di bosco, delle mele rosse, dell’uva rossa, delle ciliegie, e di molti altri frutti, della lattuga rossa, del cavolo rosso, della cipolla o delle melanzane, ma anche del vino rosso.
Insieme ai carotenoidi sono responsabili del colore delle foglie in autunno e sono utilizzati a fini commerciali come coloranti alimentari.
Inoltre le antocianine concorrono ad attrarre gli animali quando il fiore è pronto per l’impollinazione o il frutto è pronto per essere mangiato.
Sono composti bioattivi presenti nei cibi di origine vegetale che hanno un duplice interesse per l’uomo:
tecnologico, conseguente al loro impatto sulle caratteristiche sensoriali del prodotto;
salutare, essendo implicati nella protezione nei confronti del rischio cardiovascolare.
in vitro, le antocianine hanno un’attività antiossidante, grazie alla loro capacità di delocalizzare gli elettroni e formare strutture di risonanza, e un ruolo protettivo nei confronti dell’ossidazione delle LDL;
al pari di altri polifenoli, come le catechine, le proantocianidine e altri flavonoidi non colorati, possono regolare diverse vie di segnalazione coinvolte nella sopravvivenza, crescita e differenziazione della cellula.
La struttura chimica di base è il catione flavilio o 2-fenilbenzopirilio cui si legano gruppi idrossilici (-OH), metossilici (-OCH3), e uno o più zuccheri.
La molecola priva di zucchero è detta antocianidina.
Catione Flavilio
In base al numero e alla posizione dei gruppi idrossilici e metossilici sono state descritte varie antocianidine, e di queste, sei si trovano comunemente nella frutta e verdura:
pelargonidina
cianidina
delfinidina
petunidina
peonidina
malvidina
Antociani o Antocianine
Le antocianine, come la maggior parte degli altri flavonoidi, sono presenti nelle piante e nei cibi derivati in forma di glicosidi, ossia legati a una o più unità glucidiche.
I tipi più comuni di carboidrati presenti in questi pigmenti naturali sono:
Gli zuccheri sono legati principalmente in posizione C3 come 3-monoglicosidi, in C3 e C5 come diglicosidi, con le possibili forme 3-diglicoside, 3,5-diglicosidi e 3-diglicoside-5-monoglicoside.
Sono state osservate anche glicosilazioni in posizione C7, C3’ e C5’.
La struttura di queste molecole è ulteriormente complicata dal legame allo zucchero di diversi tipi di sostituenti acilici quali:
acidi alifatici, come l’acido acetico, malico, succinico e malonico;
acidi cinnamici, aromatici, come l’acido sinapico, ferulico e p-cumarico;
infine, si ritrovano pigmenti con sostituenti sia alifatici che aromatici.
Inoltre in alcuni antociani si osserva la presenza di diversi zuccheri acilati nella struttura; questi antociani sono talvolta definiti come poliglicosidi.
Sulla base del tipo di idrossilazione, metossilazione e glicosilazione, come dei diversi sostituenti legati allo zucchero, sono state individuate oltre 500 antocianine differenti che si basano su 31 monomeri di antocianidine. Tra questi 31 monomeri:
il 30% deriva dalla cianidina;
il 22% dalla delfinidina;
il 18% dalla pelargonidina.
I derivati metilati delle sopracitate antocianidine, ossia peonidina, malvidina e petunidina, insieme rappresentano il 20% degli antociani.
Quindi il 90% degli antociani che si incontrano più di frequente sono relati alla cianidina, delfinidina e pelargonidina più i loro derivati metilati.
Ruolo del pH
Il colore delle antocianine dipende dal pH del vacuolo cellulare dove sono immagazzinate, variando dal:
rosso, in condizioni molto acide;
viola-blu, in condizioni di pH intermedio;
giallo-verde, in ambiente alcalino.
Oltre che dal pH, il colore di questi flavonoidi può essere influenzato dal grado di idrossilazione o dal tipo di metilazione degli anelli aromatici, come dal tipo di glicosilazione.
Infine il colore di certi pigmenti vegetali deriva da complessi tra antocianine, flavoni e ioni metallici.
Da notare che le antocianine sono spesso utilizzati come indicatori di pH grazie alle differenze nella struttura chimica che si verificano in risposta a cambiamenti di pH.
Bibliografia
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
de Pascual-Teresa S., Moreno D.A. and García-Viguera C. Flavanols and anthocyanins in cardiovascular health: a review of current evidence. Int J Mol Sci 2010;11:1679-1703. doi:10.3390/ijms11041679
Escribano-Bailòn M.T., Santos-Buelga C., Rivas-Gonzalo J.C. Anthocyanins in cereals. J Chromatogr A 2004:1054;129-141. doi:10.1016/j.chroma.2004.08.152
Han X., Shen T. and Lou H. Dietary polyphenols and their biological significance. Int J Mol Sci 2007;9:950-988. doi:10.3390/i8090950
Manach C., Scalbert A., Morand C., Rémésy C., and Jime´nez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr 2004;79(5):727-747. doi:10.1093/ajcn/79.5.727
Ottaviani J.I., Kwik-Uribe C., Keen C.L., and Schroeter H. Intake of dietary procyanidins does not contribute to the pool of circulating flavanols in humans. Am J Clin Nutr 2012;95:851-858. doi:10.3945/ajcn.111.028340
Tsao R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010;2:1231-1246. doi:10.3390/nu2121231
Le proantocianidine o tannini condensati, chiamate anche picnogenoli o leucocianidine, sono una sottogruppo di polifenoli, e in particolare di flavonoidi, ampiamente distribuito nel regno vegetale, e seconde solo alla lignina come fenolo più abbondante in natura.
Sono presenti in elevata concentrazione in varie parti delle piante come i fiori, i frutti, le bacche, i semi, come i semi d’uva, e la corteccia, ad esempio quella del pino.
Insieme agli antociani e i loro prodotti di ossidazione, e alle catechine, sono i flavonoidi più abbondanti nella dieta dell’uomo ed è stato suggerito che costituiscano una frazione significativa dei polifenoli ingeriti nella dieta occidentale.
Dunque i tannini condensati vanno presi in considerazione quando si studia l’associazione epidemiologica tra l’assunzione di polifenoli, in particolare dei flavonoidi, e le malattie croniche.
Le proantocianidine hanno una struttura chimica complessa essendo oligomeri, da dimeri a pentameri, o polimeri, da sei o più unità fino a 60, delle catechine o flavanoli, legate tra di loro da ponti carbonio-carbonio.
(epi)afzelechina, e sono definite propelargonidine;
(epi)gallocatechina, e sono definite prodelfinidine.
Propelargonidine e prodelfinidine sono meno frequenti in natura e nei cibi rispetto alle procianidine.
In base ai legami che si stabiliscono tra i monomeri le proantocianidine posso avere una struttura definita:
di tipo B se la polimerizzazione avviene tramite legame carbonio-carbonio tra la posizione 8 dell’unità terminale e la 4 della successiva o tra le posizioni 4 e 6;
di tipo A, meno frequente, se i monomeri sono doppiamente legati tramite un legame etere C2-O-C7 o C2-O-C5 e un legame di tipo B.
Procianidine
I dimeri più comuni sono procianidine di tipo B, da B1 a B8, formati da catechina o epicatechina, unite da legami C4-C8, da B1 a B4, o C4-C6,da B5 a B8.
Procianidine B1-B4
La procianidina C1 è un trimero di tipo B.
La procianidina A2 è un esempio di procianidina di tipo A.
Assorbimento intestinale
I tannini condensati sono scarsamente assorbiti a livello intestinale e, con gli antociani e i derivati esteri con l’acido gallico delle catechine del tè, sono i polifenoli meno ben assorbiti.
Sembra che gli oligomeri a basso peso molecolare, formati da 2-3 monomeri, possano essere assorbiti come tali mentre i polimeri non lo sono.
Nella circolazione sistemica i dimeri raggiungono concentrazioni di due ordini di grandezza inferiori rispetto a quelle delle catechine.
Le proantocianidine con un grado di polimerizzazione maggiore di tre sembra attraversino lo stomaco e l’intestino tenue senza significative modificazioni, per poi essere catabolizzate dal microbiota intestinale, che è parte del più ampio microbiota umano. I prodotti di degradazione includono gli acidi fenilacetico, fenilpropionico e fenilvalerico, acidi fenolici che sono stati suggeriti essere i principali metaboliti delle proantocianidine oligomeriche e polimeriche negli esseri umani sani.
Procianidine e catechine
In passato era stato proposto che il catabolismo intestinale delle procianidine portasse alla liberazione di catechine monomeriche, contribuendo così al loro pool sistemico negli esseri umani.
In realtà è stato dimostrato che ciò non accade in quanto le procianidine non contribuiscono in maniera significativa:
alla concentrazione dei metaboliti delle catechine nella circolazione sistemica;
al totale dei metaboliti delle catechine escreti con le urine;
infine, non influenzano in modo significativo il profilo dei metaboliti plasmatici derivanti dall’attività della catecol-O-metiltransferasi.
Pertanto quando si vanno ad analizzare i potenziali effetti benefici sulla salute associati all’assunzione di cibi contenenti questi fitochimici, catechine e procianidine debbono essere considerate classi distinte di composti correlati.
Bibliografia
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
Gu L., Kelm M.A., Hammerstone J.F., Beecher G., Holden J., Haytowitz D., Gebhardt S., and Prior R.L. Concentrations of proanthocyanidins in common foods and estimations of normal consumption. J Nutr 2004;134(3):613-617. doi:10.1093/jn/134.3.613
Han X., Shen T. and Lou H. Dietary polyphenols and their biological significance. Int J Mol Sci 2007;9:950-988. doi:10.3390/i8090950
Manach C., Scalbert A., Morand C., Rémésy C., and Jime´nez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr 2004;79(5):727-747. doi:10.1093/ajcn/79.5.727
Nandakumar V., Singh T., and Katiyar S.K. Multi-targeted prevention and therapy of cancer by proanthocyanidins. Cancer Lett 2008;269(2):378-387. doi:10.1016/j.canlet.2008.03.049
Ottaviani J.I., Kwik-Uribe C., Keen C.L., and Schroeter H. Intake of dietary procyanidins does not contribute to the pool of circulating flavanols in humans. Am J Clin Nutr 2012;95:851-858. doi:10.3945/ajcn.111.028340
Tsao R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010;2:1231-1246. doi:10.3390/nu2121231
Wang Y.,Chung S., Song W.O., and Chun O.K. Estimation of daily proanthocyanidin intake and major food sources in the U.S. diet. J Nutr 2011;141(3):447-452. doi:10.3945/jn.110.133900
Le catechine o flavanoli, con i flavonoli come la quercetina, e i flavoni come la luteolina, sono un sottogruppo di flavonoidi tra più diffusi in natura.
Flavanoli e proantocianidine, insieme con le antociani e i loro prodotti di ossidazione sono i flavonoidi più abbondanti nella dieta dell’uomo.
Chimicamente si distinguono da molti altri flavonoidi in quanto:
mancano del doppio legame tra la posizione 2 e 3 dell’anello C;
non presentano un gruppo chetonico in posizione 4;
in posizione 3 hanno un gruppo ossidrilico e per questo sono anche chiamati flavan-3-oli.
Scheletro di Base delle Catechine
Altra caratteristica distintiva dei flavan-3-oli è quella di formare oligomeri, formati da 2 a 10 unità, e polimeri, formati da 10 fino a 60 unità, detti proantocianidine o tannini condensati.
Fonti alimentari
Nei prodotti di origine vegetale si trovano di solito catechina, epicatechina, gallocatechina, epigallocatechina e i loro derivati esteri con l’acido gallico: catechina gallato, gallocatechina gallato, epicatechina gallato ed epigallocatechina gallato o EGCG.
Catechine
I flavanoli presenti con frequenza maggiore sono la catechina e la epicatechina, che sono anche tra i flavonoidi più comuni conosciuti, e quasi altrettanto diffusi come il flavonolo correlato quercetina.
Le fonti di gran lunga più ricche di flavanoli sono il cacao e il tè verde, dove i principali flavonoidi sono, oltre che catechina ed epicatechina, anche i loro derivati esteri con l’acido gallico, le gallocatechine.
Il cacao è anche una buona fonte anche di epigallocatechina.
Derivati Gallati delle Catechine
Sono comunque presenti anche in molti tipi di frutta, soprattutto nelle bucce di mele, mirtilli neri (Vaccinium myrtillus) e uva, nelle verdure, nel vino rosso, nella birra e nelle arachidi.
Poiché in molti casi i flavanoli sono presenti nelle bucce o nei semi di frutta e verdura, la loro assunzione può essere limitata dal fatto che queste parti sono eliminate prima di mangiare frutta e verdura o durante la loro lavorazione.
Inoltre rispetto alle altre classi di flavonoidi, le catechine presenti nei cibi non sono glicosilate.
Nei cibi di origine vegetale si trovano comunemente anche flavan-3-oli polimerici, le proantocianidine; è stata riportata la loro presenza nella buccia di arachidi e mandorle, come nei frutti di bosco.
Tè verde e nero
Il tè verde rappresenta un’ottima fonte di flavonoidi, e, come per i semi del cacao, anche nelle foglie del tè i principali flavonoidi presenti sono i flavanoli monomerici, catechina ed epicatechina, insieme con i loro derivati gallati, come la EGCG.
La epigallocatechina gallato è la catechina più abbondante nel tè verde e sembra avere un ruolo importante nel determinare gli effetti salutari della bevanda, come:
la riduzione dell’infiammazione vascolare;
l’abbassamento della pressione;
la riduzione della concentrazione delle LDL ossidate.
Il tè nero, che è un tè fermentato, contiene meno flavanoli monomerici, in quanto vengono ossidati durante il processo di fermentazione delle foglie con produzione di polifenoli più complessi come le teaflavine teaflavina digallato, teaflavina-3-gallato, e teaflavina-3’-gallato, tutte dimeri, e le tearubigine, che invece sono polimeri.
Teaflavine e tearubigine sono catechine presenti solamente nel tè, le cui concentrazioni misurate nell’infuso sono circa 50-100 volte più basse rispetto a quelle presenti nelle foglie.
Degno di nota è il fatto che le epicatechine del tè sono notevolmente stabili quando esposte al calore fin quando il pH è acido: solamente il 15% è degradato dopo 7 ore in acqua bollente a pH 5. Ne consegue che l’aggiunta di succo di limone all’infuso di tè non determina alcuna riduzione del loro contenuto.
Cacao e prodotti derivati
Il cacao ha il più alto contenuto in polifenoli e flavanoli per porzione, una concentrazione maggiore rispetto a quelle trovate nel tè verde e nel vino rosso. La maggior parte dei flavonoidi che si trovano nei semi di cacao e nei prodotti derivati come il cioccolato nero sono flavanoli monomerici, catechina ed epicatechina, ma anche epigallocatechina, ei loro derivati come le gallocatechine; tra i polimeri sono importanti anche le proantocianidine.
Frutta, verdura e legumi
La catechina e la epicatechina sono i principali flavanoli presenti nella frutta; si ritrovano in molti frutti in concentrazioni variabili rispettivamente tra 5-3 e 0,5-6 mg/100 g di peso fresco.
Al contrario gallocatechina, epicatechina gallato, epigallocatechina e epigallocatechina gallato sono presenti in diversi frutti come uva rossa, frutti di bosco, mele, pesche e prugne, ma in concentrazioni molto basse, inferiori a 1mg/100 g di peso fresco.
Con l’eccezione di lenticchie e fave, pochi legumi e verdure contengono catechine e in concentrazioni molto basse, inferiori a 1,5 mg/100 g di peso fresco.
Bibliografia
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
de Pascual-Teresa S., Moreno D.A. and García-Viguera C. Flavanols and anthocyanins in cardiovascular health: a review of current evidence. Int J Mol Sci 2010;11:1679-1703. doi:10.3390/ijms11041679
Han X., Shen T. and Lou H. Dietary polyphenols and their biological significance. Int J Mol Sci 2007;9:950-988. doi:10.3390/i8090950
Manach C., Scalbert A., Morand C., Rémésy C., and Jime´nez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr 2004;79(5):727-747. doi:10.1093/ajcn/79.5.727
Tsao R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010;2:1231-1246. doi:10.3390/nu2121231
I flavonoidi sono i polifenoli più abbondanti nella dieta dell’uomo, rappresentandone circa i 2/3 di tutti i quelli assunti.
Come gli altri fitochimici sono il prodotto del metabolismo secondario delle piante e, al momento, non è possibile stabilire precisamente il loro numero, anche se ne sono stati identificati oltre 4000.
Nella frutta e nella verdura si trovano generalmente in forma di glicosidi e in alcuni casi acilglicosidi, mentre meno frequentemente, e in concentrazioni minori, in forme acilate, metilate e solfate.
Sono molecole idrosolubili e si accumula all’interno dei vacuoli cellulari.
La loro struttura di base è costituita da uno scheletro di difenilpropano, ossia due anelli benzenici collegati da una catena di tre atomi di carbonio che forma un anello piranico, ossia un anello eterociclico contenente ossigeno, chiuso con l’anello benzenico A, che è detto anello C. La loro struttura è pertanto definita anche come C6-C3-C6.
Scheletro di Base dei Flavonoidi
Nella maggior parte dei casi l’anello B si lega all’anello C in posizione 2, ma può legarsi anche in posizione 3 o 4; questo, insieme con le caratteristiche strutturali dell’anello B e gli schemi di glicosilazione e idrossilazione dei tre anelli, fa si che i flavonoidi siano il gruppo di fitochimici, quindi non solo di polifenoli, più ampio e diversificato presente in natura.
Le attività biologiche di questi composti, ad esempio sono dei potenti antiossidanti, dipendono sia dalle caratteristiche strutturali che dallo schema di glicosilazione.
Classificazione dei flavonoidi
Possono essere suddivisi in diverse sottoclassi sulla base del carbonio dell’anello C su cui va a legarsi l’anello B, e del grado di insaturazione e ossidazione dell’anello C.
I flavonoidi in cui l’anello B si lega in posizione 3 dell’anello C sono detti isoflavoni; quelli in cui l’anello B si lega in posizione 4 neoflavonoidi, mentre quelli in cui l’anello B si lega in posizione 2 a loro volta essere suddividi in sei sottogruppi sulla base delle caratteristiche strutturali dell’anello C: flavoni, flavonoli, flavanoni, flavanonoli, flavanoli o catechine e antociani.
Infine, i flavonoidi con l’anello C aperto sono detti calconi.
Sottogruppi di Flavonoidi
Flavoni
Hanno un doppio legame tra la posizione 2 e 3 e un chetone in posizione 4 dell’anello C. La maggior parte dei flavoni della verdura e frutta presenta un gruppo idrossilico in posizione 5 dell’anello A, mentre l’idrossilazione in altre posizioni, per la maggior parte in posizione 7 dell’anello A o 3’ e 4’ dell’anello B, possono variare a seconda della classificazione tassonomica della particolare verdura o frutta.
La glicosilazione si verifica per la maggior parte sulle posizione 5 e 7, la metilazione e l’acilazione sui gruppi idrossilici dell’anello B.
Alcuni flavoni, come la nobiletina e la tangerina, sono polimetossilati.
Flavonoli
I flavonoli rispetto ai flavoni presentano un gruppo ossidrilico in posizione 3 dell’anello C, gruppo ossidrilico che può essere anche glicosilato. Di nuovo, al pari dei flavoni, anche i flavonoli sono molto vari per quello che riguarda l’idrossilazione e la metilazione, e, considerando i vari schemi di glicosilazione, i sono forse il sottogruppo più comune e ampio di flavonoidi nella frutta e verdura. Ad esempio, la quercetina è presente in moltissimi alimenti vegetali.
Flavanoni
I flavanoni, anche detti diidroflavoni, hanno l’anello C saturo; quindi, a differenza dei flavoni, mancano del doppio legame tra le posizione 2 e 3 e questa è l’unica differenza strutturale tra i due sottogruppi di flavonoidi. I flavanoni possono essere multi-idrossilati, e diversi gruppi idrossilici possono essere metilati e/o glicosilati. Alcuni hanno modelli unici di sostituzione, ad esempio, flavanoni prenilati, furanoflavanoni, piranoflavanoni o flavanoni benzilati, dando un gran numero di derivati sostituiti.
Negli ultimi 15 anni il numero dei flavanoni scoperti è notevolmente aumentato.
Flavanonoli
I flavanonoli, anche detti diidroflavonoli, sono i 3-idrossi derivati dei flavanoni; sono un sottogruppo altamente diversificato e multisostituito.
Isoflavoni
Come anticipato, gli isoflavoni sono flavonoidi in cui l’anello B si lega in posizione 3 dell’anello C. Hanno analogie strutturali con gli estrogeni, come l’estradiolo, e per questo sono anche detti fitoestrogeni.
Catechine
Le catechine o flavanoli sono dette anche flavan-3-oli poiché il gruppo ossidrilico è quasi sempre legato in posizione 3 dell’anello C; altro nome comune è catechine.
A differenza di molti flavonoidi non presentano un doppio legame tra le posizione 2 e 3; altro carattere distintivo, ad esempio rispetto ai flavanonoli, con cui condividono un ossidrile in posizione 3, è l’assenza di un carbonile, un gruppo chetonico, in posizione 4. Questa particolare struttura chimica permette ai flavanoli di avere due centri di chiralità nella molecola, sulle posizioni 2 e 3, quindi quattro possibili diastereoisomeri. La catechina è l’isomero con configurazione trans e la epicatechina è quello con configurazione cis. Ciascuna di queste due configurazioni ha due stereoisomeri, cioè, (+)-catechina e (-)-catechina, (+)-epicatechina e (-)-epicatechina.
La (+)-catechina e (-)-epicatechina sono i due isomeri spesso presenti nelle piante commestibili.
Un’altra importante caratteristica dei flavanoli, in particolare della catechina e della epicatechina, è quella di formare polimeri, detti proantocianidine o tannini condensati. Il nome “proantocianidine” deriva dal fatto che un clivaggio acido catalizzato produce antocianidine.
Le proantocianidine in genere contengono da 2 a 60 monomeri di flavanolo, in particolare catechina o epicatechina.
Sia i flavanoli monometrici che quelli oligomerici, formati da 2 a 7 monomeri, sono potenti antiossidanti.
Antocianidine
Chimicamente, le antocianidine sono cationi di flavilio e per la maggior parte si trovano come sali di cloruro.
Le antocianine o antociani sono i glicosidi delle antocianidine; lo zucchero si lega per la maggior parte dei casi in posizione 3 dell’anello C, zucchero che spesso si coniuga con acidi fenolici come l’acido ferulico.
Sono l’unico gruppo di flavonoidi dotati di colore, e , con i carotenoidi, colorano i vegetali. Essendo dotati di colore, sono utilizzati anche a fini commerciali come coloranti alimentari.
Il colore degli antociani dipende dal pH e dall’acilazione o metilazione dei gruppi idrossilici sugli anelli A e B.
Calconi
I calconi e gli diidrocalconi vengono considerati flavonoidi con struttura aperta; sono classificati tra i flavonoidi in quanto hanno vie di sintesi simili.
Bibliografia
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
Han X., Shen T. and Lou H. Dietary polyphenols and their biological significance. Int J Mol Sci 2007;9:950-988. doi:10.3390/i8090950
Manach C., Scalbert A., Morand C., Rémésy C., and Jime´nez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr 2004;79(5):727-747. doi:10.1093/ajcn/79.5.727
Panche A.N., Diwan A.D., and Chandra S.R. Flavonoids: an overview. J Nutr Sci. 2016;5:e47. doi:10.1017/jns.2016.41
Tsao R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010;2:1231-1246. doi:10.3390/nu2121231
I polifenoli sono tra i più importanti, e sicuramente i più numerosi, fitochimici presenti nel regno vegetale.
Attualmente sono note oltre 8000 strutture fenoliche, di cui più di 4000 appartenenti alla classe dei flavonoidi, e diverse centinaia sono presenti nei vegetali commestibili.
Si ritiene però che il contenuto totale dei polifenoli nei vegetali sia sottostimato in quanto molti dei composti fenolici presenti nella frutta, verdura e derivati non sono stati ancora identificati, sfuggendo alle tecniche di analisi utilizzate, e la composizione in polifenoli per la maggior parte dei frutti e alcune varietà di cereali non è ancora nota.
Queste molecole sono presenti in molti vegetali commestibili sia per gli uomini che per gli animali. e si ritiene che sia alla loro presenza, insieme a quella di altre molecole come i carotenoidi, la vitamina C o la vitamina E, che si debbano gli effetti salutari di frutta e verdura.
Nella dieta umana sono gli antiossidanti naturali più abbondanti, e le principali fonti sono frutta, verdura, cereali integrali, ma anche altri tipi di alimenti e bevande da essi derivati come il vino rosso, ricco di resveratrolo, l’olio di oliva extravergine, ricco di idrossitirosolo, il cioccolato e il tè, in particolare il tè verde, ricco di epigallocatechina gallato o EGCG.
Con il termine di polifenoli si indica una grande varietà di molecole che possono essere suddivise in molte sottoclassi, suddivisioni che possono essere fatte sulla base della loro origine, funzione biologica svolta o struttura chimica.
Chimicamente sono composti con caratteristiche strutturali fenoliche, che si possono associare a carboidrati differenti.
Nelle piante la maggior parte si trova legata a zuccheri, e quindi in forma di glicosidi, e ad acidi organici; in entrambe i casi i sostituenti si possono posizionare in posizioni differenti sugli scheletri polifenolici.
Tra i polifenoli si trovano molecole semplici, come gli acidi fenolici, o strutture complesse come le proantocianidine o tannini condensati, molecole altamente polimerizzate.
Classificazione
Possono essere suddivisi in diverse sottoclassi in base al numero di anelli fenolici presenti nella loro struttura, agli elementi strutturali che legano questi anelli tra di loro, e ai sostituenti legati agli anelli. Possono quindi essere individuati due grandi gruppi: i flavonoidi e i non flavonoidi.
I flavonoidi, che condividono una struttura formata da due anelli aromatici, indicati come A e B, legati insieme da 3 atomi di carbonio che formano un eterociclo ossigenato, l’anello C, possono essere ulteriormente suddivisi in 6 sottoclassi principali, in funzione del tipo di eterociclo coinvolto, ossia:
Variabilità del contenuto in polifenoli dei prodotti vegetali
Anche se diverse classi di molecole fenoliche, come la quercetina, un flavonolo, si trovano nella maggior parte dei cibi vegetali, come tè, vino, cereali, legumi, frutta, succhi di frutta, altre classi si trovano solo in un particolare tipo di cibo, ad esempio i flavanoni negli agrumi, gli isoflavoni nella soia, o la florizina nelle mele.
Tuttavia in natura è comune che diversi tipi di polifenoli si trovino nello stesso prodotto; e tale è il caso delle mele che contengono flavanoli, acido clorogenico, acidi idrossicinnamici, glicosidi della floretina, glicosidi della quercetina e antociani.
La composizione in polifenoli può essere influenzata anche da altri parametri come il grado di maturazione al momento del raccolto, fattori ambientali, la lavorazione, sia casalinga che industriale, la conservazione e la varietà vegetale.
Dai dati attualmente disponibili sembra che i frutti con il contenuto più alto in polifenoli siano le fragole, i litchi e l’uva, mentre le verdure con concentrazione maggiore sono i carciofi, il prezzemolo e i cavoletti di Bruxelles. Meloni e avocado hanno le concentrazioni più basse.
Bibliografia
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
Han X., Shen T. and Lou H. Dietary polyphenols and their biological significance. Int J Mol Sci 2007;9:950-988. doi:10.3390/i8090950
Manach C., Scalbert A., Morand C., Rémésy C., and Jime´nez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr 2004;79(5):727-747. doi:10.1093/ajcn/79.5.727
Tsao R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010;2:1231-1246. doi:10.3390/nu2121231
L’assunzione di carboidrati può migliorare la capacità di resistenza e la prestazione.
L’ingestione di diversi tipi di carboidrati, che utilizzano trasportatori intestinali differenti, può:
aumentare l’assorbimento totale dei carboidrati;
aumentare l’ossidazione dei carboidrati assunti;
migliorare la prestazione.
Dagli studi condotti la migliore combinazione è risultata essere maltodestrine e fruttosio nel rapporto 2:1.
Glucosio e fruttosio
Quando durante l’esercizio fisico prolungato viene assunta una miscela di glucosio e fruttosio, nella letteratura analizzata rispettivamente 1,2 e 0,6 g/min, rapporto 2:1, per una velocità di assunzione complessiva pari a 1,8 g/min, c’è una minor competizione per l’assorbimento intestinale rispetto all’ingestione di una quantità isoenergetica di solo glucosio o solo fruttosio, essendo coinvolti due trasportatori intestinali differenti. Inoltre, l’assorbimento del fruttosio è stimolato dalla presenza del glucosio.
Tutto ciò può:
aumentare la disponibilità di carboidrati esogeni nel sangue;
produrre una velocità di ossidazione dei carboidrati esogeni maggiore rispetto al solo glucosio.
Dalla coingestione di glucosio e fruttosio si ottiene una velocità di ossidazione dei carboidrati esogeni di circa 1,26 g/min, quindi maggiore rispetto a quella osservata con l’assunzione del solo glucosio (1 g/min) anche in alte concentrazioni.
La differenza osservata, ossia +0,26 g/min, può essere attribuita per intero all’ossidazione del fruttosio ingerito.
Saccarosio e glucosio
L’ingestione di saccarosio e glucosio, nelle stesse condizioni dell’ingestione di glucosio e fruttosio, quindi rispettivamente 1,2 e 0,6 g/min, in rapporto 2:1, per apporto complessivo di carboidrati pari a 1,8 g/min, dà risultati simili.
Glucosio, saccarosio e fruttosio
Con la combinazione di glucosio, saccarosio e fruttosio si ottengono velocità di ossidazione molto elevate (nella letteratura analizzata rispettivamente 1,2, 0,6 e 0,6 g/min, in rapporto 2:1:1, per apporto complessivo di carboidrati pari a 2,4 g/min; tuttavia, notare la maggiore quantità di carboidrati assunta).
Maltodestrine e fruttosio
Velocità di ossidazione elevate si osservano anche con combinazioni di maltodestrine e fruttosio, nelle stesse condizioni dell’ingestione di glucosio e fruttosio (quindi rispettivamente 1,2 e 0,6 g/min, in rapporto 2:1, per apporto complessivo di carboidrati pari a 1,8 g/min).
Queste elevate velocità di ossidazione possono essere raggiunte con carboidrati disciolti in una bevanda, presenti in un gel o in barrette a basso contenuto di grasso, proteina e fibra.
La migliore combinazione di carboidrati da assumere durante l’esercizio fisico prolungato è probabilmente la miscela di maltodestrine e fruttosio in rapporto 2:1, in una soluzione al 5%, per un apporto di circa 80-90 g/h.
Ossidazione dei Carboidrati Ingeriti
Perche?
Questa miscela ha il miglior rapporto tra la quantità di carboidrati ingerita e la loro velocità di ossidazione e questo significa che quantità più piccole di carboidrati rimangono nello stomaco o nell’intestino riducendo il rischio di complicanze/disturbi gastrointestinali durante esercizio prolungato.
Una soluzione che contenga diversi tipi di carboidrati e che ne abbia un contenuto non superiore al 5% ottimizza lo svuotamento gastrico e migliora l’apporto di liquidi.
Esempi di soluzioni di carboidrati al 5% contenenti circa 80-90 g di maltodestrine e fruttosio in rapporto 2:1; tempo di ingestione di circa un’ora:
1,5 L di soluzione: 80 g di carboidrati, rispettivamente circa 55 g di maltodestrine e circa 25 g di fruttosio.
1,8 L di soluzione: 90 g di carboidrati, rispettivamente 60 g di maltodestrine e 30 g di fruttosio.
Conclusioni
Durante l’esercizio fisico prolungato, quando sono necessarie elevate velocità di ossidazione dei carboidrati esogeni, è preferibile l’ingestione di carboidrati differenti rispetto a quella di grandi quantità di un singolo carboidrato.
La migliore combinazione sembra essere quella tra maltodestrine e fruttosio, in rapporto di 2:1, in una soluzione al 5%, e con una velocità di ingestione di circa 80-90 g/h.
Bibliografia
Burke L.M., Hawley J.A., Wong S.H.S., & Jeukendrup A. Carbohydrates for training and competition. J Sport Sci 2011;29:Sup1,S17-S27. doi:10.1080/02640414.2011.585473
Jentjens R.L.P.G., Moseley L., Waring R.H., Harding L.K., and Jeukendrup A.E. Oxidation of combined ingestion of glucose and fructose during exercise. J Appl Physiol 2004:96;1277-1284. doi:10.1152/japplphysiol.00974.2003
Jentjens R.L.P.G., Venables M.C., and Jeukendrup A.E. Oxidation of exogenous glucose, sucrose, and maltose during prolonged cycling exercise. J Appl Physiol 2004:96;1285-1291. doi:10.1152/japplphysiol.01023.2003
Jeukendrup A.E. Carbohydrate feeding during exercise. Eur J Sport Sci 2008:2;77-86. doi:10.1080/17461390801918971
Jeukendrup A.E. Nutrition for endurance sports: marathon, triathlon, and road cycling. J Sport Sci 2011:29;sup1, S91-S99. doi:10.1080/02640414.2011.610348
I carotenoidi sono una classe di pigmenti liposolubili ampiamente presenti in natura.
Sono composti organici di colore giallo, arancio e rosso, sono costituiti da 8 unità isopreniche e hanno numerosi doppi legami coniugati. La loro catena idrocarburica può andare incontro a modificazioni che ne influenzano significativamente le proprietà biologiche.
Scoperti nella prima metà del 1800, i carotenoidi sono prodotti da tutti gli organismi fotosintetici, comprese piante, macroalghe e microalghe. Inoltre sono sintetizzati anche da organismi non fotosintetici quali alcuni tipi di funghi, batteri e insetti come gli Afidi dei piselli, il ragnetto rosso, una specie di acaro, e alcune specie di moscerini.
Con polifenoli e glucosinolati, costituiscono il gruppo dei fitochimici.
Nel corso dell’evoluzione, grazie alle loro proprietà chimiche e fisiche, si sono dimostrati estremamente versatili, essendo in grado di svolgere molte funzioni sia nelle piante che negli animali, tra le quale è molto importante l’azione antiossidante. Inoltre, nell’uomo molti carotenoidi sono precursori della vitamina A.
Si contano più di 750 carotenoidi differenti, più di 100 sono stati trovati nella frutta e verdura, e circa 40 sono assunti in quantità significative dall’uomo.
Grazie al loro colore, i carotenoidi sono utilizzati come additivi alimentari.
I carotenoidi sono stati scoperti nella prima metà del diciannovesimo secolo.
Il primo carotenoide a essere isolato fu il beta-carotene nel 1831, grazie al lavoro del farmacista tedesco Heinrich Wilhelm Ferdinand Wackenroder, il quale chiamò carotene il pigmento giallo che riuscì a cristallizzare dalla radice della carota.
Jöns Jacob Berzelius, considerato uno dei padri della chimica moderna, chiamò xantofille, dal greco xanthos, che significa giallo, e phyllon che significa foglia, i pigmenti gialli estratti dalle foglie autunnali.
Mikhail Semyonovich Tsvet , botanico italo-russo, che nel 1906 ideò la cromatografia, riuscì a separare i pigmenti fogliari, ossia le clorofille dai caroteni e xantofille, grazie alla cromatografia su colonna, e chiamò carotenoidi i pigmenti dal giallo all’arancio.
Struttura chimica
I carotenoidi sono una classe di lipidi aventi come unità strutturale di base una sequenza lineare di otto unità isopreniche. Sono quindi terpeni, precisamente tetraterpeni, per un totale di 40 atomi di carbonio.
Le unità isopreniche sono unite con un relazione posizionale 1,5, ossia testa-coda, tranne al centro della catena idrocarburica, dove la relazione posizionale è 1,6, quindi coda-coda, il che, invertendo l’ordine, rende la molecola simmetrica.
I carotenoidi sono caratterizzati dalla presenza di un esteso sistema di doppi legami, dei quali da 3 a 13 possono essere coniugati.
La catena idrocarburica dei carotenoidi può andare incontro a modificazioni chimiche quali idrogenazione, deidrogenazione, introduzione di atomi di ossigeno, ciclizzazione di una o entrambe le estremità a formare anelli ionone, ed esterificazione con acidi grassi. Tali modificazioni permettono la formazione di molte strutture differenti, la maggior parte delle quali sono descritte dalla formula chimica C40H56On, con 0≤n≤6.
Tradizionalmente, ai carotenoidi sono stati assegnati nomi comuni provenienti dalla fonte biologica da cui sono stati isolati, sebbene possano essere denominati anche utilizzando la nomenclatura IUPAC.
Isomeria
I carotenoidi presentano molti doppi legami che possono avere isomeriacis–trans, anche detta isomeria geometrica. Sebbene ognuno dei doppi legami possa essere in configurazione cis o trans, in natura la configurazione prevalente è la trans, termodinamicamente più stabile rispetto alla configurazione cis grazie al minor ingombro sterico tra i sostituenti.
La rotazione intorno ai legami semplici, o legami sigma, presenti lungo la catena carboniosa permette anche la formazione di isomeri conformazionali. Quando i doppi legami sono presenti sullo stesso lato rispetto al legame sigma si parla di conformazione s-cis. Al contrario, se i doppi legami sono su lati opposti rispetto al legame sigma si parla di conformazione s-trans.
In natura i carotenoidi aciclici sono principalmente in conformazione s-trans, in quanto è quella a minor ingombro sterico. In presenza di strutture cicliche alle estremità della catena del carotenoide, la rotazione intorno al legame sigma C6-C7 da luogo alla formazione di un isomero 6-s-cis a minor ingombro sterico e quindi energeticamente favorito.
Si noti che l’ingombro sterico e le dimensioni dei carotenoidi influenzano la capacità di interagire con enzimi o di formare strutture sovramolecolari.
La maggior parte dei carotenoidi presenta anche un centro di chiralità o un asse di chiralità, che fa si che esistano isomeri ottici. In questi casi, per dare un nome univoco alla molecola si ricorre al sistema RS, e non alle regole della convenzione di Fischer, utilizzata solo per carboidrati e aminoacidi.
Classificazione
I carotenoidi possono essere classificati sulla base della presenza o meno di ossigeno o di strutture cicliche.
Sulla base della presenza o meno di ossigeno sono suddivisi in xantofille e caroteni.
I caroteni, privi di ossigeno, sono formati da soli atomi di carbonio e idrogeno, e hanno formula chimica C40H56. Esempi di caroteni sono l’alfa-carotene, il beta-carotene, il delta-carotene, lo zeta-carotene, il fitoene, e il licopene.
Le xantofille contengono anche atomi di ossigeno, hanno formula chimica C40H56On, con 0˂n≤6, e possono a loro volta essere suddivise in:
idrossicarotenoidi, che contengono almeno un gruppo idrossilico, come l’alfa-criptoxantina, la beta-criptoxantina, la zeaxantina e la luteina;
epossicarotenoidi, che presentano almeno un gruppo epossidico, come l’anteraxantina, l’auroxantina e la luteoxantina;
chetocarotenoidi, che presentano uno o più gruppi carbonilici, come l’astaxantina e la cantaxantina.
La presenza o meno di strutture cicliche nella molecola permette la suddivisione in carotenoidi ciclici e aciclici.
I carotenoidi ciclici contengono una o due strutture cicliche, e, rispetto agli aciclici sono più corti, ma con un ingombro sterico maggiore e un maggior spazio occupato. Esempi sono l’alfa-carotene, il beta-carotene, il gamma-carotene e il delta-carotene.
I carotenoidi aciclici sono molecole formate da una catena carboniosa lineare. Esempi sono il licopene, lo zeta-carotene, il fitoene, e il fitofluene.
Infine esistono anche carotenoidi non comuni o specie-specifici, come la bixina, la capsantina e la capsorubina.
Solubilità
I carotenoidi sono molecole idrofobiche, quindi solubili in solventi organici, solubilità che varia in funzione dei sostituenti presenti. Data lo loro idrofobicità, nelle cellule si ritrovano nelle membrane cellulari. Nella maggior parte dei casi le xantofille presentano i gruppi polari ai lati opposti della catena, e, quando presenti nelle membrane cellulari, al fine di minimizzare l’energia del sistema, si dispongono in modo tale che i gruppi polari siano in contato con i gruppi polari del doppi strato lipidico.
I carotenoidi possono avere accesso anche all’ambiente acquoso se in associazione a proteine, cui si legano in modo non covalente. Nel plasma sono trasportati dalle lipoproteine.
Colore
Il sistema di doppi legami coniugati, agendo come un cromoforo, è responsabile del colore dei carotenoidi. Tuttavia, perché il colore sia percettibile, devono essere presenti almeno sette doppi legami coniugati, per cui molecole come il fitofluene, che ha cinque legami coniugati, o il fitoene, che ha tre doppi legami coniugati, sono incolori.
All’aumentare del numero dei doppi legami coniugati il colore vira dal giallo, all’arancio, fino al rosso; ad esempio, la luteina, l’alfa-criptoxantina e la violaxantina sono gialle, l’alfa-carotene, il beta-carotene, e il gamma-carotene sono arancio, e il licopene è rosso.
Ruolo nelle piante
Nelle piante i carotenoidi svolgono molte funzioni.
Contribuiscono alla protezione della cellula nei confronti del danno ossidativo, aumentano l’assorbimento della luce solare, sono i precursori di fitormoni, come l’acido abscissico, e intervengono nelle vie di segnalazione.
Concorrono inoltre, con la clorofilla e gli antociani, una classe di flavonoidi, che sono il principale tipo di polifenoli, a determinare il colore delle foglie, dei frutti, delle verdure, dei cereali, colorazione che ha la funzione di attirare gli animali così da promuovere l’impollinazione e la dispersione dei semi.
Possono infine fungere anche da agenti repellenti per fitofagi e patogeni.
Quenching dell’ossigeno singoletto
Negli organismi che svolgono la fotosintesi ossigenica i carotenoidi concorrono alla protezione dei danni conseguenti all’eccessiva irradiazione solare.
Quando la luce che arriva complesso antenna del fotosistema II eccede la capacità di conversione dei centri di reazione, il surplus energetico può far si che la clorofilla permanga nel suo stato eccitato, il che può portare alla formazione del suo stato di tripletto. L’energia in eccesso può essere ceduta dalla clorofilla all’ossigeno molecolare determinando la formazione dell’ossigeno singoletto, una delle specie reattive dell’ossigeno, che è in grado di danneggiare l’apparato fotosintetico stesso e, più in generale, gli acidi nucleici, le proteine, gli acidi grassi insaturi di membrana, nonché i cofattori enzimatici.
I carotenoidi possono prevenire il danno ossidativo causato dall’ossigeno singoletto in due modi.
Possono reagire direttamente con l’ossigeno singoletto, un processo noto come quenching dell’ossigeno singoletto. In questo caso l’energia viene trasferita al carotenoide dall’ossigeno singoletto, che torna allo stato fondamentale di tripletto, mentre il carotenoide passa allo stato di tripletto, dissipando poi l’energia in eccesso in forma di calore. La capacità di disattivazione dell’ossigeno singoletto aumenta all’aumentare del numero dei doppi legami coniugati, divenendo massima da nove o più doppi legami coniugati.
Studi in vitro sembrano indicare che i carotenoidi monociclici siano in grado di disattivare l’ossigeno singoletto con maggiore efficienza rispetto alle strutture acicliche, e che la presenza di gruppi carbonilici, come nell’astaxantina, aumenti il potenziale antiossidante del carotenoide rispetto a quelli con gruppi idrossilici, probabilmente grazie al più esteso sistema di doppi legami coniugati. Sembra che ogni carotenoide, prima di incorrere in reazioni di degradazione, sia in grado di interagire e disattivare circa 1000 molecole di ossigeno singoletto.
I carotenoidi sono anche in grado di prevenire la formazione dell’ossigeno singoletto assorbendo l’energia della clorofilla eccitata, passando a loro volta in uno stato di tripletto. Essendo il loro livello energetico inferiore a quello dell’ossigeno singoletto, una volta formatisi decadono spontaneamente allo stato fondamentale.
Scavenging dei radicali liberi
I carotenoidi sono in grado di agire da scavenger dei radicali liberi, quali il radicale idrossilico, i radicali perossidici, il radicale superossido, o il radicale dell’ossido nitrico. La loro azione può avvenire secondo quattro modalità differenti.
Può verificarsi il trasferimento di un elettrone dal carotenoide al catione radicale, a formare il catione radicale del carotenoide, mentre il radicale diventa una specie neutra. Il catione del radicale carotenoide può essere rigenerato nel carotenoide di partenza grazie all’azione di altri antiossidanti cellulari come la vitamina E, la vitamina C e il glutatione.
Si può avere il trasferimento di un protone dal carotenoide al radicale, che diventa una specie neutra, mentre il carotenoide diviene un radicale anione.
La neutralizzazione del radicale può avvenire a seguito di reazioni di addizione, come nel caso della neutralizzazione dei radicali idrossilici e perossidici.
Un atomo di idrogeno può essere trasferito dal carotenoide al radicale, con formazione di un radicale carotenoide stabilizzato per risonanza.
Ruolo fotoassorbente
I carotenoidi sono presenti nei complessi antenna in qualità di pigmenti fotoassorbenti accessori, principalmente associati alle proteine antenna.
Grazie al sistema di doppi legami coniugati sono in grado di assorbire la luce solare nell’intervallo 400-500 nm. L’energia assorbita viene quindi convogliata ai centri di reazione.
Gli spettri di assorbimento dei singoli carotenoidi sono determinati dalla lunghezza della coniugazione e dal tipo di gruppi funzionali presenti.
Il principale carotene coinvolto nell’assorbimento della radiazione solare è il beta-carotene, presente nel nucleo del fotosistema I e II di tutti gli organismi che operano una fotosintesi ossigenica, mentre la luteina, la violaxantina, la neoxantina e la zeaxantina, le principali xantofille nelle piante, sono legate ai complessi di raccolta della luce.
Fonti alimentari
Gli esseri umani non sono in grado di sintetizzare i carotenoidi, li assumono dagli alimenti, li accumulano principalmente nel fegato e nel tessuto adiposo, ma anche nei polmoni, nei reni, nel cervello e nelle ossa, dove svolgono molte funzioni.
Di tutti i carotenoidi attualmente identificati, un centinaio sono presenti negli alimenti consumati dall’uomo, mentre, se si considera il singolo alimento, in genere ne sono presenti da uno a cinque principali, accompagnati da altri presenti in tracce o comunque piccole quantità.
Frutta e verdura sono la principale fonte di carotenoidi nella dieta umana. Tra le verdure, quelle a foglia verde sono ricche di beta-carotene, il carotenoide più abbondante nella dieta e nei tessuti umani, e luteina, seguiti da violaxantina e neoxantina, la radice delle carote è ricca di beta-carotene, la zucca di alfa-carotene, il pomodoro di licopene, e i peperoni di capsantina e capsorubina. I carotenoidi della frutta sono molto vari, e quelli dei frutti maturi possono essere diversi da quelli dei frutti acerbi. Inoltre, la maggior parte dei carotenoidi presenti nei frutti maturi sono esterificati con acidi grassi. Tuttavia, quelli di alcuni frutti, in particolare dei frutti che a maturazione terminata rimangono verdi, come il kiwi, subiscono una limitata o assente esterificazione.
Le uova di gallina, grazie al fatto che le galline ovaiole mangiano per la maggior parte il mais, possono essere una buona fonte di alcuni carotenoidi, come la luteina e zeaxantina, responsabili del colore giallo-arancio del tuorlo.
Infine, molluschi e crostacei, che con i pesci sono le fonti per eccellenza di acidi grassi polinsaturi omega-3 a catena lunga, quali l’acido eicosapentaenoico e l’acido docosaesaenoico, sono anche la principale fonte dei carotenoidi prodotti dalle microalghe e che raramente si trovano nelle piante, quali l’astaxantina presente nei gamberetti, ma anche nel salmone.
Additivi alimentari
I carotenoidi aggiunti ai cibi come additivi alimentari, a differenza dei carotenoidi naturalmente presenti, sono relativamente instabili, essendo suscettibili all’azione dell’ossigeno e della luce solare, all’auto-ossidazione, e alla dispersione nell’alimento, che ne facilita la degradazione. Inoltre, alla temperatura corporea e a quella di conservazione degli alimenti i carotenoidi aggiunti sono presenti in forma cristallina a causa del loro elevato punto di fusione; per ovviare a questo problema sono utilizzati in forma di emulsione olio in acqua.
Grazie al loro potere colorante sono utilizzati come coloranti in prodotti che, a causa della lavorazione o della conservazione tendono a perdere parte del loro colore naturale, o per uniformare il colore del prodotto, come nel caso delle bevande, dei succhi di frutta o degli insaccati, o per intensificarne il colore, migliorandone quindi l’aspetto.
Essendo precursori di composti responsabili del sapore e aroma di alcuni alimenti, possono essere utilizzati come esaltatori di sapidità.
Poiché alcuni carotenoidi sono precursori della vitamina A, sono utilizzati per fortificare gli alimenti.
Va infine sottolineato che, quando aggiunti agli alimenti, i carotenoidi non sono dei potenti antiossidanti.
Carotenoidi e salute dell’uomo
Nell’uomo i carotenoidi svolgono importanti funzioni.
Il ruolo più importante è quello di precursori della vitamina A, che da sola controlla l’espressione di quasi 700 geni. Non tutti i carotenoidi ne sono precursori; solo quelli che posseggono l’anello beta-ionone non sostituito possono essere convertiti nella vitamina, conversione che avviene a livello intestinale o nei tessuti di destinazione. Il più importante è il beta-carotene, che è la provitamina A per eccellenza e, avendo due anelli beta-ionone non sostituiti, viene convertito in due molecole di vitamina A. L’alfa-carotene e la beta-criptoxantina, avendo un solo anello beta-ionone non sostituito, possono dare una sola molecola di vitamina A, quindi hanno il 50% di attività di provitamina A.
L’importanza dell’attività di provitamina A svolta da alcuni carotenoidi è sottolineata dal fatto che la deficienza della vitamina può portare a cecità notturna e xeroftalmia, rallentare la crescita e la rigenerazione delle mucose, aumentare il tasso di mortalità causato dalle malattie infettive a seguito di disfunzioni della risposta immunitaria, ed è la singola causa più importante di cecità infantile nei paesi in via di sviluppo.
I carotenoidi contribuiscono alla salute dell’uomo anche concorrendo alla prevenzione di numerose patologie croniche attraverso meccanismi differenti rispetto all’azione di provitamina A, come l’azione antiossidante, antinfiammatoria e di fotoprotezione. Di seguito alcuni esempi.
E’ stato osservato che una supplementazione con luteina e zeaxantina, due xantofille che si accumulano nella macula lutea, è associata a un miglioramento della funzione visiva e a una riduzione del rischio di progressione della degenerazione maculare in tarda età.
Luteina e zeaxantina sembrano avere anche un effetto preventivo sullo sviluppo della cataratta e di retinopatie, e la luteina anche un effetto positivo sulle funzioni cognitive.
Tra i caroteni, il consumo di cibi ricchi di licopene è stato associato a una riduzione dell’incidenza di certi tipi di tumore, come il tumore della prostata e dello stomaco, e a una riduzione del rischio cardiovascolare.
Carotenoidi nel plasma
Alfa-carotene, beta-carotene, licopene, luteina, beta-criptoxantina e zeaxantina costituiscono oltre il 90% dei carotenoidi presenti nel plasma.
Le concentrazioni plasmatiche dei carotenoidi dipendono da diversi fattori tra cui i metodi di cottura dei cibi, la quantità di lipidi nella dieta, variabili individuali riguardanti i processi di digestione dei lipidi e assorbimento dei lipidi medesimi, con i sali biliari a svolgere un ruolo essenziale, e i livelli di vitamina A nell’organismo. Considerando quest’ultimo fattore, un soggetto che abbia bassi livelli di vitamina A avrà probabilmente tassi di conversione dei carotenoidi precursori della vitamina A più elevati, e ciò potrebbe riflettersi in una minore concentrazione plasmatica.
Bibliografia
Amengual J. Bioactive properties of carotenoids in human health. Nutrients 2019;11(10):2388. doi:10.3390/nu11102388
Archetti, M., Döring T.F., Hagen S.B., Hughes N.M., Leather S.R., Lee D.W., Lev-Yadun S., Manetas Y., Ougham H.J. Unravelling the evolution of autumn colours: an interdisciplinary approach. Trends Ecol Evol 2009;24(3):166-73. doi:10.1016/j.tree.2008.10.006
Böhm V., Lietz G., Olmedilla-Alonso B., Phelan D., Reboul E., Bánati D., Borel P., Corte-Real J., de Lera A.R., Desmarchelier C., Dulinska-Litewka J., Landrier J.F., Milisav I., Nolan J., Porrini M., Riso P., Roob J.M., Valanou E., Wawrzyniak A., Winklhofer-Roob B.M., Rühl R., Bohn T. From carotenoid intake to carotenoid blood and tissue concentrations – implications for dietary intake recommendations. Nutr Rev 2021;79(5):544-573. doi:10.1093/nutrit/nuaa008
Burrows T.L., Williams R., Rollo M., Wood L., Garg M.L., Jensen M., Clare Collins C.E. Plasma carotenoid levels as biomarkers of dietary carotenoid consumption: a systematic review of the validation studies. J Nutr Intermed Metab 2015;2(1-2):15-64. doi:10.1016/j.jnim.2015.05.001
de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, 2010
Desmarchelier C., Borel P. Overview of carotenoid bioavailability determinants: from dietary factors to host genetic variations. Trends Food Sci Technol 2017;69:270-280 doi:10.1016/j.tifs.2017.03.002
Eggersdorfer M., Wyss A. Carotenoids in human nutrition and health. Arch Biochem Biophys 2018;652:18-26. doi:10.1016/j.abb.2018.06.001
Gruszecki W.I., Strzałka K. Carotenoids as modulators of lipid membrane physical properties. Biochim Biophys Acta 2005;1740(2):108-15. doi:10.1016/j.bbadis.2004.11.015
Leyva-Porras C., Román-Aguirre M., Cruz-Alcantar, P., Pérez-Urizar, J.T., Saavedra-Leos M.Z. Application of antioxidants as an alternative improving of shelf life in foods. Polysaccharides 2021;2:594-607. doi:10.3390/polysaccharides2030036
Namitha K.K., Negi P.S. Chemistry and biotechnology of carotenoids. Crit Rev Food Sci 2010;50:728-760. doi:10.1080/10408398.2010.499811
Ramel F., Birtic S., Cuiné S., Triantaphylidès C., Ravanat J.L., Havaux M. Chemical quenching of singlet oxygen by carotenoids in plants. Plant Physiol 2012;158(3):1267-78. doi:10.1104/pp.111.182394
Rodriguez-Amaya D.B. Food carotenoids: chemistry, biology and technology. Wiley J. & Sons, 2015.
Saini R.K., Prasad P., Lokesh V., Shang X., Shin J., Keum Y.S., Lee J.H. Carotenoids: dietary sources, extraction, encapsulation, bioavailability, and health benefits – A review of recent advancements. Antioxidants 2022;11(4):795. doi:10.3390/antiox11040795
Sourkes T. L. The discovery and early history of carotene. Bull Hist Chem 2009;34(1)
Swapnil P., Meena M., Kumar Singh S., Praveen Dhuldhaj U., Harish, Marwal A. Vital roles of carotenoids in plants and humans to deteriorate stress with its structure, biosynthesis, metabolic engineering and functional aspects. Curr Plant Biol 2021;26: 100203. doi:10.1016/j.cpb.2021.100203
Xu P., Chukhutsina V.U, Nawrocki W.J., Schansker G, Bielczynski L.W., Lu Y., Karcher D., Bock R., Croce R. Photosynthesis without β-carotene. eLife 2020;9:e58984. doi:10.7554/eLife.58984
Il carico di carboidrati è un’ottima strategia per ottimizzare le riserve energetiche nei muscoli prima dell’inizio di una competizione di resistenza come la maratona, l’ironman, il nuoto in acque libere o una gara di ciclismo su strada.
Cosa “mangia” il muscolo durante gli sport di resistenza?
Negli sport di resistenza le cause più probabili alla base dell’insorgenza della fatica sono la disidratazione e la deplezione dei carboidrati, in particolare del glicogeno muscolare ed epatico.
Per evitare la “crisi” dovuta alla deplezione dei carboidrati muscolari ed epatici è fondamentale avere alla partenza ottimi depositi di glicogeno.
Cosa influenza i depositi di glicogeno?
L’alimentazione nei giorni precedenti la gara.
Il livello di allenamento: chi è più allenato sintetizza più glicogeno e ha depositi potenzialmente maggiori perché ha enzimi più efficienti;
L’attività il giorno della gara e i giorni precedenti, in quanto se il muscolo non lavora non perde glicogeno. Quindi nei giorni che precedono la gara è bene fare allenamenti leggeri, così da non intaccarne le riserve, e curare l’alimentazione.
L’origine “svedese” del carico di carboidrati
Negli eventi che durano più di 90 minuti, avere riserve muscolari di glicogeno molto elevate (si parla di supercompensazione del glicogeno) può migliorare la performance, ossia il tempo necessario per completare una data distanza, di un 2-3% in confronto con una situazione in cui le riserve di glicogeno sono normali o basse. Nelle competizioni con durata inferiore ai 90 minuti i benefici della supercompensazione sembrano essere piccoli o assenti.
Gli atleti ben allenati possono ottenere la supercompensazione delle riserve di glicogeno anche senza ricorrere alla fase di deplezione dei carboidrati precedente al carico degli stessi, vecchia tecnica messa a punto da due ricercatori svedesi, Saltin e Hermansen, negli anni ’60 del secolo scorso.
I due ricercatori scoprirono che la concentrazione muscolare del glicogeno poteva essere raddoppiata seguendo nei sei giorni precedenti la gara una dieta di questo tipo:
tre giorni di dieta ipoglucidica (poverissima di carboidrati);
tre giorni di dieta iperglucidica, il cosiddetto carico di carboidrati (dieta ricchissima di carboidrati).
Questa dieta crea un sacco di problemi: i primi tre giorni senza carboidrati, ossia senza pasta, riso, pane, patate, legumi, frutta ecc., sono durissimi, ci possono essere anche sintomi simili alla depressione dovuti al carente apporto di glucosio al cervello, mentre i vantaggi che si ottengono sono pochi.
Inoltre, con le tecniche di preparazione attuali, tipo e quantità di lavoro svolto, già si riescono a ottenere livelli di glicogeno elevati, oltre i 2,5 g/kg.
Il carico di carboidrati “moderno”
Immaginando di avere la gara la domenica un possibile schema per ottenere la supercompensazione delle riserve di glicogeno può essere il seguente:
mercoledì, ossia 4 giorni prima della competizione, allenamento discreto e poi cena senza carboidrati;
da giovedì, quindi 3 giorni prima della competizione, dieta iperglucidica, ossia il carico di carboidrati e allenamenti leggeri.
Carico di Carboidrati: Dieta da 2500 kcal
La quantità di carboidrati necessaria per ripristinare le scorte di glicogeno o per promuoverne il carico varia in funzione della durata e dell’intensità del programma di allenamento, ed è compresa tra 5 e 12 g/kg/d a seconda dell’atleta e della sua attività. Con apporti di carboidrati maggiori si possono ottenere scorte più elevate di glicogeno, ma non sempre questo determina prestazioni migliori. Inoltre c’è anche da considerare il fatto che l’accumulo di glicogeno si accompagna a un aumento di peso dovuto alla ritenzione di acqua, circa 3 grammi di acqua per ogni grammo di glicogeno, e per alcuni sport questo potrebbe non essere vantaggioso.
Bibliografia
Burke L.M., Hawley J.A., Wong S.H.S., & Jeukendrup A. Carbohydrates for training and competition. J Sport Sci 2011;29:Sup1,S17-S27. doi:10.1080/02640414.2011.585473
Hargreaves M., Hawley J.A., & Jeukendrup A.E. Pre-exercise carbohydrate and fat ingestion: effects on metabolism and performance. J Sport Sci 2004;22:31-38. doi10.1080/0264041031000140536
Jeukendrup A.E., C. Killer S.C. The myths surrounding pre-exercise carbohydrate feeding. Ann Nutr Metab 2010;57(suppl 2):18-25. doi:10.1159/000322698
Moseley L., Lancaster G.I, Jeukendrup A.E. Effects of timing of pre-exercise ingestion of carbohydrate on subsequent metabolism and cycling performance. Eur J Appl Physiol 2003;88:453-458. doi:10.1007/s00421-002-0728-8
L’ipotesi alla base della dieta alcalina afferma gli alimenti proteici e i cereali, con un basso apporto di potassio, portano ad una dieta acida, a una escrezione acida netta a livello renale, a un aumento del calcio nelle urine, e al rilascio di calcio dallo scheletro: tutto ciò causerebbe osteoporosi.
E’ vero?
Il calcio presente nelle ossa in forma di carbonati e fosfati rappresenta un grande serbatoio di basi, cioè di sostanze con cui tamponare l’acidità nel corpo. In risposta a un carico acido, come quello derivante da diete ricche di proteine, questi sali vengono rilasciati in circolo per mantenere costante il pH. Il minerale è quindi eliminato nelle urine. E’ stato stimato che la quantità persa da un soggetto che segua questo tipo di diete potrebbe arrivare a circa 480 g in 20 anni o quasi metà della massa scheletrica di calcio!
Anche se queste perdite di calcio possono essere tamponate mangiando alimenti ricchi di sostanze alcaline, come frutta e verdura, e molte informazioni on-line così come un certo numero di libri promuovono una dieta alcalina per la salute delle ossa, una recente meta-analisi ha mostrato che l’associazione causale tra malattia dell’osso da osteoporosi e carico acido dietetico non è supportata da prove e non vi è alcuna evidenza che una dieta alcalina sia protettiva nei confronti della salute delle ossa, mentre protegge contro il rischio di calcoli renali.
Nota: è possibile che la frutta e la verdura siano benefiche per la salute delle ossa attraverso meccanismi diversi da quanto supposto dall’ipotesi della dieta alcalina.
Qual è il ruolo delle proteine?
L’eccesso di proteine nella dieta, a causa del loro elevato carico acido renale, può ridurre la densità ossea se non tamponato dalla ingestione di alimenti che ricchi di alcali, cioè frutta e verdura. Tuttavia, un adeguato apporto di proteine è necessario per il mantenimento dell’integrità ossea. Pertanto, potrebbe essere necessario l’aumento della quantità di frutta e verdura, invece della eccessiva riduzione delle proteine.
E’ quindi consigliabile consumare una dieta normo-proteica ricca di frutta e verdura e povera di sodio, come la dieta mediterranea. Un corretto abbinamento dei cibi prevede l’ingestione di alimenti con un carico acido negativo accompagnati da altri un carico acido positivo. Esempio: pasta più verdure o carne più verdura e frutta, le ultime due, in particolare la verdura, in porzioni generose.
Dieta alcalina e massa muscolare
Con l’avanzare dell’età vi è una perdita di massa muscolare che predispone a cadute e fratture. Una dieta ricca di potassio, ottenuto da frutta e verdura, così come un carico acido ridotto, protegge la massa muscolare sia negli uomini che nelle donne anziane.
Dieta alcalina e ormone della crescita
Nei bambini gravi forme di acidosi metabolica sono associate a bassi livelli di ormone della crescita, con conseguente bassa statura: la correzione dell’acidosi con bicarbonato o citrato di potassio aumenta in modo significativo l’ormone della crescita migliorando la crescita.
Nelle donne in postmenopausa, l’uso di sufficiente bicarbonato di potassio nella dieta per neutralizzare il carico acido netto giornaliero ha determinato un aumento significativo dell’ormone della crescita e dell’osteocalcina risultante.
Migliorare i livelli di ormone della crescita può ridurre i fattori di rischio cardiovascolare, migliorare la qualità della vita, la composizione corporea, e anche la memoria e la cognizione.
Conclusioni
La dieta alcalina può comportare un certo numero di benefici per la salute.
L’aumento del consumo di frutta e verdura migliorerebbe il rapporto potassio/sodio e ciò potrebbe essere di beneficio per la salute delle ossa, potrebbe ridurre la perdita muscolare, così come mitigare altre malattie croniche come l’ipertensione e ictus.
L’aumento dell’ormone della crescita può avere molti effetti positivi, dalla salute cardiovascolare alla memoria e cognizione.
L’aumento del magnesio intracellulare è un altro vantaggio ulteriore della dieta alcalina; ad esempio, il magnesio, necessario per attivare la vitamina D, comporterebbe numerosi benefici aggiunti nei sistemi ormonali in cui la vitamina è implicata.
Va notato che una delle prime considerazioni riguardanti una dieta alcalina, che comprende più frutta e verdura, è sapere in che tipo di terreno questi cibi sono state coltivati poiché può influenzare in modo significativo il loro contenuto in minerali e quindi il loro potere tamponante.
Bibliografia
Fenton T.R., Lyon A.W., Eliasziw M., Tough S.C., Hanley D.A. Meta-analysis of the effect of the acid-ash hypothesis of osteoporosis on calcium balance. J Bone Miner Res 2009;24(11):1835-1840. doi:10.1359/jbmr.090515
Fenton T.R., Lyon A.W., Eliasziw M., Tough S.C., Hanley D.A. Phosphate decreases urine calcium and increases calcium balance: a meta-analysis of the osteoporosis acid-ash diet hypothesis. Nutr J 2009;8:41. doi:10.1186/1475-2891-8-41
Fenton T.R., Tough S.C., Lyon A.W., Eliasziw M., Hanley D.A. “Causal assessment of dietary acid load and bone disease: a systematic review and meta-analysis applying Hill’s epidemiologic criteria for causality.” Nutr J 2011;10:41. doi:10.1186/1475-2891-10-41
Schwalfenberg G.K. The alkaline diet: is there evidence that an alkaline pH diet benefits health? J Environ Public Health 2012; Article ID 727630. doi:10.1155/2012/727630
La vita dipende da adeguati valori di pH sia all’interno dell’organismo e delle cellule che lo compongono che nell’ambiente circostante.
Gli esseri umani per evitare l’acidosi metabolica e sopravvivere necessitano di uno strettissimo controllo del valore del pH del sangue, che è pari a circa 7,4. A titolo di confronto, negli ultimi 100 anni il pH del mare è sceso da 8,2 a 8,1 a causa della crescente dissoluzione al suo interno di anidride carbonica, con un impatto negativo sulla vita degli oceani.
Anche il contenuto in minerali del cibo che mangiamo è notevolmente influenzato dal pH del terreno in cui le piante sono coltivate.
La scala del pH
Il pH ideale del terreno per ottenere la migliore disponibilità complessiva di nutrienti essenziali è compresa tra 6 e 7: un terreno acido, con pH inferiore a 6, può avere un ridotto contenuto in magnesio e calcio, mentre valori superiori a pH 7 possono rendere chimicamente non disponibili zinco, ferro, rame e manganese.
Acidosi metabolica e rivoluzione agricola e industriale
Nella dieta umana, dalla società dei cacciatori-raccoglitori alla presente, c’è stato un notevole cambiamento nel pH e nel carico acido netto della dieta umana. Con la rivoluzione agricola, e ancora più di recente, con l’industrializzazione, si è osservato:
un incremento della quantità di sodio rispetto a quella di potassio, con il rapporto potassio/sodio che si è invertito passando da 10 a 1 a 1 a 3, e del cloruro rispetto al bicarbonato;
Ne risulta una dieta che può portare ad acidosi metabolica, una condizione che non corrisponde a quelle che sono le esigenze nutrizionali geneticamente determinate.
Inoltre, con l’invecchiamento, si assiste a una progressiva perdita della funzione regolatrice renale dell’equilibrio acido-base e al conseguente aumento dell’acidosi metabolica indotto dalla dieta.
Infine, una dieta povera di carboidrati ma ad alto contenuto proteico, con il suo aumentato carico acido, determina cambiamenti molto piccoli nella chimica del sangue, e nei valori del suo pH, ma causa molti cambiamenti nella chimica delle urine. Si osserva infatti un aumento del calcio, dell’acido urico indissociato e del fosfato, mentre si riducono il magnesio, il citrato e il pH delle urine.
Tutto ciò aumenta il rischio di calcoli renali.
Il pH come scudo acido
Il corpo umano ha una straordinaria capacità di mantenere un pH stabile nel sangue, con i principali meccanismi di compensazione presenti a livello renale e respiratorio.
Il pH nel corpo varia notevolmente da una zona all’altra.
L’acidità massima si trova nello stomaco (pH 1,35-3,5), dove aiuta nella digestione e ci protegge nei confronti dei microrganismi opportunisti.
La pelle è abbastanza acida (pH 4-6,5), e questo fornisce un “scudo” acido che funge da barriera protettiva verso l’ambiente nei confronti della proliferazione microbica. Ciò si osserva anche nella vagina, dove un pH inferiore a 4,7 protegge dalla proliferazione microbica.
L’urina ha un pH variabile da acido ad alcalino a seconda delle necessità di equilibrare l’ambiente interno.
Neutralizza l’acidità gastrica, aiuto nella digestione
Succo pancreatico
8,8
Neutralizza l’acidità gastrica, aiuto nella digestione
Fluido vaginale
<4,7
Limita la sovracrescita dei microbi opportunistici
Liquido cerebrospinale
7,3
Bagna l’esterno del cervello
Liquido intracellulare
Tra 6,0 e 7,2
Dovuto alla produzione di acido nelle cellule
Siero venoso
7,35
Strettamente regolato
Siero arterioso
7,4
Strettamente regolato
Modificato da: Schwalfenberg G.K.; vedere nella Bibliografia
Bibliografia
Fenton T.R., Lyon A.W., Eliasziw M., Tough S.C., Hanley D.A. Meta-analysis of the effect of the acid-ash hypothesis of osteoporosis on calcium balance. J Bone Miner Res 2009;24(11):1835-1840. doi:10.1359/jbmr.090515
Fenton T.R., Lyon A.W., Eliasziw M., Tough S.C., Hanley D.A. Phosphate decreases urine calcium and increases calcium balance: a meta-analysis of the osteoporosis acid-ash diet hypothesis. Nutr J 2009;8:41. doi:10.1186/1475-2891-8-41
Fenton T.R., Tough S.C., Lyon A.W., Eliasziw M., Hanley D.A. Causal assessment of dietary acid load and bone disease: a systematic review and meta-analysis applying Hill’s epidemiologic criteria for causality. Nutr J 2011;10:41. doi:10.1186/1475-2891-10-41
Schwalfenberg G.K. The alkaline diet: is there evidence that an alkaline pH diet benefits health? J Environ Public Health 2012; Article ID 727630. doi:10.1155/2012/727630
Una delle funzioni più importanti del fegato è quella di partecipare alla regolazione della glicemia, ossia al mantenimento dei livelli di glucosio nel sangue entro un intervallo ben definito, che in condizioni normali è pari a 60-100 mg/dL o 3,33-5,56 mmol/L. Per fare ciò rilascia glucosio nel sangue:
nel digiuno;
nell’intervallo tra i pasti;
durante l’attività muscolare.
Regolazione della glicemia: ruolo della glucosio-6-fosfatasi epatica
Nel fegato il glicogeno è la forma di deposito del glucosio il quale non viene rilasciato come tale ma in forma fosforilata, ossia dotata di carica, il glucosio-1-fosfato, nel processo detto glicogenolisi.
La molecola fosforilata non può diffondere liberamente dalla cellula: a livello epatico è presente l’enzima glucosio-6-fosfatasi che idrolizza il glucosio-6-fosfato, prodotto dal glucosio-1-fosfato nella reazione catalizzata dalla fosfoglucomutasi, a glucosio, una defosforilazione irreversibile.
glicogeno(n residui di glucosio) + Pi → glucosio-1-fosfato + glicogeno(n-1 residui di glucosio)
glucosio-1-fosfato ↔ glucosio-6-fosfato
glucosio-6-fosfato + H2O → glucosio + Pi
Il glucosio potrà così lasciare l’epatocita, per mezzo di un trasportatore di membrana detto GLUT2, e riversarsi nel circolo sanguigno per essere distribuito alle cellule extraepatiche, in primis neuroni e globuli rossi per i quali rappresenta la principale, e per i globuli rossi l’unica fonte di energia. I neuroni, con l’esclusione di quelli presenti in alcune zone del cervello che non possono fare a meno del glucosio, sono in grado di utilizzare un’altra fonte di energia che diviene preponderante nei periodi di digiuno prolungato, i corpi chetonici.
Il fegato ricava la maggior parte dell’energia di cui necessita dall’ossidazione degli acidi grassi, una classe di lipidi, e non dal glucosio.
La glucosio-6-fosfatasi oltre che nel fegato è presente anche nel rene e nell’intestino mentre è assente nel muscolo e nel cervello. Pertanto in queste sedi il glucosio-6-fosfato non potrà essere rilasciato dalla cellula.
La glucosio-6-fosfatasi ha un ruolo importante anche nella gluconeogenesi.
La glucosio-6-fosfatatasi è presente nella membrana del reticolo endoplasmatico. L’idrolisi del glucosio-6-fosfato avviene nel lume del reticolo, quindi questa reazione è separata dalla via glicolitica, ed è necessaria la presenza di uno specifico trasportatore, la glucosio-6-fosfato traslocasi, per portare lo zucchero fosforilato dal citosol nel lume del reticolo stesso. Sebbene nella membrana del reticolo endoplasmatico sia presente un trasportatore per il glucosio, la maggior parte del glucosio liberato non è ritrasportato nel citosol della cellula ma secreto nel circolo ematico. Infine un trasportatore anionico trasloca il fosfato inorganico liberato dal lume del reticolo endoplasmatico nel citosol.
Bibliografia
Nelson D.L., M. M. Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
Roach P.J., Depaoli-Roach A.A., Hurley T.D and Tagliabracci V.C. Glycogen and its metabolism: some new developments and old themes. Biochem J 2012;441:763-787. doi:10.1042/BJ20111416
Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
La letteratura scientifica internazionale è concorde nello stabilire come limite inferiore per l’apporto calorico giornaliero 1200 kcal per la donna e 1500 kcal per l’uomo.
Per rendere il bilancio calorico giornaliero negativo, perdere peso, ma soprattutto perdere massa grassa, alla valutazione delle reali necessità caloriche del soggetto sarà da affiancarsi:
la corretta suddivisione dei pasti nella giornata;
un’aumentata attività fisica, grazie alla quale il bilancio negativo potrà essere ottenuto senza particolari sacrifici a tavola.
Bilancio Calorico Giornaliero
Questo renderà il dimagrimento più facile e proteggerà dai successivi aumenti di peso, in particolare da quello che è conosciuto come effetto yo-yo.
In definitiva ci deve essere un cambio nello stile di vita.
Dunque, la strategia migliore per perdere massa grassa non è una drastica riduzione dell’apporto calorico e neppure seguire regimi alimentari costrittivi, ad es. diete che promettono miracoli come la “dieta del minestrone”, la dieta Plank, la dieta Master Cleanse e molte altre che obbligano a eliminare o ridurre fortemente l’apporto di determinati macronutrienti, quasi sempre i carboidrati, con apporti spesso esagerati di proteine. Condotte simili possono essere:
molto stressanti dal punto di vista psicologico;
non percorribili per lunghi periodi;
rischiose per la salute a causa di inevitabili carenze di nutrienti.
Infine non assicurano affatto che i chili persi siano solo o quasi solo massa grassa, e spesso sono seguite da aumenti del peso corporeo sostanziali ossia da quello che è conosciuto come effetto yo-yo.
Perché?
Effetti sulla massa grassa della restrizione calorica
Una riduzione eccessiva dell’apporto calorico vuol dire mangiare molto poco. Ciò determina il rischio, elevato, di non assumere in quantità adeguate i diversi nutrienti essenziali, quelli cioè che non possiamo sintetizzare, come le vitamine, alcuni aminoacidi, gli acidi grassi essenziali, che sono una sottoclasse di lipidi, e i minerali, tra cui il calcio, indispensabile per il metabolismo osseo in ogni fase della vita, o il ferro che è utilizzato in molte funzioni del nostro organismo come il trasporto di ossigeno ai tessuti. Tutto ciò si traduce anche in una depressione del metabolismo e quindi una riduzione dei consumi. Se poi la riduzione dell’apporto calorico è eccessiva o addirittura ci sono periodi di digiuno al danno si aggiunge la beffa in quanto si perderà una quota di massa magra. In che modo?
Riduzione delle calorie e ruolo dei carboidrati
Il glucosio rappresenta l’unica fonte di energia per i globuli rossi e alcune zone del cervello, mentre altre zone possono ricavare energia anche dai corpi chetonici, un prodotto del metabolismo degli acidi grassi.
A riposo il cervello estrae il 10% del glucosio dal circolo, una quantità non trascurabile, circa 75 mg/min., se si considera che il suo peso è di circa 1,5 kg. Per mantenere costante la glicemia, e quindi assicurare un costante rifornimento di glucosio ai vari tessuti, è necessario assumere carboidrati, o in alternativa aminoacidi, entrambe facilmente ottenibili dagli alimenti.
In caso di un apporto dietetico di carboidrati scarso o assente, considerando che dopo circa 18 ore si esaurisce il glicogeno epatico che rilascia in circolo glucosio, l’organismo sintetizza de novo glucosio a partire da alcuni aminoacidi attraverso un processo detto gluconeogenesi. Questa via metabolica è attiva anche dopo un pasto normale, ma in caso di digiuno la sua importanza aumenta.
Ma qual è la principale fonte di aminoacidi nell’organismo nel caso in cui anche il loro apporto dietetico sia scarso o assente? Le proteine endogene, ed esiste una sorta di gerarchia nella loro utilizzazione cioè prima si consumano quelle che servono di meno e solo in seguito quelle più importanti. Per primi saranno utilizzati gli enzimi della digestione, pepsina, chimotripsina, elastasi, carbossipeptidasi e aminopeptidasi, in tutto 35-40 g; in seguito fegato e pancreas rallentano la loro attività di sintesi di proteine da esportare e gli aminoacidi inutilizzati sono avviati verso la gluconeogenesi. E’ evidente che queste riserve di aminoacidi sono abbastanza modeste, e sarà il muscolo a farsi carico di fornirne le quantità necessarie, cioè ha inizio la proteolisi delle proteine del muscolo.
Da notare che comunque non esiste una sequenzialità assoluta nella degradazione delle diverse proteine, c’è invece un intreccio in cui, procedendo, certe vie perdono di importanza ed altre ne acquistano.
Quindi per mantenere la glicemia costante viene ridotta la componente proteica del muscolo, compreso quello scheletrico, un tessuto che rappresenta una discreta quota del valore del metabolismo basale e che con l’attività fisica è in grado di aumentare considerevolmente il suo dispendio energetico: dunque fondamentale ai fini della perdita di peso, cioè di massa grassa, e del successivo mantenimento. E’ come se si riducesse la cilindrata del motore.
Una cosa a cui non si pensa è che anche il cuore è un muscolo per cui potrà essere soggetto agli stessi processi visti per il muscolo scheletrico.
In definitiva produrre glucosio a partire dalle proteine, anche di origine alimentare, è come scaldarsi al camino bruciando il mobilio del settecento, gli aminoacidi, avendo a disposizione legna da ardere, i carboidrati alimentari.
Pertanto un adeguato apporto di carboidrati con l’alimentazione previene la perdita eccessiva delle proteine ossia c’è un effetto di risparmio delle proteine svolto dai carboidrati.
Nota: i mammiferi, e quindi gli esseri umani, non hanno la capacità di sintetizzare glucosio a partire dai grassi.
Carboidrati: cosa entra quando loro escono
L’eliminazione o la forte riduzione dell’apporto di carboidrati con la dieta si traduce in un aumentato apporto di proteine, lipidi, incluso il colesterolo, in quanto sarà aumentata l’assunzione di prodotti di origine animale, uno dei principali difetti delle diete iperproteiche. Infatti, nell’organismo non esistono riserve di aminoacidi per cui questi vengono metabolizzati e, come sottoprodotto della loro utilizzazione, si forma ammoniaca che dovrà essere eliminata in quanto tossica.
Per questo motivo le diete iperproteiche comportano un lavoro extra per fegato e reni, e anche per questo non sono esenti da potenziali rischi per la salute.
Un aumentato apporto di grassi molto spesso si traduce in un aumento dell’assunzione di acidi grassi saturi, acidi grassi trans e colesterolo, con tutte le conseguenze che ciò può avere a livello cardiovascolare.
Quanto detto non deve incitare ad assumere quantità elevate di carboidrati. Questa classe di macronutrienti dovrebbe rappresentare il 55-60% della quota calorica giornaliera, i grassi il 25-30% (olio di olivain primis) e la restante quota alle proteine: dunque una composizione in macronutrienti che si rifà alla dieta prudente o alla dieta mediterranea.
Massa grassa e ingresso nella fase di carestia/malattia
Una riduzione eccessiva dell’apporto calorico viene registrata a livello dei nostri meccanismi di difesa come un “ingresso” in una fase di carestia/malattia.
L’abbondanza di cibo è una caratteristica della nostra epoca, almeno nei paesi industrializzati, mentre il nostro organismo si è evoluto nel corso di centinaia di migliaia di anni durante i quali non c’era l’attuale abbondanza: dunque è stato programmato per cercare di superare con il minimo dei danni periodi di carestia. Se l’apporto calorico viene ridotto drasticamente si mima una carestia: quello che l’organismo fa è di abbassare i consumi, ridurre il metabolismo basale ossia consuma di meno e quindi anche mangiando poco non si otterranno grandi perdite di massa grassa. E’ come se a una macchina si abbassasse la cilindrata, consumerà meno, e nel nostro caso brucerà meno massa grassa.
Effetto yo-yo
Il weight cycling o effetto yo-yo, ossia fasi ripetute di perdita e acquisto di peso, appare correlato a un eccesso di peso e a un accumulo di grasso a livello addominale.
Diversi studi suggeriscono l’esistenza nelle donne di un legame tra effetto yo-yo e diverse condizione quali:
un aumento importante del binge eating disorder o sindrome da alimentazione incontrollata;
un senso di depressione riguardo al peso
Infine l’effetto yo-yo si associa anche a una maggiore facilità di prendere peso rispetto alle donne che non sono soggette. A questo riguardo c’è da sottolineare che questa condizione si verifica nell’arco di anni durante i quali, invecchiando, la velocità del metabolismo inevitabilmente tende a diminuire: questo potrebbe rendere più difficoltose le perdite successive.
Bibliografia
Cereda E., Malavazos A.E., Caccialanza R., Rondanelli M., Fatati G. and Barichella M. Weight cycling is associated with body weight excess and abdominal fat accumulation: a cross-sectional study. Clin Nutr 2011;30(6):718-723. doi:10.1016/j.clnu.2011.06.009
Montani J-P., Viecelli A.K., Prévot A. & Dulloo A.G. Weight cycling during growth and beyond as a risk factor for later cardiovascular diseases: the ‘repeated overshoot’ theory. Int J Obes (Lond) 2006;30:S58-S66. doi:10.1038/sj.ijo.0803520
Ravussin E., Lillioja S., Knowler W.C., Christin L., Freymond D., Abbott W.G.H., Boyce V., Howard B.V., and Bogardus C. Reduced rate of energy expenditure as a risk factor for body-weight gain. N Engl J Med 1988;318:467-472. doi:10.1056/NEJM198802253180802
Sachiko T. St. Jeor S.T. St., Howard B.V., Prewitt T.E., Bovee V., Bazzarre T., Eckel T.H., for the AHA Nutrition Committee. Dietary Protein and Weight Reduction. A Statement for Healthcare Professionals From the Nutrition Committee of the Council on Nutrition, Physical Activity, and Metabolism of the American Heart Association. Circulation 2001;104:1869-1874. doi:10.1161/hc4001.096152
Nel caso in cui si consumi un eccesso di calorie in forma di carboidrati o proteine, tale surplus è utilizzato per la sintesi degli acidi grassi e quindi trigliceridi, cosa che non accade se l’eccesso di calorie proviene dai grassi stessi.
Biosintesi degli Acidi Grassi Saturi e Insaturi a Catena Lunga
Sintesi de novo degli acidi grassi negli animali e piante
La sintesi de novo degli acidi grassi, molecole appartenenti al gruppo die lipidi, è in gran parte simile tra le piante e gli animali.
Avviene nei cloroplasti delle cellule fotosintetiche delle piante superiori, e nel citosol delle cellule animali, attraverso l’azione concertata di due enzimi: l’acetil-CoA carbossilasi (EC 6.4.1.2) e l’acido grasso sintetasi (EC 2.3.1.85).
L’acido grasso sintetasi catalizza una sequenza ripetuta di 4 passaggi tramite la quale la catena acilica è allungata di due atomi di carbonio, all’estremità carbossilica, a ogni passaggio attraverso il ciclo; questo processo in quattro tappe è il medesimo in tutti gli organismi.
Negli animali il sito primario per il metabolismo lipidico non è il tessuto adiposo ma il fegato. Il tessuto adiposo è comunque un organo importante in cui si verifica la sintesi degli acidi grassi, anche se negli esseri umani è meno attivo rispetto a molte altre specie animali.
Da notare che in alcune piante, quali la palma e la noce di cocco, si verifica la terminazione della catena acilica prima che sia rilasciato l’acido palmitico: fino al 90% degli acidi grassi prodotti, e quindi presenti negli oli ricavati da questi vegetali, ha tra gli 8, atomi di carbonio, l’acido caprilico, un degli acidi grassi a catena media, e i 14 atomi di carbonio, ossia l’acido miristico.
Sintesi degli acidi grassi a catena lunga saturi e insaturi
L’acido palmitico è, tra gli acidi grassi saturi, il più comune nei lipidi di piante e animali, sebbene generalmente non sia presente in grandi quantità poiché può entrare in diverse vie metaboliche.
Infatti:
è il precursore dell’acido stearico;
può essere desaturato a dare acido palmitoleico, il precursore di tutti gli acidi grassi della famiglia omega-7, in una reazione catalizzata dall’enzima Δ9-desaturasi (EC 1.14.19.1). Questo enzima è ubiquitario sia nel regno animale che vegetale, è il più attivo del metabolismo lipidico nei tessuti di mammifero, ed è lo stesso che catalizza la desaturazione dell’acido stearico ad acido oleico.
La Δ9-desaturasi inserisce un doppio legame nella posizione 9-10 della catena carboniosa dell’acido grasso, posizione numerata a partire dall’estremità carbossilica della molecola. Se il substrato è l’acido palmitico, il doppio legame apparirà tra la posizione posizione n-7 e n-8 della catena, stavolta numerate a partire dall’estremità metilica, producendo acido palmitoleico, dunque il capostipite della famiglia omega-7. Se il substrato è l’acido stearico il doppio legame apparirà tra la posizione posizione n-9 e n-10 della catena, e verrà prodotto l’acido oleico.
Può essere esterificato in lipidi complessi.
Naturalmente nelle piante e negli animali ci sono acidi grassi sia con catene più lunghe sia con gradi di insaturazione maggiori rispetto a quelli visti, il tutto grazie a sistemi enzimatici (di nuovo di desaturazione e allungamento), che catalizzano reazioni di sintesi degli acidi grassi che sono organismo-, tessuto- e cellula specifiche.
Ad esempio l’acido stearico può essere:
allungato ad acido arachidico, acido beenico e acido lignocerico, tutti acidi grassi saturi, in reazioni catalizzate da elongasi.
Anche in questo caso, l’allungamento della catena si verifica, sia nei mitocondri che nel reticolo endoplasmatico liscio, attraverso l’aggiunta di unità a due atomi di carbonio alla volta all’estremità carbossilica dell’acido grasso, tramite l’azione dei sistemi di allungamento degli acidi grassi (in particolare gli acidi grassi a catena lunga e molto lunga, da 18 a 24 atomi di carbonio, sono sintetizzati sulla superficie citosolica del reticolo endoplasmatico liscio);
desaturato, come visto, nella reazione catalizzata dalla Δ9-desaturasi, a dare acido oleico, un acido grasso omega-9.
Diversi ricercatori hanno supposto che la ragione per cui l’acido stearico non concorra allo sviluppo della ipercolesterolemia sia la sua rapida conversione in acido oleico.
L’acido oleico è il punto di partenza per la sintesi di molti acidi grassi insaturi attraverso reazioni di allungamento e/o desaturazione.
Infatti:
l’allungamento della sua catena acilica per mezzo di un altro sistema enzimatico, l’acido grasso elongasi, produce acido gadoleico, acido erucico e acido nervonico, tutti acidi grassi monoinsaturi.
Gli acidi grassi saturi e i monoinsaturi della famiglia omega-9, in genere l’acido oleico, ma anche l’acido palmitoleico e gli altri omega-7, sono gli unici acidi grassi prodotti attraverso la sintesi de novo nei mammiferi.
Grazie all’azione consecutiva degli enzimi Δ12-desaturasi (1.14.19.6) e Δ15-desaturasi (EC 1.14.19.25), che inseriscono un doppio legame rispettivamente nella posizione posizione 12-13 e nella posizione 15-16 della catena carboniosa dell’acido grasso, l’acido oleico è convertito prima in acido linoleico, precursore di tutti gli acidi grassi polinsaturi omega-6, e quindi in acido alfa-linolenico, precursore di tutti gli acidi grassi polinsaturi omega-3 (acidi grassi polinsaturi che saranno prodotti a partire da questi precursori attraverso reazione ripetute di allungamento e desaturazione).
In caso di deficienza di acidi grassi essenziali, l’acido oleico può essere desaturato e allungato a dare la serie di acidi grassi polinsaturi omega-9, con accumulo soprattutto di acido Mead.
Sintesi degli omega-3 e omega-6
I tessuti animali possono desaturare gli acidi grassi in posizione 9-10 della catena, grazie alla presenza della Δ9 desaturasi; come visto in precedenza se il substrato della reazione è l’acido palmitico il doppio legame apparirà tra le posizioni n-7 e n-8 della catena, con l’acido stearico tra le posizioni n-9 e n-10 portando così alla formazione rispettivamente di acido palmitoleico e oleico.
Gli animali non possiedono la Δ12- e la Δ15-desaturasi, enzimi in grado desaturare i legami carbonio carbonio oltre la posizione 9-10 della catena, e non sono quindi in grado di sintetizzare de novo gli acidi grassi polinsaturi omega-3 e omega-6, che posseggono doppi legami anche oltre la posizione 9-10, che sono pertanto acidi grassi essenziali.
La Δ12- e la Δ15-desaturasi sono presenti nelle piante; sebbene molte piante terrestri manchino della Δ15-desaturasi, detta anche omega-3 desaturasi, il plancton e le piante acquatiche delle acque fredde la posseggono e producono quantità abbondanti di acidi grassi omega-3.
Bibliografia
Akoh C.C. and Min D.B. “Food lipids: chemistry, nutrition, and biotechnology”. CRC Press Taylor & Francis Group, 2008 3th ed. 2008
Bender D.A. “Benders’ dictionary of nutrition and food technology”. 2006, 8th Edition. Woodhead Publishing. Oxford
Burr G.O. and Burr M.M. A new deficiency disease produced by the rigid exclusion of fat from the diet. Nutr Rev 1973;31(8):148-149. doi:10.1111/j.1753-4887.1973.tb06008.x
Chow Ching K. “Fatty acids in foods and their health implication”. 3rd Edition. CRC Press Taylor & Francis Group, 2008
Rosenthal M.D., Glew R.H. Mediacal biochemistry. Human metabolism in health and disease. John Wiley & Sons, Inc. 2009
Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
Il glicogeno muscolare è un’importante riserva di energia negli esercizi prolungati di intensità medio-alta, importanza che aumenta nel caso di esercizi intervallati di alta intensità, comuni negli allenamenti di nuotatori, corridori, vogatori e negli sport di squadra, o in lavori di resistenza contro pesi. Se ad esempio si considera la maratona, circa l’80% dell’energia necessaria deriva dall’ossidazione dei carboidrati, per la maggior parte glicogeno muscolare.
La deplezione dei livelli di glicogeno muscolare è correlata con l’insorgenza della fatica, sebbene non siano ancora chiari i meccanismi molecolari alla base di questa relazione. Una delle ipotesi è che esista una concentrazione minima di glicogeno che viene “protetta” ed è resistente all’utilizzo durante l’esercizio, forse per assicurare una riserva di energia in caso di estrema necessità.
Data la stretta relazione tra deplezione del glicogeno muscolare e fatica, la sua velocità di ripristino nel post-esercizio è uno dei fattori più importanti nel determinare il tempo necessario al recupero.
Infine l’atleta maggiormente allenato non solo ha depositi di glicogeno muscolare potenzialmente maggiori, ma è anche in grado di sintetizzarlo più velocemente grazie a enzimi più efficienti.
Per produrre glicogeno è indispensabile assumere carboidrati; ma quanti, quali, quando e con che frequenza?
Nel tentativo di ripristinare il più velocemente possibile le riserve di glicogeno muscolare è utile conoscere l’andamento bifasico che può assumere la sua velocità di sintesi a seguito di allenamenti o gare che comportino deplezioni delle sue riserve muscolari pari ad almeno il 75% del valore a riposo e non a digiuno.
Conoscere e quindi sfruttare la tale bifasicità è importante per quegli atleti che siano impegnati in più allenamenti giornalieri, o che abbiano poco tempo a disposizione per il recupero tra un allenamento impegnativo e il successivo, ossia meno di 8 ore, al fine di massimizzarne la sintesi e ottenere la migliore performance nella sessione successiva.
Le due fasi sono caratterizzate da:
una diversa sensibilità all’insulina circolante;
una diversa velocità di sintesi.
La prima fase
La prima fase, immediatamente successiva al termine dell’attività e della durata di 30-60 minuti, è insulino-indipendente, ossia l’uptake del glucosio da parte della cellula muscolare come la sintesi della molecola al suo interno sono indipendenti dall’azione dell’ormone.
Questa fase è caratterizzata da una elevata velocità di sintesi che però si riduce rapidamente se non si assumono carboidrati: la massima velocità si registra nei primi 30 minuti, per poi ridursi a circa 1/5 dal 60° minuto, e a circa 1/9 al 120° minuto dal termine dell’esercizio.
Come è possibile sfruttare questa prima fase per ripristinare quanto più possibile le scorte muscolari di glicogeno? Facendo si che al muscolo arrivi la maggior quantità possibile di glucosio nei momenti immediatamente successivi al termine dell’attività, meglio se entro i primi 30 minuti.
Cosa assumere?
Carboidrati a alto indice glicemico, ma di facile digestione e assorbimento.
E’ quindi consigliabile sostituire cibi, magari anche ad alto indice glicemico, che necessitano di un certo tempo per la digestione e il successivo assorbimento, con soluzioni/gel contenenti ad esempio glucosio e/o saccarosio. Queste soluzioni/gel assicurano la massima velocità possibile di assorbimento e rifornimento di glucosio al muscolo in quanto contengono solo glucosio e sono prive di fibre o altro che rallenterebbero la digestione dei carboidrati e il successivo assorbimento dei monosaccaridi, sono cioè in grado di produrre elevate glicemie in un tempo relativamente breve.
Da sottolineare ulteriormente che il ricorso nell’immediato post-esercizio a tali soluzioni/gel contenenti carboidrati a rapida disponibilità è consigliabile solo quando il tempo di recupero tra un esercizio che causa una forte deplezione del glicogeno muscolare e il successivo è breve, meno di 8 ore.
Sarà possibile giocare anche sulla temperatura e concentrazione della soluzione per accelerarne il transito gastrico.
Assumere carboidrati, ma in che quantità?
Sono stati condotti molti studi per cercare di definire la quantità ideale di carboidrati da assumere.
Se nel post-esercizio l’atleta non si alimenta la velocità di sintesi del glicogeno è molto bassa, mentre se immediatamente dopo il termine del lavoro assume quantità adeguate di carboidrati la velocità può raggiungere valori oltre 20 volte maggiori.
Dal confronto della letteratura sembra ragionevole affermare che, a seguito di allenamenti che riducano le scorte di glicogeno muscolare a valori inferiori al 75% dei valori a riposo e non a digiuno, la massima velocità di sintesi si ottenga con assunzioni di carboidrati, ad alto indice glicemico ed elevata velocità di digestione e assorbimento, pari a circa 1,2 g/kg di peso corporeo/h per le 4-5 ore successive dal termine dell’esercizio stesso.
In questo modo si determina la produzione di una quantità di glicogeno maggiore del 150% rispetto all’ingestione di 0,8 g/kg/h.
Poiché aumenti fino a 1,6 g/kg/h non hanno portato ulteriori incrementi, la quantità di carboidrati pari a 1,2 g/kg/h può essere considerata quella ottimale per massimizzare la velocità di risintesi delle scorte di glicogeno muscolare nel post-esercizio.
Con che frequenza?
Riguardo alla frequenza di assunzione è stato osservato che se i carboidrati sono assunti di frequente, ogni 15-30 minuti, sembra ci sia un’ulteriore stimolazione dell’uptake del glucosio da parte del muscolo, come della ricostituzione del glicogeno muscolare rispetto ad assunzioni a intervalli di due ore. In particolare, le assunzioni nelle prime ore del post-esercizio sembrano ottimizzare livelli di glicogeno.
La seconda fase
La seconda fase delle sintesi del glicogeno muscolare ha inizio dalla fine della prima, perdura sino all’inizio del pasto precedente l’impegno successivo, dunque da alcune ore a giorni, ed è insulino-dipendente, ossia l’uptake del glucosio da parte della cellula muscolare come la sintesi del glicogeno al suo interno sono sensibili ai livelli circolanti dell’ormone.
Inoltre si osserva una significativa riduzione della velocità di sintesi del glicogeno muscolare: con un’assunzione adeguata di carboidrati la velocità si attesta su valori inferiori di circa il 10-30% rispetto a quelli della prima fase.
Questa fase può perdurare per diverse ore, ma tende a essere più breve se:
l’apporto di carboidrati è elevato;
la sintesi del glicogeno è più attiva;
i livelli di glicogeno muscolare sono aumentati.
Come è possibile sfruttare questa fase per ottimizzare la velocità di sintesi del glicogeno muscolare?
Le evidenze sperimentali indicano che pasti con carboidrati ad alto indice glicemico sono più efficaci di quelli con carboidrati a basso indice glicemico. Ma se tra un allenamento e il successivo passano giorni e non ore, non ci sono evidenze a favore di carboidrati ad alto indice glicemico rispetto a quelli a basso indice purché ne sia assunta una quantità adeguata.
Velocità di sintesi del glicogeno e assunzione di carboidrati e proteine
La contemporanea assunzione di carboidrati e proteine, o aminoacidi insulino-tropici liberi, permette di ottenere velocità di sintesi del glicogeno nel post-esercizio che non differiscono significativamente da quelle raggiunte con quantità maggiori di soli carboidrati. Questo potrebbe essere un vantaggio per l’atleta che ne potrà assumere quantità più contenute, limitando così l’insorgenza di eventuali complicazioni gastrointestinali comuni durante l’allenamento/gara dopo un loro consumo elevato.
Dall’analisi della letteratura sembra ragionevole affermare che, dopo un esercizio che comporti la deplezione di almeno il 75% delle riserve muscolari di glicogeno, si possano ottenere velocità di sintesi del glicogeno analoghe a quelle raggiunte con 1,2 g/kg/h di soli carboidrati, le maggiori ottenibili, con la coingestione di 0,8 g/kg/h di carboidrati e 0,4 g/kg/h di proteine, mantenendo le stesse tempistiche di ingestione, ossia ogni 15-30 minuti per le prime 4-5 ore del post-esercizio.
Le due fasi: meccanismi molecolari
In entrambe le fasi l’aumento della sintesi del glicogeno è conseguenza di un aumento:
della velocità di trasporto del glucosio nella cellula;
dell’attività della glicogeno sintetasi, l’enzima che catalizza la sintesi del glicogeno.
Tuttavia i meccanismi molecolari che sottostanno a queste modificazioni sono differenti.
Nella prima fase l’aumento della velocità di trasporto del glucosio, indipendente dalla presenza dell’insulina, è mediato dalla traslocazione, indotta dalla contrazione, dei trasportatori del glucosio, detti GLUT4, sulla membrana plasmatica della cellula muscolare.
In aggiunta, anche i bassi livelli di glicogeno agiscono da stimolo al trasporto in quanto si ritiene che gran parte delle vescicole contenenti il trasportatore siano legate al glicogeno, e dunque potrebbero divenire disponibili quando i suoi livelli sono ridotti.
Infine bassi livelli di glicogeno muscolare vanno anche a stimolare l’attività della glicogeno sintetasi: è stato dimostrato che il livello del glicogeno muscolare è un regolatore dell’attività dell’enzima molto più potente di quanto sia l’insulina.
Nella seconda fase l’aumento della sintesi è dovuto all’azione dell’insulina sui trasportatori del glucosio e sull’attività della glicogeno sintetasi della cellula muscolare. Questa sensibilità all’azione dell’insulina circolante, che può persistere per 48 ore a seconda dell’assunzione di carboidrati e della quantità di glicogeno muscolare risintetizzato, ha suscitato grande attenzione: è infatti possibile, tramite opportuni interventi nutrizionali, incrementarne la secrezione al fine di migliorare la sintesi del glicogeno stesso ma anche l’anabolismo proteico, riducendo al contempo la velocità di degradazione delle proteine stesse.
Insulina e velocità di sintesi del glicogeno muscolare
La contemporanea assunzione di carboidrati e proteine, o aminoacidi liberi, aumenta la secrezione di insulina postprandiale rispetto ai soli carboidrati; in alcuni studi sono stati osservati incrementi nella secrezione dell’ormone di 2-3 volte rispetto ai soli carboidrati.
E’ stato supposto che, data la maggior quantità di insulina circolante, si potessero ottenere ulteriori aumenti della velocità di sintesi del glicogeno rispetto a quelli osservati con i soli carboidrati, ma in realtà non sembra essere così. Se infatti la quantità di carboidrati viene portata a 1,2 g/kg/h, più 0,4 g/kg/h di proteine, non si osservano ulteriori aumenti nella velocità di sintesi se paragonati a quelli ottenuti con l’ingestione dei soli carboidrati nella stessa quantità, ossia 1,2 g/kg/h, che come detto, al pari della coingestione di 0,8 g/kg/h di carboidrati e 0,4 g/kg/h di proteine, danno la massima velocità raggiungibile nel post-esercizio, o in quantità isoenergetica, quindi 1,6 g/kg.
Insulina e accumulo preferenziale dei carboidrati
I livelli più elevati di insulina circolante raggiunti con la coingestione di carboidrati e proteine, o aminoacidi liberi, potrebbero stimolare un accumulo dei carboidrati ingeriti nei tessuti maggiormente sensibili all’azione dell’ormone, quali il fegato e il muscolo che ha precedentemente lavorato.
In questo modo si verificherebbe un loro deposito più efficiente ai fini dell’attività sportiva, in quanto i carboidrati verrebbero accumulati preferenzialmente anche nel muscolo, dove saranno in seguito utilizzati.
Bibliografia
Beelen M., Burke L.M., Gibala M.J., van Loon J.C. Nutritional strategies to promote postexercise recovery. Int J Sport Nutr Exerc Metab 2010:20(6);515-532. doi:10.1123/ijsnem.20.6.515
Berardi J.M., Noreen E.E., Lemon P.W.R. Recovery from a cycling time trial is enhanced with carbohydrate-protein supplementation vs. isoenergetic carbohydrate supplementation. J Intern Soc Sports Nutrition 2008;5:24. doi:10.1186/1550-2783-5-24
Betts J., Williams C., Duffy K., Gunner F. The influence of carbohydrate and protein ingestion during recovery from prolonged exercise on subsequent endurance performance. J Sports Sciences 2007;25(13):1449-1460. doi:10.1080/02640410701213459
Howarth K.R., Moreau N.A., Phillips S.M., and Gibala M.J. Coingestion of protein with carbohydrate during recovery from endurance exercise stimulates skeletal muscle protein synthesis in humans. J Appl Physiol 2009:106;1394–1402. doi:10.1152/japplphysiol.90333.2008
Jentjens R., Jeukendrup A. E. Determinants of post-exercise glycogen synthesis during short-term recovery. Sports Medicine 2003:33(2);117-144. doi:10.2165/00007256-200333020-00004
Millard-Stafford M., Childers W.L., Conger S.A., Kampfer A.J., Rahnert J.A. Recovery nutrition: timing and composition after endurance exercise. Curr Sports Med Rep 2008;7(4):193-201. doi:10.1249/JSR.0b013e31817fc0fd
Price T.B., Rothman D.L., Taylor R., Avison M.J., Shulman G.I., Shulman R.G. Human muscle glycogen resynthesis after exercise: insulin-dependent and –independent phases. J App Physiol 1994:76(1);104–111. doi:10.1152/jappl.1994.76.1.104
Schweitzer G.G., Kearney M.L., Mittendorfer B. Muscle glycogen: where did you come from, where did you go? J Physiol 2017;595(9):2771-2772. doi:10.1113/JP273536
van Loon L.J.C., Saris W.H.M., Kruijshoop M., Wagenmakers A.J.M. Maximizing postexercise muscle glycogen synthesis: carbohydrate supplementation and the application of amino acid or protein hydrolysate mixtures. Am J Clin Nutr 2000;72: 106-111. doi:10.1093/ajcn/72.1.106
Gli acidi grassi polinsaturi omega-6 sono i principali acidi grassi polinsaturi, detti anche PUFA, acronimo dell’inglese polyunsaturated fatty acids, nella dieta occidentale, circa il 90% dei tutti quelli presenti, essendo tra i costituenti della maggior parte dei grassi animali e vegetali.
All’interno della famiglia omega-6 uno degli acidi grassi più importanti e diffusi è l’acido linoleico, il precursore di tutti gli acidi grassi polinsaturi omega-6. È prodotto de novo dall’acido oleico, un acido grasso omega-9, solamente nelle piante in una reazione catalizzata dalla Δ12-desaturasi, dunque l’enzima che forma la famiglia degli acidi grassi polinsaturi omega-6 a partire da quella degli omega-9.
La Δ12-desaturasi catalizza l’inserimento di un doppio legame tra gli atomi di carbonio 6 e 7, numerati a partire dall’estremità metilica della molecola.
L’acido linoleico, insieme con con l’acido alfa-linolenico, è un prodotto primario della sintesi degli acidi grassi polinsaturi nelle piante.
Metabolismo degli Acidi Grassi Polinsaturi Omega-6
Gli animali, essendo privi della Δ12-desaturasi, non sono in grado di sintetizzare de novo ne l’acido linoleico ne tutta la famiglia degli omega-6 e sono obbligati ad assumerlo dagli alimenti vegetali e/o da animali che se ne cibino; per questo motivo gli omega-6 sono considerati acidi grassi essenziali, i cosiddetti EFA, acronimo dell’inglese essential fatty acids.
La loro essenzialità, in particolare proprio l’essenzialità dell’acido linoleico, è stata riportata per la prima volta nel 1929 da Burr e Burr.
Omega-6: da acido linoleico ad acido arachidonico
Gli animali sono in grado di allungare e desaturare l’acido linoleico ottenuto con la dieta in una cascata di reazioni che porta alla formazione di PUFA omega-6 a catena molto lunga.
L’acido linoleico viene prima desaturato ad acido gamma-linolenico, un altro importante acido grasso omega-6 con significativi effetti fisiologici, nella reazione catalizzata dalla Δ6-desaturasi. Si pensa che la velocità di questa reazione in certe condizioni, come negli anziani, in alcune situazioni patologiche e nei neonati prematuri, sia il fattore limitante della cascata; per questo motivo, e per il fatto che l’acido gamma-linolenico si trova in quantità relativamente piccole nella dieta, alcuni oli che lo contengono, quali gli oli di semi ottenuti da ribes, primula della sera e borragine, hanno attirato l’attenzione.
A sua volta l’acido gamma-linolenico può essere allungato ad acido diomo-gamma-linolenico in una reazione catalizzata da un elongasi. L’enzima catalizza l’aggiunta di due atomi di carbonio provenienti dal metabolismo del glucosio per allungare la catena dell’acido grasso, che può essere ulteriormente desaturato in quantità molto limitata a dare acido arachidonico, in una reazione catalizzata da un altro enzima limitante la velocità della cascata, la Δ5-desaturasi.
L’acido arachidonico può essere allungato e desaturato ad acido adrenico.
Va notato che gli acidi grassi polinsaturi della famiglia omega-6, e di ogni altra famiglia, possono essere interconvertiti attraverso processi enzimatici solo all’interno della stessa famiglia, non tra famiglie.
Gli acidi grassi polinsaturi con 20 atomi di carbonio appartenenti alle famiglie omega-6 e omega-3 sono i precursori degli eicosanoidi (prostaglandine, prostacicline, trombossani, e leucotrieni), potenti ormoni ad azione breve e locale.
Mentre la privazione di acidi grassi polinsaturi omega-3 causa la disfunzione in una vasta gamma di modalità comportamentali e fisiologiche, l’omissione dalla dieta degli acidi grassi polinsaturi omega-6 determina una palese disfunzione sistemica.
Negli oli di semi gli acidi grassi polinsaturi omega-6 con catena più lunga di 18 atomi di carbonio sono presenti solo in tracce mentre l’acido arachidonico si trova in tutti i tessuti animali e nei cibi di origine animale.
Bibliografia
Akoh C.C. and Min D.B. “Food lipids: chemistry, nutrition, and biotechnology” 3th ed. 2008
Aron H. Uber den Nahvert (On the nutritional value). Biochem Z. 1918;92:211–233 (German)
Bender D.A. “Benders’ dictionary of nutrition and food technology”. 2006, 8th Edition. Woodhead Publishing. Oxford
Bergstroem S., Danielsson H., Klenberg D. and Samuelsson B. The enzymatic conversion of essential fatty acids into prostaglandins. J Biol Chem 1964;239:PC4006-PC4008.
Burr G.O. and Burr M.M. A new deficiency disease produced by the rigid exclusion of fat from the diet. Nutr Rev 1973;31(8):148-149. doi:10.1111/j.1753-4887.1973.tb06008.x
Chow Ching K. “Fatty acids in foods and their health implication” 3th ed. 2008
Rosenthal M.D., Glew R.H. Mediacal biochemistry. Human metabolism in health and disease. John Wiley & Sons, Inc. 2009
Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
Van D., Beerthuis R.K., Nugteren D.H. and Vonkeman H. Enzymatic conversion of all-cis-polyunsaturated fatty acids into prostaglandins. Nature 1964;203:839-841
Gli acidi grassi polinsaturi omega-3 sono acidi grassi insaturi, dunque lipidi, che presentano un doppio legame in posizione 3 rispetto all’estremità metilica, detta anche estremità n, della catena carboniosa.[3]
Per gli esseri umani gli omega-3 più importanti sono:
l’acido alfa-linolenico o ALA o 18:3n-3, il più semplice, con 18 atomi di carbonio e tre doppi legami;
l’acido eicosapentaenoico o EPA o 20:5n-3, con 20 atomi di carbonio e 5 doppi legami;
l’acido docosaesaenoico o DHA o 22:6n-3, che con i suoi 22 atomi di carbonio e 6 doppi legami è il più complesso.
EPA e DHA sono definiti acidi grassi polinsaturi a catena lunga o LC-PUFA.
A causa della mancanza di due specifiche desaturasi, la delta-12 desaturasi (EC 1.14.19.6) e la delta-15 desaturasi (EC 1.14.19.13), che inseriscono doppi legami rispettivamente in posizione 6 e 3 dall’estremità metilica della catena carboniosa, i mammiferi non sono in grado di produrre l’acido linoleico o LA, capostipite della famiglia degli acidi grassi polinsaturi omega-6, e l’acido alfa-linolenico, a sua volta capostipite degli omega-3 e che deriva dall’acido linoleico.[9] L’acido linoleico e l’acido alfa-linolenico risultano pertanto essere acidi grassi essenziali.[34]
Molti animali, compreso l’uomo, hanno il corredo enzimatico necessario per sintetizzare, a partire dall’acido alfa-linolenico di origine alimentare, tutti gli altri omega-3, che quindi risultano essenziali solamente in assenza di ALA di origine alimentare, e per questo sono definiti acidi grassi semiessenziali.[9]
EPA e DHA sono componenti strutturali delle membrane cellulari che rappresentano la loro principale sede di deposito, in particolare nei tessuti muscolare e nervoso.[35] In ciò si differenziano dalla maggior parte degli altri acidi grassi che sono immagazzinati principalmente nei trigliceridi del tessuto adiposo.
Il DHA è il principale componente dei fosfolipidi delle membrane cellulari dei tessuti nervosi dei vertebrati, compresi i fotorecettori della retina, dove svolge importanti funzioni.[22][26][29][32] Oltre alla funzione strutturale, gli LC-PUFA omega-3 sono substrati per la produzione di mediatori lipidici bioattivi ad azione antiinfiammatoria quali eicosanoidi, maresine, resolvine, e protectine.[10][21]
Gli acidi grassi polinsaturi omega-3 sono essenziali nel corso dello sviluppo neurologico del feto, e la loro assunzione da parte della gestante è particolarmente importante nel terzo trimestre di gravidanza, fase in cui si verifica una crescita cerebrale significativa.[8] Nel corso della vita la loro assunzione è stata associata a una riduzione del rischio di sviluppare molte malattie croniche, in particolare le malattie cardiovascolari.[1][27][33]
Le fonti migliori per l’uomo sono i prodotti della pesca, specialmente quelli ottenuti da mari freddi.[3][9][36]
L’acido alfa-linolenico è prodotto dall’acido linoleico, un omega-6, nei plastidi del fitoplancton e delle piante terrestri vascolari, dove è presente la delta-15 desaturasi che inserisce un doppio legame tra gli atomi di carbonio 3 e 4, numerati a partire dall’estremità metilica, di LA.[3][9]
ALA va di seguito incontro a successive reazioni di desaturazione, catalizzate dalla delta-5 desaturasi (EC 1.14.19.44) e dalla delta-6 desaturasi (EC 1.14.19.3), e di allungamento, catalizzate dalla elongasi 5 (EC 2.3.1.199) e dalla elongasi 5 e/o dalla elongasi 2 (EC 2.3.1.199), e a una limitata beta-ossidazione nei perossisomi, a dare infine DHA.[6][8]
Va sottolineato che le piante terrestri vascolari non hanno la capacità di sintetizzare LC-PUFA omega-3, come EPA e DHA.[23]
Metabolismo degli Acidi Grassi Polinsaturi Omega-3
Gli enzimi che catalizzano la conversione dell’acido alfa-linolenico in DHA sono condivisi con le vie che portano alla sintesi degli acidi grassi polinsaturi omega-6, omega-7 e omega-9.[9]
Sembra che la delta-5 desaturasi e dalla delta-6 desaturasi abbiano come substrati preferenziali gli omega-3. Tuttavia, considerando che nella dieta occidentale il rapporto tra l’assunzione di ALA e LA è molto spostato a favore del secondo, ne risulterebbe favorita la via degli omega-6 rispetto alle altre.[38] Questa potrebbe essere una delle spiegazioni che giustificano il basso tasso di conversione dell’acido alfa-linolenico negli altri omega-3, sebbene anche la sintesi di acido arachidonico o ARA dall’acido linoleico sembra essere molto contenuta.
Da notare che sia la famiglia degli acidi grassi polinsaturi omega-3 che quella degli omega-6 inibiscono la formazione degli acidi grassi polinsaturi omega-9.
Biosintesi degli Omega-3 nell’uomo
L’uomo, così come molti altri animali, è in grado di allungare e desaturare l’acido alfa-linolenico sino a formare il DHA, via metabolica presente principalmente nel fegato e nel microcircolo cerebrale della barriera ematoencefalica, ma anche nell’endotelio celebrale e negli astrociti.[18] E’ tuttavia opinione comune che, al pari degli altri animali terrestri, abbia una limitata capacità di produrre gli LC-PUFA, e dunque necessiti di EPA e DHA di origine alimentare.[7]
E’ stato evidenziato che l’efficienza della sintesi diminuisce lungo la via: la conversione dell’acido alfa-linolenico a EPA è limitata, e il fattore limitante sarebbe l’attività della delta-6 desaturasi. La sintesi del acido docosaesaenoico sarebbe ancora più limitata della precedente.[7] Tuttavia recenti studi hanno dimostrato l’esistenza di un marcato polimorfismo globale nel cluster dei geni FADS, i geni che codificano per le desaturasi, in particolare per quelli che codificano per la delta-5 desaturasi e delta-6 desaturasi, rispettivamente FADS1 e FADS2, geni contigui presenti sul cromosoma 11q12.2.[6]
Dal confronto tra materiale genetico di soggetti vissuti nell’Età del Bronzo con quello di popolazioni contemporanee è stato possibile ottenere un quadro chiaro riguardo ai cambiamenti della frequenza allelica nel cluster dei geni FADS.[25] Differenze genotipiche che si sono riflesse in differenze fenotipiche.
Nelle popolazioni europee, il passaggio da una società di cacciatori-raccoglitori a una di agricoltori avrebbe determinato un aumento dell’assunzione di acido linoleico e acido alfa-linolenico e una riduzione dell’assunzione di EPA e ARA. La selezione avrebbe quindi favorito l’aplotipo associato all’aumento dell’espressione di FADS1 e alla diminuzione dell’espressione di FADS2.[6]
Questo è esattamente il pattern opposto a quello osservato negli Inuit, dove si ipotizza che la selezione abbia agito per favorire alleli che riducano la conversione dell’acido linoleico e dell’acido alfa-linolenico in LC-PUFA al fine di compensare il loro apporto relativamente alto con la dieta osservato in queste popolazioni.[13]
Gli altri animali hanno bisogno di EPA e DHA?
Gli organismi privi della delta-15 desaturasi non sono in grado di sintetizzare l’acido alfa-linolenico, e quindi tutta la famiglia degli omega-3, e, se necessario, sono obbligati a ottenerlo dai vegetali e/o da animali che se ne cibano. Ma non tutti gli animali hanno necessità di ottenere EPA e DHA dal cibo.
I vertebrati erbivori terrestri, come le mucche, soddisfano le loro necessità di LC-PUFA omega-3 producendoli a partire dall’acido alfa-linolenico ottenuto mangiando le parti verdi delle piante.[19]
E ci sono animali che non necessitano di EPA e DHA, e ne sono praticamente privi. Tra questi gli insetti terrestri, nei quali si ritrovano solo bassissimi livelli di EPA, paragonabili a quelli delle vitamine. In essi l’EPA è sintetizzato a partire dall’ALA di origine alimentare e utilizzato per la produzione di eicosanoidi.[16]
Invece, gli insetti acquatici presentano elevati livelli di EPA, mentre il DHA è praticamente assente.[31]
Sono molto ricchi in EPA e DHA diverse classi di fitoplancton come le Cryptophyceae, le Dinophyceae, mentre sono molto ricche in EPA le Bacillariophyceae o diatomee. In generale le microalghe rappresentano i produttori primari di EPA e DHA. Gli ecosistemi acquatici sono quindi la principale fonte di LC-PUFA omega-3 nella biosfera.[2][28] EPA e DHA sintetizzati in queste alghe unicellulari sono trasferiti lungo la catena alimentare, passando dagli invertebrati ai pesci è poi agli animali terrestri, esseri umani inclusi. Dunque, dalle microalghe agli esseri umani.[16]
Benefici
Gli acidi grassi polinsaturi omega-3 sono componenti essenziali di una dieta salutare e bilanciata in quanto necessari nel corso dello sviluppo, a partire dalla vita fetale, e associati, nel corso della vita, a miglioramenti della salute e alla riduzione del rischio di malattia.[8][26][36]
Studi epidemiologici hanno ad esempio associato elevati livelli di assunzione di EPA e DHA con tassi di mortalità per malattie cardiovascolari, specialmente cardiache, più bassi di quelli attesi in soggetti sani, probabile conseguenza del miglioramento di molti fattori di rischio, in particolare dei livelli plasmatici dei trigliceridi, del colesterolo HDL e della proteina C-reattiva, e miglioramenti dei valori della pressione, sia sistolica che diastoliche, e della frequenza cardiaca.[1][27]
EPA e DHA si sono dimostrati utili anche nel trattamento di malattie quali l’artrite reumatoide, e potrebbero essere utili nel trattamento di altre condizioni infiammatorie quali l’asma, la psoriasi o le malattie infiammatorie croniche intestinali, grazie alla loro capacità di modulare molti aspetti dei processi infiammatori.[33]
Al contrario, gli LC-PUFA omega-3 sembrano avere effetti minimi o nulli sulla prevenzione e il trattamento del diabete mellito di tipo 2.[5]
Rapporto Omega-3/Omega-6
Molte prove epidemiologiche suggeriscono che il consumo di una dieta con un basso valore del rapporto omega-3/omega-6 abbia influenzato negativamente la salute umana, concorrendo alla promozione, probabilmente assieme a fattori quali il fumo e la sedentarietà, dello sviluppo delle principali classi di patologie.[8]
Sono stati infatti osservati tassi più bassi di incidenza di cancro, autoimmunità e malattia coronarica nelle popolazioni che consumano una dieta con un alto valore del rapporto omega-3/omega-6, in particolare negli Eschimesi, Giapponesi e in altre società dove è elevato il consumo di pesce.[20][37]
Nonostante ciò, la dieta di tipo occidentale è divenuta via via più ricca in acidi grassi saturi, povera in acidi grassi polinsaturi omega-3 e ricca in acidi grassi polinsaturi omega-6, con una rapporto omega-3/omega-6 compreso tra 1/10 e 1/20, quindi molto lontano dal rapporto raccomandato di 1:5.[33]
Il basso valore del rapporto omega-3/omega-6 è dovuto a diversi fattori, alcuni dei quali sono di seguito elencati.
Sebbene gli alimenti vegetali selvatici siano in genere ricchi in omega-3, nell’agricoltura industriale le colture ricche in omega-6 hanno avuto molto più successo di quelle ricche in omega-3.[3]
Il basso consumo di prodotti della pesca e di olio di pesce.[33]
L’elevato consumo di animali allevati con mangimi a base di mais, quali polli, bovini e suini.
A questo va aggiunto anche il fatto che il contenuto in omega-3 di alcune specie di pesci allevate è più basso rispetto a quello delle controparti selvatiche.[9]
L’elevato consumo di oli ricchi in omega-6 e poveri in omega-3, come l’olio di cartamo, girasole, di semi di soia, e di mais.[4]
Nota: il rapporto omega-3/omega-6 non sembra avere influenza ne sulla prevenzione ne sul trattamento del diabete di tipo 2.[5]
Effetti a livello molecolare di EPA e DHA
Negli ultimi anni si stanno chiarendo i meccanismi molecolari alla base degli effetti funzionali attribuiti agli acidi grassi polinsaturi omega-3, in particolare a EPA e DHA, e la maggior parte di questi richiedono l’incorporazione dei due acidi grassi nei fosfolipidi di membrana.[23]
Gli omega-3 sono componenti strutturali delle membrane cellulari, dove svolgono un ruolo essenziale nel regolarne la fluidità.[35] Grazie a questo effetto gli omega-3, in particolare EPA e DHA, sono in grado di modulare le risposte cellulari dipendenti da proteine di membrana. Questo si è rivelato essere particolarmente importante nell’occhio, dove la presenza del DHA permette un’attività ottimale della rodopsina.[29] L’azione sulla fluidità di membrana è poi essenziale per gli animali che vivono in acque fredde, nei quali EPA e DHA svolgono anche una funzione di antigelo naturali.
EPA e DHA sono in grado di modificare la formazione delle zattere lipidiche, microdomini con una composizione lipidica specifica che possono agire come piattaforme per l’azione dei recettori e per l’inizio di vie di segnalazione intracellulare. Modificandone la formazione influenzano le vie di segnalazione intracellulare in tipi cellulari differenti, come i neuroni, le cellule del sistema immunitario e le cellule cancerose. In questo modo EPA e DHA sono in grado di modulare l’attivazione di fattori di trascrizione, come NF-κB, PPARs e SREBPs, e di conseguenza i pattern di espressione genica da essi regolati. Questo ruolo è centrale rispetto alla loro funzione di controllo del metabolismo degli acidi grassi e dei trigliceridi, dell’infiammazione e della differenziazione adipocitaria.[8]
ARA, EPA, e DHA sono substrati per la produzione di numerosi mediatori lipidici, tra cui spiccano gli eicosanoidi, molecole con un ruolo ben definito nella regolazione dell’infiammazione, immunità, aggregazione piastrinica, funzione renale e contrazione della muscolatura liscia.[21]
Gli eicosanoidi prodotti dall’acido arachidonico, che, dal punto di vista quantitativo, è il maggior substrato per la loro sintesi, hanno importanti ruoli fisiologici, ma un loro eccesso è stato associato a numerosi processi patologici.[8] L’aumento del contenuto di EPA e DHA nei fosfolipidi di membrana si accompagna a una riduzione del contenuto di ARA, a una riduzione nella produzione di mediatori lipidici derivanti da ARA, e a un aumento della produzione di mediatori lipidici derivanti dai due omega-3. In aggiunta, tra le molecole derivanti da EPA e DHA ci sono alcuni eicosanoidi analoghi a quelli derivati dall’acido arachidonico, sebbene dotati di minor attività biologica, le resolvine, e, per quanto riguarda DHA, le protectine e le maresine. Queste molecole sembrano essere responsabili di molte delle azioni antiinfiammatorie immuno-modulatrici attribuite ai due acidi grassi polinsaturi omega-3.[10]
E’ stato dimostrato che EPA e DHA possono svolgere un ruolo in forma libera. Sono infatti in grado di agire direttamente attraverso recettori di membrana accoppiati alle proteine G, modulandone l’attività o legandosi a specifici recettori nucleari, come il DHA su PPARα.[30]
Infine, riducono l’assorbimento degli acidi grassi polinsaturi omega-6, e, a livello enzimatico inibiscono competitivamente la ciclo ossigenasi e la lipossigenasi, e competono per le aciltransferasi.[3]
Le fonti più importanti di EPA e DHA per l’uomo
In generale i pesci e gli invertebrati acquatici, come i crostacei e i molluschi, sono la fonte più importante di EPA e DHA per l’uomo.[3][9][36] Questi animali sono in grado di convertire l’acido alfa-linolenico in EPA e DHA oltre che di assumerlo con il cibo, quindi, in ultima analisi dal fitoplancton.[16] Inoltre, considerando il solo DHA, questi è presente in concentrazione elevata in molti oli di pesce, specialmente in quelli di pesci cresciuti in acque fredde. Si noti però che molti di questi oli sono ricchi anche di acidi grassi saturi.[24]
Per chi non mangia prodotti della pesca, il fegato degli animali terrestri ma anche la selvaggina dell’ordine dei Passeriformi rappresentano una buona fonte di EPA e DHA.[14][15]
Riguardo gli apporti giornalieri non è ancora chiaro quale siano i valori raccomandati. Nella tabella seguente sono riportati i valori suggeriti dalla FAO delle Nazioni Unite e dalla autorità europea per la sicurezza alimentare (EFSA).[11][12]
Categoria
Raccomandazioni
FAO
Uomini adulti e donne adulte non gravide e non in allattamento
Almeno 250 mg di EPA + DHA al giorno
FAO
Donne gravide o in allattamento
Almeno 300 mg di EPA + DHA al giorno di cui almeno 200 mg dovrebbero essere di DHA
FAO
Bambini di età compresa tra i 2 e i 4 anni
100-150 mg di EPA + DHA al giorno
FAO
Bambini dai 4 ai 6 anni
150-200 mg di EPA + DHA al giorno
FAO
Bambini dai 6 ai 10 anni
200-250 mg di EPA + DHA al giorno
EFSA
Uomini adulti e donne adulte non gravide
L’assunzione adeguata è di 250 mg di EPA + DHA al giorno
EFSA
Donne gravide
Ulteriori 100-200 mg di DHA al giorno
EFSA
Neonati e bambini da 6 mesi a 2 anni
100 mg di DHA al giorno
EFSA
Bambini dai 2 ai 18 anni
“Coerente con gli adulti”
Omega-3 negli alimenti
Gli acidi grassi polinsaturi sono particolarmente sensibili all’ossidazione a seguito del riscaldamento. I vari processi cui è sottoposto il cibo nella fase di preparazione potrebbero quindi ridurne il contenuto.[3] Tuttavia questo è vero solo in parte. Negli alimenti i due acidi grassi polinsaturi omega-3 non sono presenti in forma chimica pura, ma per la maggior parte esterificati nei fosfolipidi di membrana, e, in questa forma, risultano molto meno suscettibili all’ossidazione.[9]
Quando si considera il contenuto in EPA e DHA negli alimenti, esprimerlo come percentuale degli acidi grassi totali anziché come contenuto assoluto, mg/g di peso umido, induce in un errore. Se ad esempio si considera un pesce grasso come il salmone, il suo contenuto assoluto di EPA + DHA è alto, circa 8 mg/g di peso umido, mentre se si esprime in percentuale si ottiene il 20%. Di contro il merluzzo atlantico presenta una percentuale di EPA + DHA di oltre il 40%, ma un contenuto assoluto inferiore a 3 mg/g di peso umido. In questo caso l’alta percentuale è dovuta al fatto che il pesce in esame è magro, mentre nei pesci grassi il contenuto in EPA + DHA è “diluito” dalle elevate riserve di acidi grassi presenti nel tessuto adiposo dell’animale.[17]
Se invece si esprime il contenuto di EPA + DHA in mg/g di prodotto, non si osserva alcuna riduzione a seguito della maggior parte dei trattamenti cui il cibo è sottoposto nella fase di preparazione.[16]
Bibliografia
^ ab AbuMweis S., Jew S., Tayyem R. Agraib L. Eicosapentaenoic acid and docosahexaenoic acid containing supplements modulate risk factors for cardiovascular disease: a meta-analysis of randomised placebo-control human clinical. J Hum Nutr Diet 2018;31(1):67-84. doi:10.1111/jhn.12493
^ Ahlgren G., Lundstedt L., Brett M., Forsberg C. Lipid composition and food quality of some freshwater phytoplankton for cladoceran zooplankters. J Plankton Res 1990;12(4):809-18. doi:10.1093/plankt/12.4.809
^ abcdefg Akoh C.C. and Min D.B. Food lipids: chemistry, nutrition, and biotechnology. 3th ed. 2008
^ Blasbalg T.L., Hibbeln J.R., Ramsden C.E., Majchrzak S..F, Rawlings R.R. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr 2011;93(5):950-62. doi:10.3945/ajcn.110.006643
^ ab Brown T.J., Brainard J., Song F., Wang X., Abdelhamid A., Hooper L. Omega-3, omega-6, and total dietary polyunsaturated fat for prevention and treatment of type 2 diabetes mellitus: systematic review and meta-analysis of randomised controlled trials. BMJ 2019;366:l4697. doi:10.1136/bmj.l4697
^ abc Buckley M.T., Racimo F., Allentoft M.E., et al. Selection in Europeans on fatty acid desaturases associated with dietary changes. Mol Biol Evol 2017;34(6):1307-1318. doi:10.1093/molbev/msx103
^ ab Burdge G.C., Wootton S.A. Conversion of α-linolenic acid to eicosapentaenoic, docosapentaenoic and docosahexaenoic acids in young women. Br J Nutr 2002;88(4):411-420. doi:10.1079/BJN2002689
^ abcdef Calder P.C. Very long-chain n-3 fatty acids and human health: fact, fiction and the future. Proc Nutr Soc 2018 77(1):52-72. doi:10.1017/S0029665117003950
^ abcdefgh Chow Ching K. Fatty acids in foods and their health implication. 3th ed. 2008
^ ab Duvall M.G., Levy B.D. DHA- and EPA-derived resolvins, protectins, and maresins in airway inflammation. Eur J Pharmacol 2016;785:144-155. doi:10.1016/j.ejphar.2015.11.001
^ EFSA Panel on Dietetic Products, Nutrition, and Allergies (NDA). Scientific opinion on dietary reference values for fats, including saturated fatty acids, polyunsaturated fatty acids, monounsaturated fatty acids, trans fatty acids, and cholesterol. 2010. doi:10.2903/j.efsa.2010.1461
^ FAO. Global recommendations for EPA and DHA intake (As of 30 June 2014)
^ Fumagalli M., Moltke I., Grarup N., Racimo F., Bjerregaard P., Jørgensen M.E., Korneliussen T.S., Gerbault P., Skotte L., Linneberg A., Christensen C., Brandslund I., Jørgensen T., Huerta-Sánchez E., Schmidt E.B., Pedersen O., Hansen T., Albrechtsen A., Nielsen R.. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science 2015;349(6254):1343-7. doi:10.1126/science.aab2319
^ Gladyshev M.I., Makhutova O.N., Gubanenko G.A.; Rechkina E.A., Kalachova G.S., Sushchik N.N. Livers of terrestrial production animals as a source of long-chain polyunsaturated fatty acids for humans: an alternative to fish? Eur J Lipid Sci Technol 2015;117:1417-1421. doi:10.1002/ejlt.201400449
^ Gladyshev M.I., Popova O.N., Makhutova, O.N., Zinchenko, T.D., Golovatyuk, L.V., Yurchenko, Y.A., Kalachova, G.S., Krylov, A.V., Sushchik, N.N. Comparison of fatty acid compositions in birds feeding in aquatic and terrestrial ecosystems. Contemp Probl Ecol 2016;9:503-513. doi:10.1134/S1995425516040065
^ abcd Gladyshev M.I. and Sushchik N.N. Long-chain omega-3 polyunsaturated fatty acids in natural ecosystems and the human diet: assumptions and challenges. Biomolecules 2019;9(9):485. doi:10.3390/biom9090485
^ Gladyshev M.I., Sushchik N.N., Tolomeev A.P., Dgebuadze Y.Y. Meta-analysis of factors associated with omega-3 fatty acid contents of wild fish. Rev Fish Biol Fish 2018;28,:277-299. doi:10.1007/s11160-017-9511-0
^ Kim H-Y. Novel metabolism of docosahexaenoic acid in neural cells. J Biol Chem 2007;282(26):18661-18665. doi:10.1074/jbc.R700015200
^ Kouba M., Mourot J. A review of nutritional effects on fat composition of animal products with special emphasis on n-3 polyunsaturated fatty acids. Biochimie 2011;93(1):13-7. doi:10.1016/j.biochi.2010.02.027
^ Kromann N., Green A. Epidemiological studies in the Upernavik district, Greenland. Incidence of some chronic diseases 1950-1974. Acta Med Scand 1980;208(5):401-6. doi:10.1111/j.0954-6820.1980.tb01221.x
^ Lauritzen L., Hansen H.S., Jørgensen M.H., Michaelsen K.F. The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Prog Lipid Res 2001;40(1-2):1-94. doi:10.1016/s0163-7827(00)00017-5
^ ab Lee J.M., Lee H., Kang S. and Park W.J. Fatty acid desaturases, polyunsaturated fatty acid regulation, and biotechnological advances. Nutrients 2016;8(1):23. doi:10.3390/nu8010023
^ Mason R.P., Sherratt S.C.R. Omega-3 fatty acid fish oil dietary supplements contain saturated fats and oxidized lipids that may interfere with their intended biological benefits. Biochem Biophys Res Commun 2017;483(1):425-429. doi:10.1016/j.bbrc.2016.12.127
^ Mathieson I., Lazaridis I., Rohland N., Mallick S., Patterson N., Roodenberg S.A., Harney E., Stewardson K., Fernandes D., Novak M., et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 2015;528:499-503. doi:10.1038/nature16152
^ ab McCann J.C., Ames B.N. Is docosahexaenoic acid, an n-3 long-chain polyunsaturated fatty acid, required for development of normal brain function? An overview of evidence from cognitive and behavioral tests in humans and animals. Am J Clin Nutr 2005;82(2):281-95. doi:10.1093/ajcn.82.2.281
^ ab Mensink R.P., Zock P.L., Kester A.D., Katan M.B. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: a meta-analysis of 60 controlled trials. Am J Clin Nutr 2003;77(5):1146-55. doi:10.1093/ajcn/77.5.1146
^ Muller-Navarra D.C. Biochemical versus mineral limitation in Daphnia. Limnol Oceanogr 1995;40:1209-1214. doi:10.4319/lo.1995.40.7.1209
^ ab Niu S.L., Mitchell D.C., Lim S.Y., Wen Z.M., Kim H.Y., Salem N. Jr, Litman B.J. Reduced G protein-coupled signaling efficiency in retinal rod outer segments in response to n-3 fatty acid deficiency. J Biol Chem 2004;279(30):31098-104. doi:10.1074/jbc.M404376200
^ Oh D.Y., Talukdar S., Bae E.J., Imamura T., Morinaga H., Fan WQ, Li P., Lu W.J., Watkins S.M., Olefsky J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010 142(5):687-698. doi:10.1016/j.cell.2010.07.041
^ Popova O.N., Haritonov A.Y., Sushchik N.N., Makhutova O.N., Kalachova G.S., Kolmakova A.A., Gladyshev M.I. Export of aquatic productivity, including highly unsaturated fatty acids, to terrestrial ecosystems via Odonata. Sci Total Environ 2017;581-582:40-48. doi:10.1016/j.scitotenv.2017.01.017
^ Salem N. Jr, Litman B., Kim H.Y., Gawrisch K. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids 2001;36(9):945-59. doi:10.1007/s11745-001-0805-6
^ abcd Simopoulos A.P. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med 2008;233(6):6746-88. doi:10.1016/S0753-3322(02)00253-6
^ Smith W., Mukhopadhyay R. Essential fatty acids: the work of George and Mildred Burr. J Biol Chem 2012;287(42):35439-35441. doi:10.1074/jbc.O112.000005
^ ab Stubbs C.D., Smith A.D. The modification of mammalian membrane polyunsaturated fatty acid composition in relation to membrane fluidity and function. Biochim Biophys Acta 1984;779(1):89-137. doi:10.1016/0304-4157(84)90005-4
^ abc Tocher D.R., Betancor M.B., Sprague M., Olsen R.E., Napier J.A. Omega-3 long-chain polyunsaturated fatty acids, EPA and DHA: bridging the gap between supply and demand. Nutrients 2019;11(1):89. doi:10.3390/nu11010089
^ Yano K., MacLean C.J., Reed D.M., Shimizu Y., Sasaki H., Kodama K., Kato H., Kagan A. A comparison of the 12-year mortality and predictive factors of coronary heart disease among Japanese men in Japan and Hawaii. Am J Epidemiol 1988;127(3):476-87. doi:10.1093/oxfordjournals.aje.a114824
^ Williams C.M., Burdge G. Long-chain n-3 PUFA: plant v. marine sources. Proc Nutr Soc 2006;65(1):42-50. doi:10.1079/pns2005473
Gli acidi grassi essenziali o EFA sono acidi grassi insaturi che non possono essere sintetizzati dagli animali e, al pari degli altri nutrienti essenziali, devono essere ottenuti con la dieta. Nello specifico sono l’acido linoleico e l’acido alfa-linolenico.
L’incapacità degli animali di sintetizzare i due acidi grassi deriva dalla mancanza di due desaturasi, la delta-12 desaturasi (EC 1.14.19.6) e la delta-15 desaturasi (EC 1.14.19.25), enzimi in grado di inserire doppi legami cis oltre il carbonio 9.[1][9][15] Le due desaturasi sono presenti in organismi vegetali e microrganismi quali batteri, funghi e muffe.[10]
La delta-12 desaturasi catalizza la sintesi dell’acido linoleico a partire dall’acido oleico inserendo un doppio legame sul carbonio 12, quindi tra i carboni 6 e 7 se numerati dall’estremità metilica della molecola.[15]
La delta-15 desaturasi catalizza la sintesi dell’acido alfa-linolenico a partire dall’acido linoleico, inserendo un doppio legame sul carbonio 15, quindi tra i carboni 3 e 4 dall’estremità metilica della molecola. L’enzima è presente solo nel reticolo endoplasmatico e nei plastidi del fitoplancton e delle piante terrestri vascolari.[1][9]
Biosintesi degli Acidi Grassi Essenziali
L’acido linoleico e l’acido alfa-linolenico sono i capostipiti rispettivamente delle famiglie degli acidi grassi polinsaturi omega-6 e degli acidi grassi polinsaturi omega-3. Nel caso di mancanza nella dieta dei due acidi grassi essenziali, una condizione piuttosto rara, anche gli altri acidi grassi omega-6 e omega-3 diventano essenziali; per questo motivo questi lipidi da alcuni sono definiti acidi grassi semiessenziali.[9]
Nota: gli acidi grassi essenziali sono acidi grassi polinsaturi, ma non tutti gli acidi grassi polinsaturi sono essenziali, come quelli appartenenti alle famiglie degli omega-7 e omega-9.
La prima indicazione dell’esistenza degli acidi grassi essenziali risale al 1918, quando Hans Aron suggerì che il grasso assunto con la dieta potesse essere essenziale per il normale accrescimento degli animali e che, oltre al suo contributo calorico, ci fosse uno speciale valore nutritivo intrinseco derivante dalla presenza di alcune specifiche molecole lipidiche.[2]
Nel 1927, Herbert M. Evans e George Oswald Burr dimostrarono che, nonostante l’aggiunta alla dieta delle vitamine liposolubili A, D ed E, una carenza di grasso influenzava in modo grave sia la crescita che la riproduzione degli animali da esperimento, suggerendo la presenza in esso di una nuova sostanza indispensabile, che chiamarono vitamina F.[11]
Nel 1929, undici anni dopo il lavoro di Aron, Burr e sua moglie Mildred Lawson ipotizzarono che gli animali a sangue caldo non fossero in grado di sintetizzare quantità apprezzabili di certi acidi grassi e, un anno più tardi, scoprirono che l’acido linoleico era essenziale per gli animali.[7] E’ a questi due ricercatori che si deve la locuzione “acido grasso essenziale”.[8][19]
E fu solamente nel 1958, grazie al lavoro di Arild Hansen, che si ebbe la prima descrizione di una deficienza di acidi grassi essenziali nell’uomo, in particolare in neonati alimentati con una dieta a base di latte in cui tali acidi grassi erano assenti.[14]
Nel 1964, grazie alle ricerche di Van Dorp e Bergstroem, fu scoperta una delle loro funzioni biologiche, quella di essere precursori per la sintesi delle prostaglandine.[3][21]
Funzioni
Gli acidi grassi essenziali svolgono importanti funzioni biologiche.
L’acido linoleico e l’acido alfa-linolenico possono essere utilizzati a scopi energetici, quindi andare incontro a beta-ossidazione.[22]
A partire da essi gli animali, compreso l’uomo, sono in grado di sintetizzare, sebbene con efficienza variabile, gli altri membri delle famiglie degli omega-6, ad esempio l’acido arachidonico, e omega-3, ad esempio l’acido eicosapentaenoico e l’acido docosaesaenoico.[5][6][18]
Gli acidi grassi essenziali sono componenti strutturali delle membrane cellulari, di cui, tra le altre cose, ne modulano la fluidità.[20][22]
Sono essenziali, in particolare l’acido linoleico presente negli sfingolipidi dello strato corneo della pelle, per la formazione della barriera che si oppone alla perdita d’acqua dalla pelle stessa.[23]
Hanno un ruolo cruciale nella prevenzione di molte malattie, in particolare delle malattie coronariche.[12][16]
Fonti alimentari
L’acido linoleico è il più abbondante acido grasso polinsaturo nella dieta occidentale, e rappresenta l’85-90% di tutti gli omega-6 ingeriti.[13] Le principali fonti per l’uomo sono diversi oli vegetali e i semi di molte piante.[4]
Alimenti
Acido linoleico (mg/g)
Olio di cartamo
∼ 740
Olio di girasole
∼ 600
Olio di soia
∼ 530
Olio di mais
∼ 500
Olio di semi di cotone
∼ 480
Noci
∼ 340
Noci brasiliane
∼ 250
Olio di arachidi
∼ 240
Olio di colza
∼ 190
Arachidi
∼ 140
Olio di semi di lino
∼ 135
L’acido linoleico è presente in discreta quantità anche in prodotti di origine animale, come le uova di gallina o il lardo, ma solo perché gli animali sono stati allevati con mangimi che lo contengono.[9]
Da notare che alcune tra le principali fonti di acido linoleico, come le noci, l’olio di semi di lino, l’olio di soia, e l’olio di colza sono anche ricche di acido alfa-linolenico.[17]
Tra le fonti più ricche di acido alfa-linolenico presenti nell’alimentazione umana si ritrovano l’olio di semi di lino, circa 550 mg/g, l’olio di colza, circa 85 mg/g, e l’olio di soia, circa 75 mg/g.
Altri alimenti ricchi di acido alfa-linolenico sono le noci, con un contenuto di circa 70 mg/g e i semi di soia, circa 10 mg/g.[1]
Bibliografia
^ abc Akoh C.C. and Min D.B. Food lipids: chemistry, nutrition, and biotechnology. 3th Edition. CRC Press, Taylor & Francis Group, 2008
^ Aron H. Uber den Nahrwert. Biochem Z 1918; 92: 211-33
^ Bergstroem S., Danielsson H., Klenberg D., Samuelsson B. The enzymatic conversion of essential fatty acids into prostaglandins. J Biol Chem 1964;239:PC4006-8. doi:10.1016/S0021-9258(18)91234-2
^ Blasbalg T. L., Hibbeln J. R., Ramsden C. E., Majchrzak S. F. & Rawlings R. R. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr 2011;93(5):950-962. doi:10.3945/ajcn.110.006643
^ Burdge G.C, Jones A.E, Wootton S.A. Eicosapentaenoic and docosapentaenoic acids are the principal products of α-linolenic acid metabolism in young men. Br J Nutr 2002;88(4):355-364. doi:10.1079/BJN2002662
^ Burdge G.C., Wootton S.A. Conversion of α-linolenic acid to eicosapentaenoic, docosapentaenoic and docosahexaenoic acids in young women. Br J Nutr 2002;88(4):411-420. doi:10.1079/BJN2002689
^ Burr G. and Burr M. A new deficiency disease produced by the rigid exclusion of fat from the diet. J Biol Chem 1929;82:345-367. doi:10.1016/S0021-9258(20)78281-5
^ Burr G.O., Burr M.M., Miller E.S. On the fatty acids essential in nutrition. III. J Biol Chem 1932;97(1):1-9. doi:10.1016/S0021-9258(18)76213-3
^ abcd Chow Ching K. Fatty acids in foods and their health implication. 3th Edition. CRC Press, Taylor & Francis Group, 2008
^ Das U.N. Essential fatty acids: biochemistry, physiology and pathology. Biotechnol J 2006;1;420-439. doi:10.1002/biot.200600012
^ Evans H. M. and G. O. Burr. A new dietary deficiency with highly purified diets. III. The beneficial effect of fat in the diet. Proc Soc Exp Biol Med 1928;25:390-397. doi:10.3181/00379727-25-3867
^ Farvid M.S., Ding M., Pan A., Sun Q., Chiuve S.E., Steffen L.M., Willett W.C., Hu F.B. Dietary linoleic acid and risk of coronary heart disease: a systematic review and meta-analysis of prospective cohort studies. Circulation 2014;130:1568-1578. doi:10.1161/CIRCULATIONAHA.114.010236
^ Guyenet S.J., Carlson S.E. Increase in adipose tissue linoleic acid of US adults in the last half century. Adv Nutr 2015;6(6):660-664. doi:https://DOI.org/10.3945/an.115.009944
^ Hansen A.E., Haggard M.E., Boelsche A.N., Adam D.J., Wiese H.F. Essential fatty acids in infant nutrition. III. Clinical manifestations of linoleic acid deficiency. J Nutr 1958;66(4):565-76. doi:10.1093/jn/66.4.565
^ ab Malcicka M., Visser B., Ellers J. An evolutionary perspective on linoleic acid synthesis in animals. Evol Biol 2018;45:15-26. doi:10.1007/s11692-017-9436-5
^ Mensink R.P., Zock P.L., Kester A.D., Katan M.B. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: a meta-analysis of 60 controlled trials. Am J Clin Nutr 2003;77(5):1146-55. doi:10.1093/ajcn/77.5.1146
^ Rajaram S. Health benefits of plant-derived α-linolenic acid. Am J Clin Nutr 2014;100 Suppl 1:443S-8S. doi:10.3945/ajcn.113.071514
^ Rett B.S., Whelan J. Increasing dietary linoleic acid does not increase tissue arachidonic acid content in adults consuming Western-type diets: a systematic review. Nutr Metab (Lond) 2011;8:36. doi:10.1186/1743-7075-8-36
^ Smith W., Mukhopadhyay R. Essential fatty acids: the work of George and Mildred Burr. J Biol Chem 2012;287(42):35439-35441. doi:10.1074/jbc.O112.000005
^ Stubbs C.D., Smith A.D. The modification of mammalian membrane polyunsaturated fatty acid composition in relation to membrane fluidity and function. Biochim Biophys Acta 1984;779(1):89-137. doi:10.1016/0304-4157(84)90005-4
^ Van Dorp D.A., Beerthuis R.K., Nugteren D.H. and Vonkeman H. Enzymatic conversion of all-cis-polyunsaturated fatty acids into prostaglandins. Nature 1964;203:839-841. doi:10.1038/203839a0
^ ab Whelan J. and Fritsche K. Linoleic acid. Adv Nutr 2013;4(3): 311-312. doi:10.3945/an.113.003772
^ Ziboh V.A. Prostaglandins, leukotrienes, and hydroxy fatty acids in epidermis. Semin Dermatol 1992;11(2):114-20
Lo zucchero invertito corrisponde a saccarosio parzialmente o totalmente scisso in fruttosio e glucosio e, a prescindere dal procedimento utilizzato, la soluzione ottenuta contiene una eguale quantità dei due carboidrati.
Inoltre, a seconda del prodotto, può essere presente anche saccarosio non scisso.
Preparazione dello zucchero invertito
La rottura della molecola del saccarosio può avvenire in una reazione catalizzata da enzimi, quali:
la saccarasi-isomaltasi (EC 3.2.1.48), attiva a livello del nostro intestino dove è coinvolta nella digestione dei carboidrati;
la invertasi, enzima secreto dalle api nel miele e utilizzato a livello industriale per ottenere lo zucchero invertito.
Un altro procedimento ricorre all’azione di acidi, come avviene in parte nel nostro stomaco e come avveniva in passato, e ancora avviene, a livello casalingo e industriale. Venivano utilizzati acido solforico o cloridrico, scaldando con cautela per un certo periodo la soluzione; la reazione infatti è tanto più veloce quanto più è acida la soluzione a prescindere dal tipo di acido utilizzato, e maggiore è la temperatura. L’acidità veniva poi tamponata con sostanze alcaline quali la soda o il bicarbonato di sodio.
Un procedimento chimicamente simile a quello appena descritto si verifica quando si preparano cibi acidi. Un esempio è la preparazione di marmellate e confetture di frutta dove sono realizzate entrambe le condizioni di acidità, naturalmente, e alte temperature, attraverso il riscaldamento. Situazione analoga quando si dolcificano i succhi di frutta con saccarosio.
La reazione procede anche a temperatura ambiente, ovviamente più lentamente.
Questo che significa dal punto di vista pratico?
Significa che anche dolci e preparazioni acide, anche quelle già viste, durante la conservazione vanno incontro a una lenta reazione di inversione del saccarosio contenuto/residuo, con conseguente modificazione della dolcezza, poiché lo zucchero invertito alle basse temperature è più dolce, grazie alla presenza del fruttosio, e assunzione di un profilo gustativo differente.
Utilizzi
E’ utilizzato soprattutto in pasticceria e nella produzione del gelato grazie ad alcune sue peculiari caratteristiche.
Ha una maggiore affinità per l’acqua rispetto al saccarosio, per cui mantiene più umidi i prodotti in cui si trova: ad esempio, le torte fatte con lo zucchero invertito si seccano meno facilmente.
Evita o ritarda la formazione di cristalli, in quanto i due monosaccaridi formano meno cristalli rispetto al saccarosio, proprietà utile in pasticceria per glasse o coperture.
Ha un punto di congelamento più basso.
Incrementa, anche se di poco, la dolcezza del prodotto in cui è stato aggiunto poiché è più dolce di una eguale quantità di saccarosio. Va comunque ricordato che la dolcezza del fruttosio dipende dalla temperatura a cui si trova.
Può partecipare alla reazione di Maillard, cosa che il saccarosio non può fare, contribuendo così al colore e al gusto di alcuni prodotti da forno.
Da notare che il miele, essendo privo di saccarosio, ha quasi la stessa composizione in fruttosio e glucosio dello zucchero invertito al 100% (il fruttosio è leggermente più abbondante del glucosio). Quindi un miele, magari non molto aromatico, diluito in acqua può fare le veci di uno prodotto ottenuto industriale.
Bibliografia
Belitz .H.-D., Grosch W., Schieberle P. “Food Chemistry” 4th ed. Springer, 2009
Bender D.A. “Benders’ Dictionary of Nutrition and Food Technology”. 8th Edition. Woodhead Publishing. Oxford, 2006
Jordan S. Commercial invert sugar. Ind Eng Chem 1924;16(3):307-310. doi:10.1021/ie50171a037
Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2013 [Google eBooks]
Biochemistry and metabolism
Utilizziamo i cookie per essere sicuri che tu possa avere la migliore esperienza sul nostro sito. Se continui ad utilizzare questo sito noi assumiamo che tu ne sia felice.OkPrivacy policy
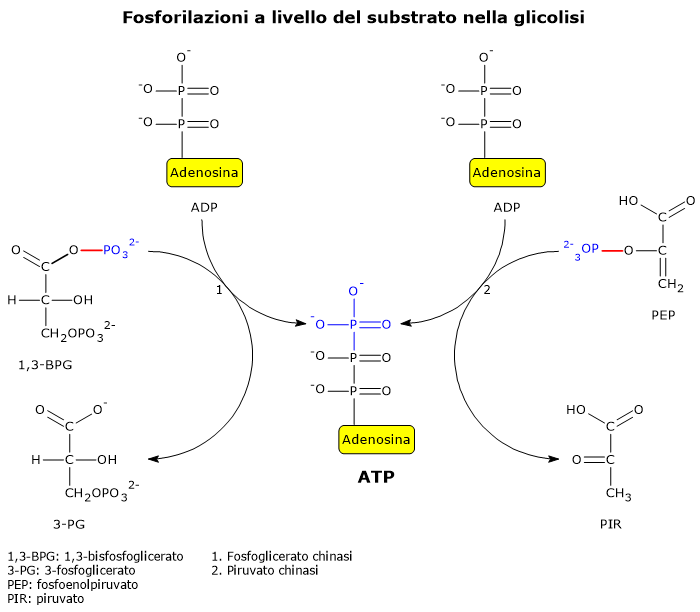 Le reazioni coinvolte sono quelle catalizzate dalla fosfoglicerato chinasi (EC 2.7.2.3), e dalla piruvato chinasi (EC 2.7.1.40).
Le reazioni coinvolte sono quelle catalizzate dalla fosfoglicerato chinasi (EC 2.7.2.3), e dalla piruvato chinasi (EC 2.7.1.40).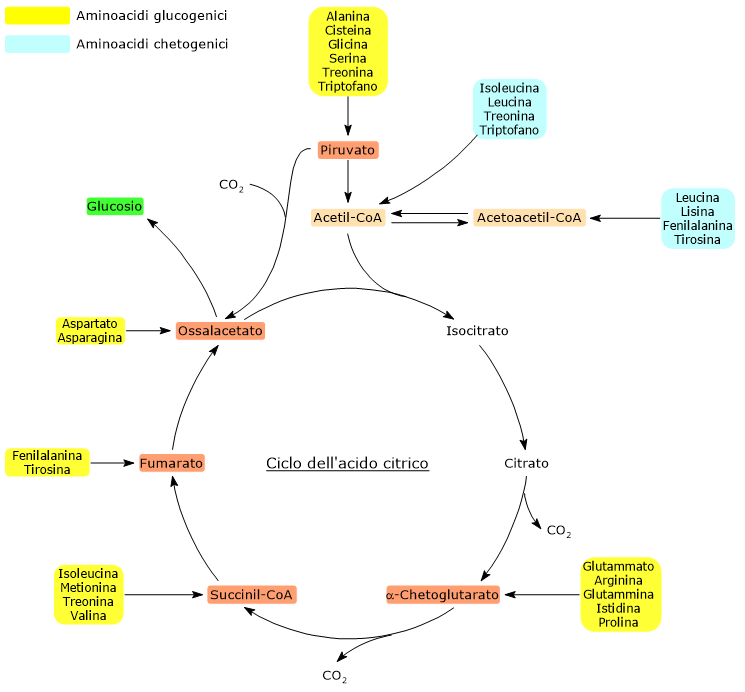
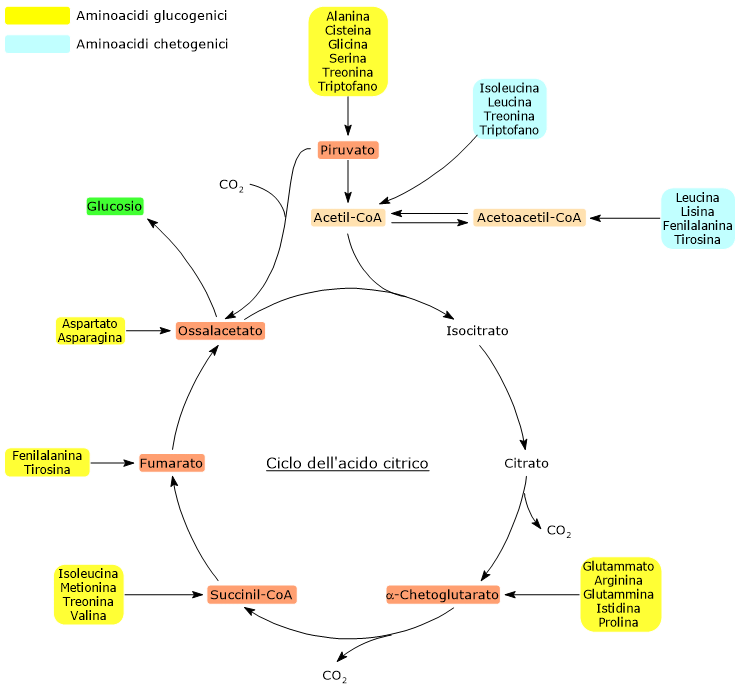
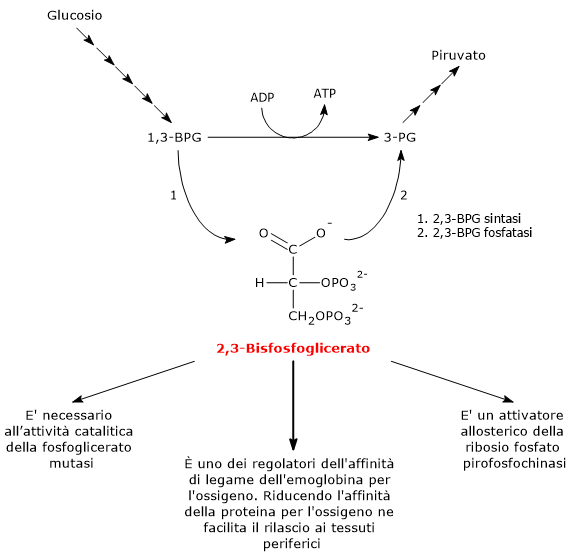 Nella seconda reazione dello shunt, il 2,3-bisfosfoglicerato viene defosforilato a 3-fosfoglicerato. La reazione è catalizzata dall’attività fosfatasica della bisfosfoglicerato mutasi. Il 3-fosfoglicerato rientra quindi nella via glicolitica a livello dell’ottava tappa, catalizzata dalla fosfoglicerato mutasi (EC 5.4.2.1).
Nella seconda reazione dello shunt, il 2,3-bisfosfoglicerato viene defosforilato a 3-fosfoglicerato. La reazione è catalizzata dall’attività fosfatasica della bisfosfoglicerato mutasi. Il 3-fosfoglicerato rientra quindi nella via glicolitica a livello dell’ottava tappa, catalizzata dalla fosfoglicerato mutasi (EC 5.4.2.1).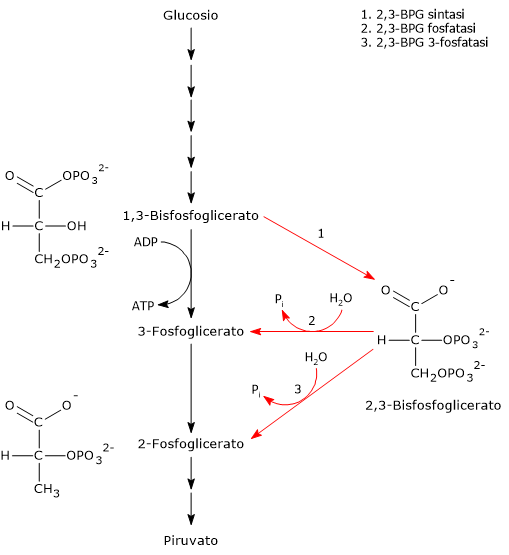 L’attività di fosfatasi è bassa, circa 1000 volte inferiore all’attività di sintasi, ed è stimolata da diversi anioni, come cloruro, fosfato, solfito e, in modo più marcato, dal 2-fosfoglicolato.
L’attività di fosfatasi è bassa, circa 1000 volte inferiore all’attività di sintasi, ed è stimolata da diversi anioni, come cloruro, fosfato, solfito e, in modo più marcato, dal 2-fosfoglicolato.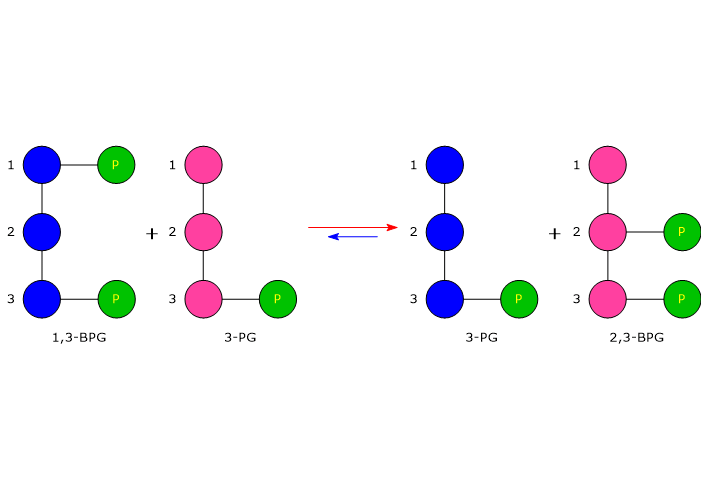 Nel secondo passaggio l’attività fosfatasica della bisfosfoglicerato mutasi catalizza la sintesi del 3-fosfoglicerato, un intermedio glicolitico, a seguito dell’idrolisi del gruppo fosforico in posizione C-2 del 2,3-BPG.Il 3-fosfoglicerato prodotto può quindi rientrare nella via glicolitica a livello della reazione catalizzata dalla fosfoglicerato mutasi, ossia l’ottava tappa della via.
Nel secondo passaggio l’attività fosfatasica della bisfosfoglicerato mutasi catalizza la sintesi del 3-fosfoglicerato, un intermedio glicolitico, a seguito dell’idrolisi del gruppo fosforico in posizione C-2 del 2,3-BPG.Il 3-fosfoglicerato prodotto può quindi rientrare nella via glicolitica a livello della reazione catalizzata dalla fosfoglicerato mutasi, ossia l’ottava tappa della via.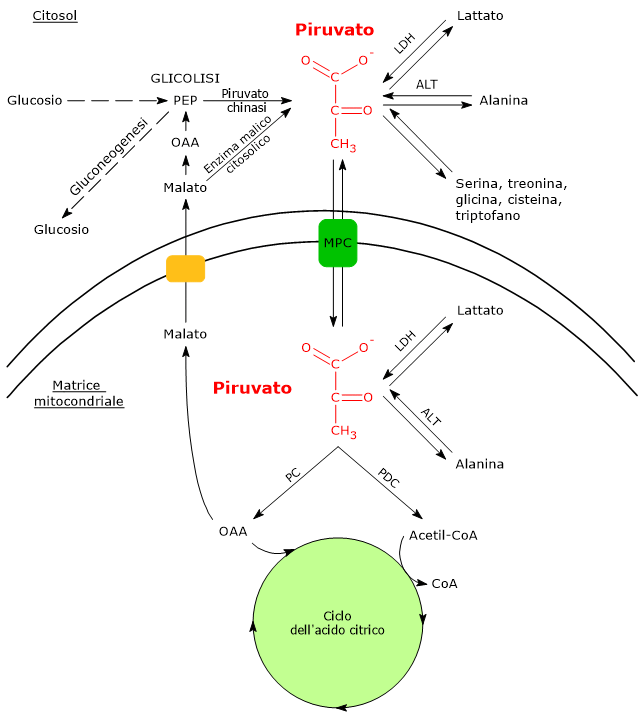 L’acido piruvico prodotto nel citosol, a mezzo di trasportatori specifici, entra nel mitocondrio dove potrà essere utilizzato a fini energetici, entrando nel ciclo dell’acido citrico, e/o biosintetici/anaplerotici, sulla base delle necessità della cellula.
L’acido piruvico prodotto nel citosol, a mezzo di trasportatori specifici, entra nel mitocondrio dove potrà essere utilizzato a fini energetici, entrando nel ciclo dell’acido citrico, e/o biosintetici/anaplerotici, sulla base delle necessità della cellula.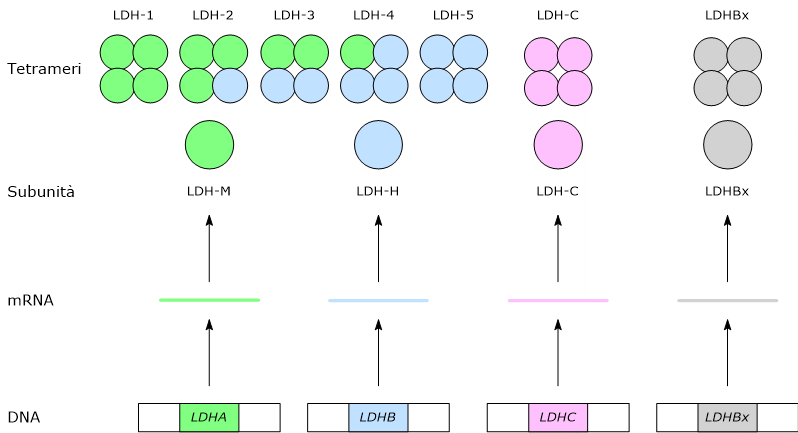 La subunità C va a formare una sesta isoforma, omotetramerica, indicata come LDH-6 o C4.
La subunità C va a formare una sesta isoforma, omotetramerica, indicata come LDH-6 o C4.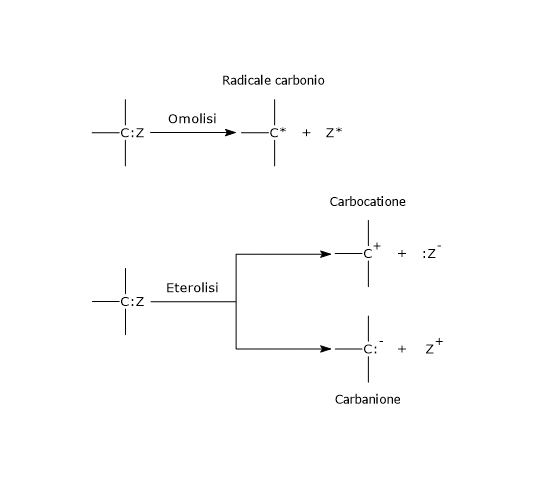 Nell’eterolisi, la rottura del legame covalente porta alla formazione di due atomi dotati di carica, ossia due ioni, un catione e un anione, poiché entrambi gli elettroni di legame vengono presi da uno dei due atomi precedentemente legati, quello più elettronegativo.
Nell’eterolisi, la rottura del legame covalente porta alla formazione di due atomi dotati di carica, ossia due ioni, un catione e un anione, poiché entrambi gli elettroni di legame vengono presi da uno dei due atomi precedentemente legati, quello più elettronegativo.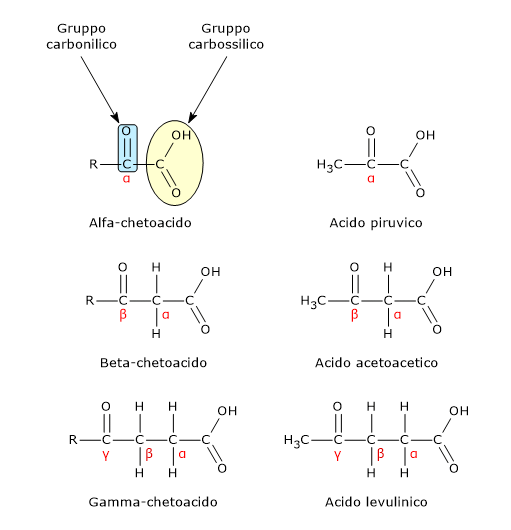 I chetoacidi, e in particolare gli alfa-chetoacidi, sono molto importanti in biochimica, essendo coinvolti in molte vie metaboliche.
I chetoacidi, e in particolare gli alfa-chetoacidi, sono molto importanti in biochimica, essendo coinvolti in molte vie metaboliche.
 L’enzima, al pari delle
L’enzima, al pari delle 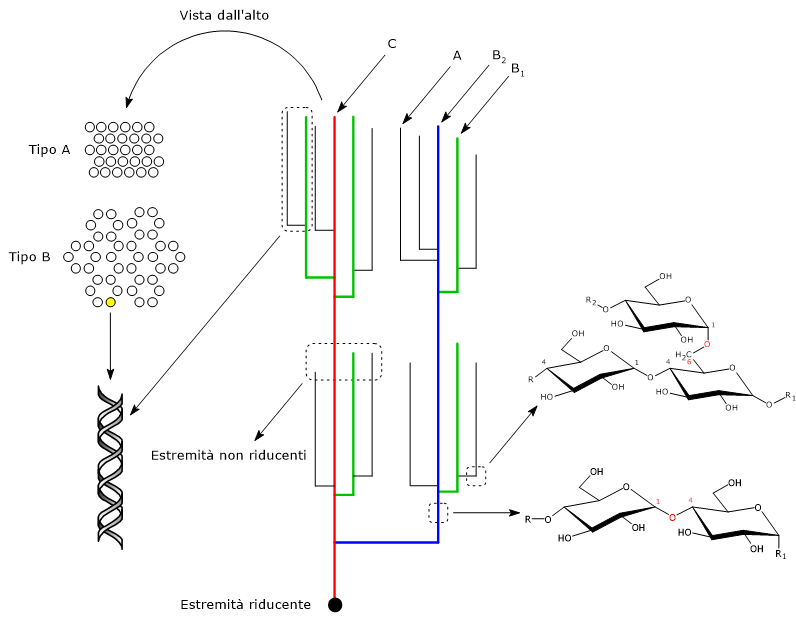

 Questi
Questi 
 Rappresentando la principale fonte di lipidi, quando associato a un basso consumo di prodotti di origine animale ad alto contenuto di grassi, assicura un elevato rapporto tra acidi grassi monoinsaturi e
Rappresentando la principale fonte di lipidi, quando associato a un basso consumo di prodotti di origine animale ad alto contenuto di grassi, assicura un elevato rapporto tra acidi grassi monoinsaturi e  Le poche ramificazioni sono connesse alla catena lineare da legami glicosidici α-(1→6), al pari di quanto accade con l’amilopectina e il
Le poche ramificazioni sono connesse alla catena lineare da legami glicosidici α-(1→6), al pari di quanto accade con l’amilopectina e il 
 In questi casi il
In questi casi il 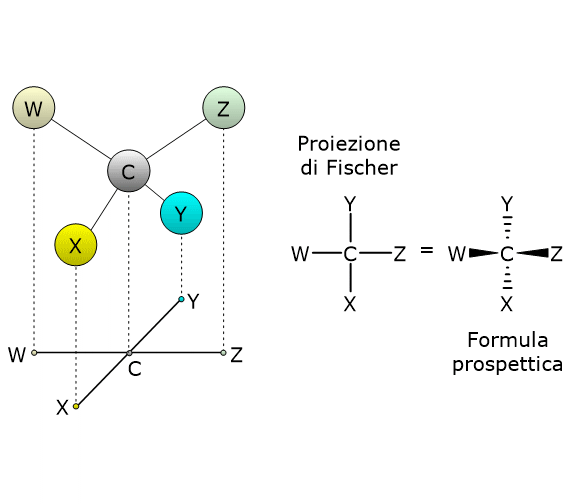
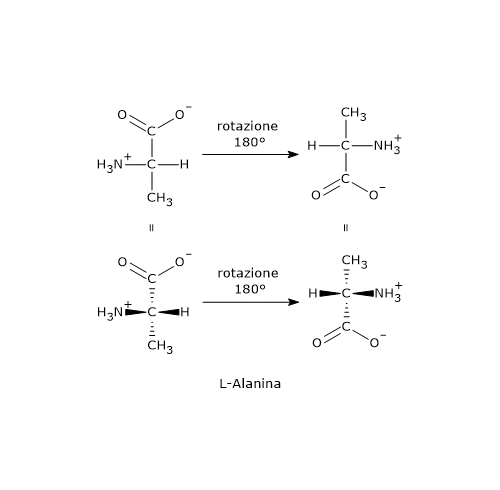
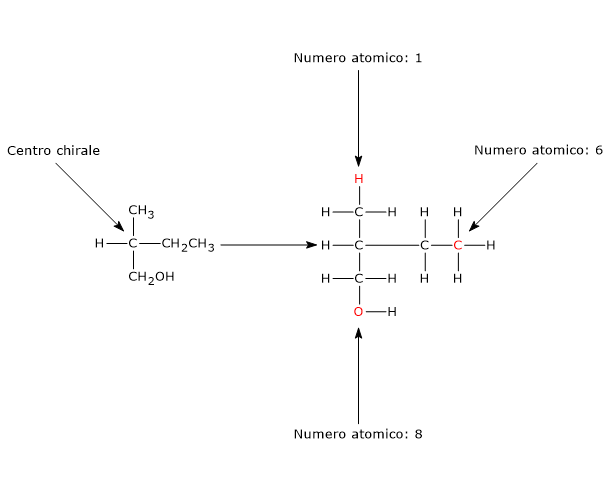
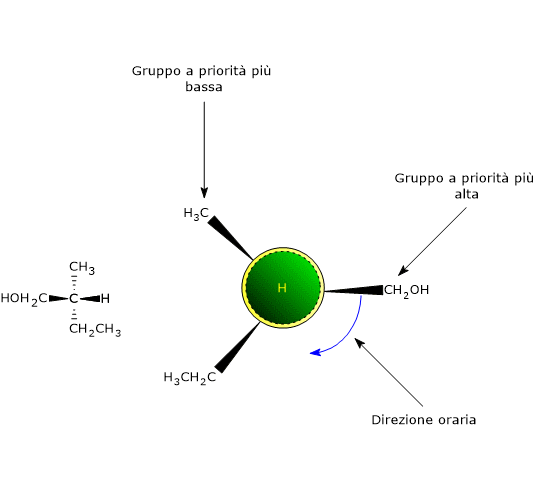
 Nel caso di doppio legame C=Y, un atomo Y sarà da legare sull’atomo di carbonio, e un atomo di carbonio sull’atomo Y.
Nel caso di doppio legame C=Y, un atomo Y sarà da legare sull’atomo di carbonio, e un atomo di carbonio sull’atomo Y. Si consideri il carbonio 2. L’ordine di priorità dei gruppi legati è: –OH > –CH2OHCH3 > –CH3 > –H. Si ruoti la molecola in modo che l’idrogeno, il gruppo con la priorità più bassa, sia diretto in direzione opposta all’osservatore. Disegnando una circonferenza partendo dal gruppo –OH, il gruppo con la priorità più alta, verso il gruppo –CH3, il gruppo a priorità più bassa, ci si muove in senso antiorario, per cui la configurazione del carbonio 2 è R. Applicando la stessa procedura al carbonio 3, si scopre che la sua configurazione è R. Quindi l’enantiomero in figura è il (2R,3R)-2,3-butandiolo.
Si consideri il carbonio 2. L’ordine di priorità dei gruppi legati è: –OH > –CH2OHCH3 > –CH3 > –H. Si ruoti la molecola in modo che l’idrogeno, il gruppo con la priorità più bassa, sia diretto in direzione opposta all’osservatore. Disegnando una circonferenza partendo dal gruppo –OH, il gruppo con la priorità più alta, verso il gruppo –CH3, il gruppo a priorità più bassa, ci si muove in senso antiorario, per cui la configurazione del carbonio 2 è R. Applicando la stessa procedura al carbonio 3, si scopre che la sua configurazione è R. Quindi l’enantiomero in figura è il (2R,3R)-2,3-butandiolo.
 Il distomero, l’enantiomero S, poteva causare gravi difetti alla nascita, in particolare la focomelia. Questo è forse l’esempio più eclatante dell’importanza delle proprietà chirali delle molecole, che ha poi spinto gli organismi di salute pubblica a promuovere, da parte dell’industria farmaceutica, la sintesi di farmaci, talidomide compresa, contenenti un singolo enantiomero.
Il distomero, l’enantiomero S, poteva causare gravi difetti alla nascita, in particolare la focomelia. Questo è forse l’esempio più eclatante dell’importanza delle proprietà chirali delle molecole, che ha poi spinto gli organismi di salute pubblica a promuovere, da parte dell’industria farmaceutica, la sintesi di farmaci, talidomide compresa, contenenti un singolo enantiomero. A temperatura ambiente, l’azoto inverte rapidamente la sua configurazione. Il fenomeno è noto come inversione dell’azoto, ossia, una rapida oscillazione dell’atomo e dei suoi ligandi, nel corso della quale l’azoto passa attraverso uno stato di transizione planare in cui ha ibridazione sp2. Come conseguenza, se l’azoto è il solo centro chirale della molecola, non c’è attività ottica in quanto si viene a formare una miscela racemica. L’inversione di configurazione viene evitata solamente in alcuni casi in cui l’azoto fa parte di una struttura ciclica che la impedisce. Dunque, la presenza di un centro chirale può non essere sufficiente per permettere la separazione dei rispettivi enantiomeri.
A temperatura ambiente, l’azoto inverte rapidamente la sua configurazione. Il fenomeno è noto come inversione dell’azoto, ossia, una rapida oscillazione dell’atomo e dei suoi ligandi, nel corso della quale l’azoto passa attraverso uno stato di transizione planare in cui ha ibridazione sp2. Come conseguenza, se l’azoto è il solo centro chirale della molecola, non c’è attività ottica in quanto si viene a formare una miscela racemica. L’inversione di configurazione viene evitata solamente in alcuni casi in cui l’azoto fa parte di una struttura ciclica che la impedisce. Dunque, la presenza di un centro chirale può non essere sufficiente per permettere la separazione dei rispettivi enantiomeri.
 Esempi di isomeria di catena si hanno anche tra
Esempi di isomeria di catena si hanno anche tra 
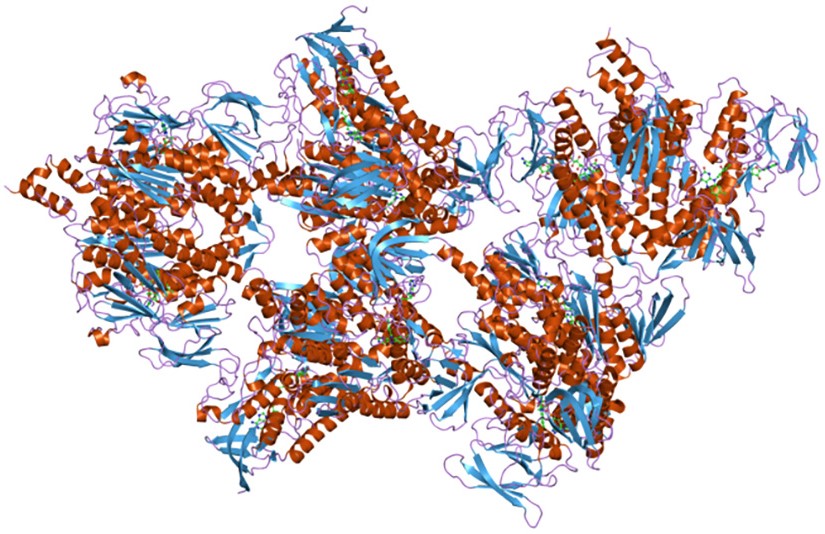
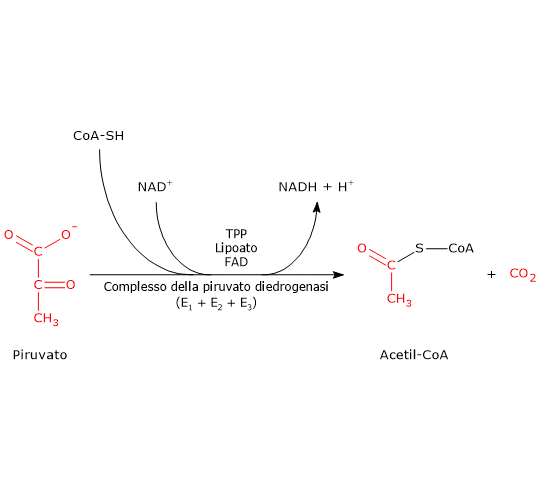
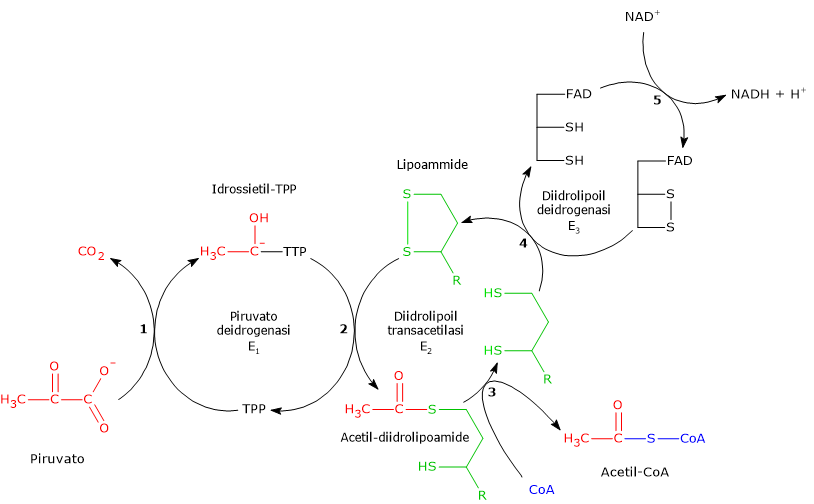
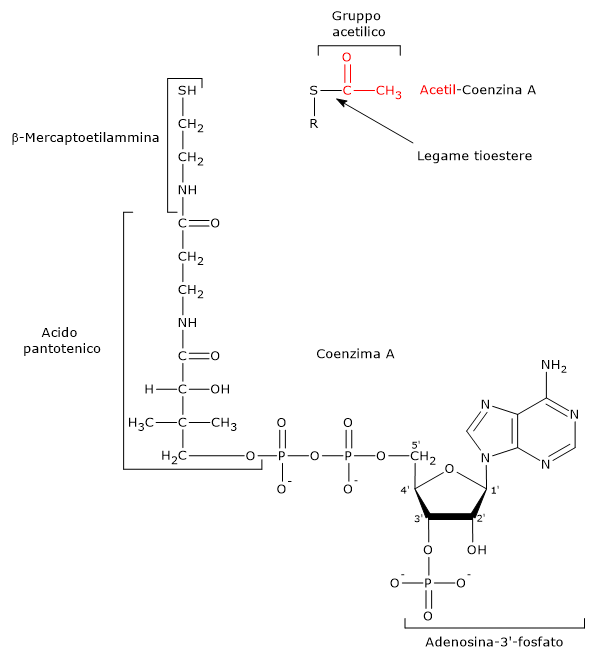
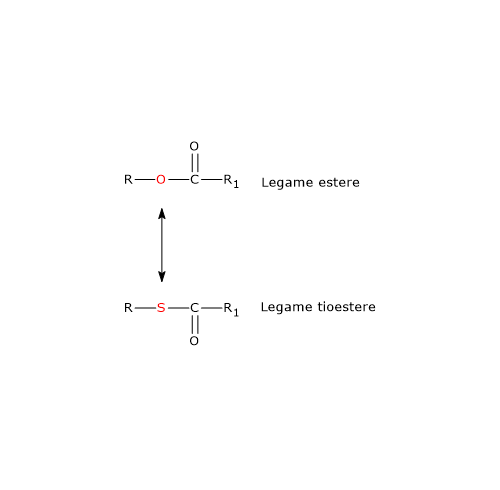
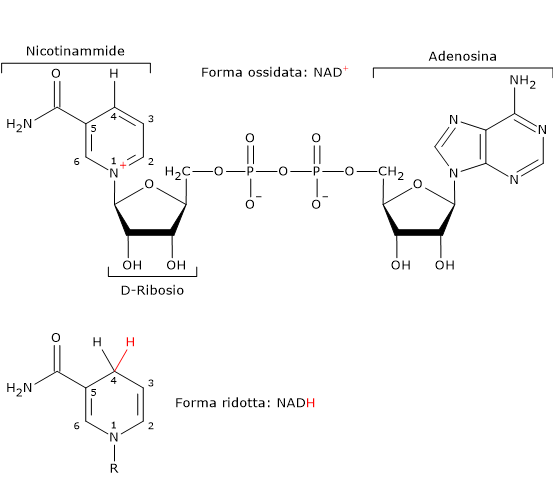
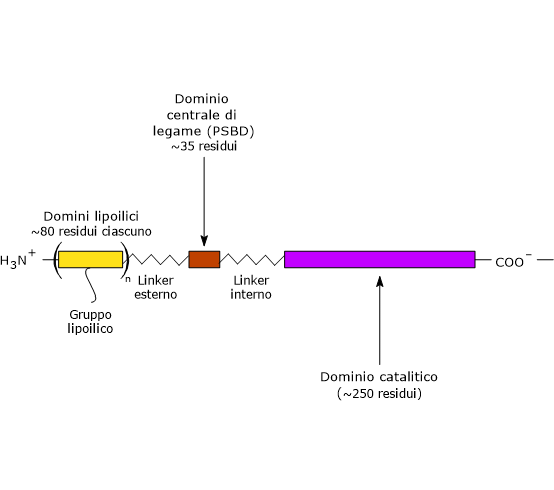

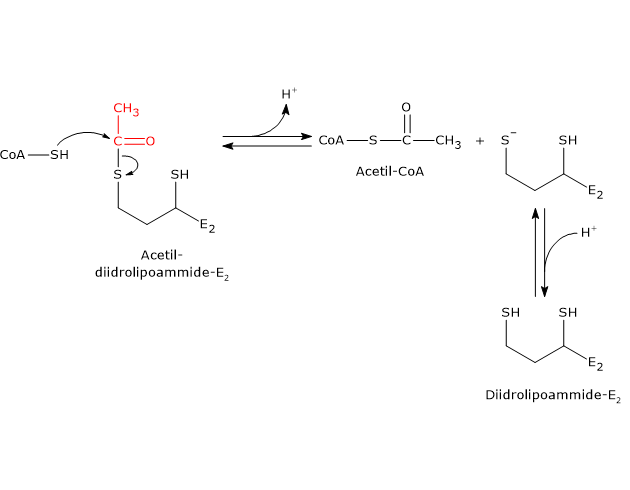 Meccanismo Catalitico della Diidrolipoil Transacetilasi
Meccanismo Catalitico della Diidrolipoil Transacetilasi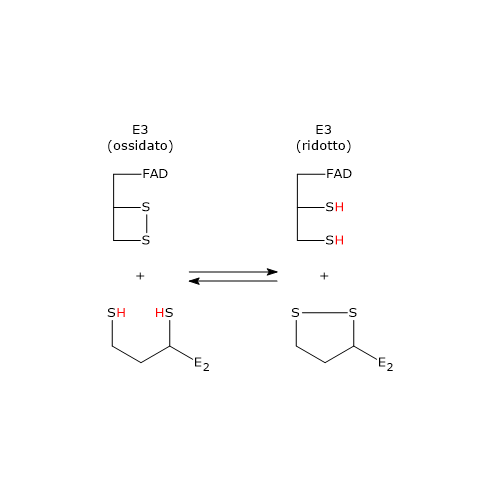
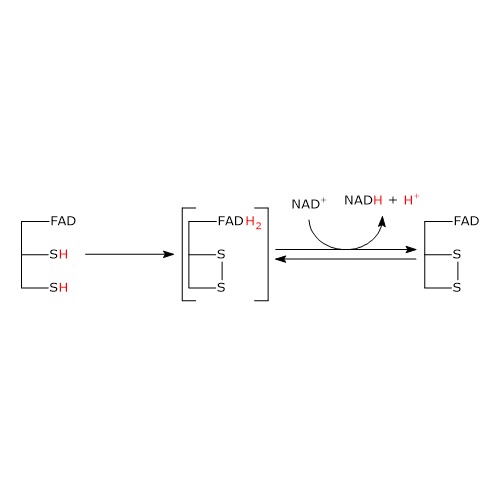
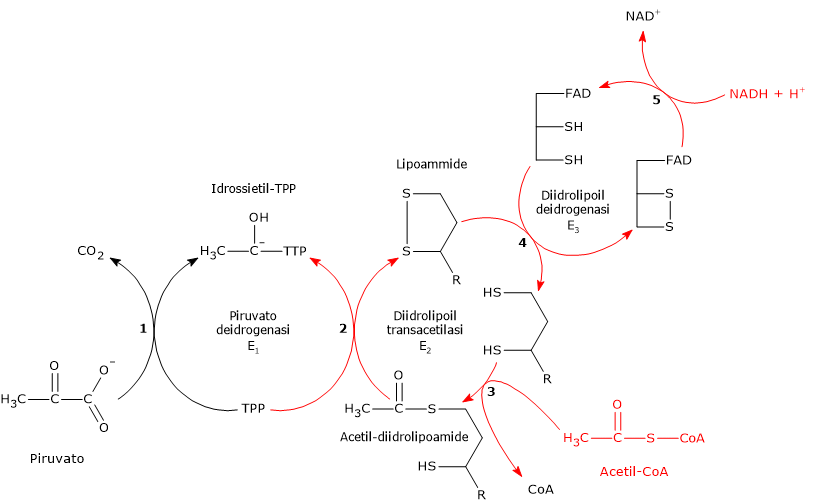
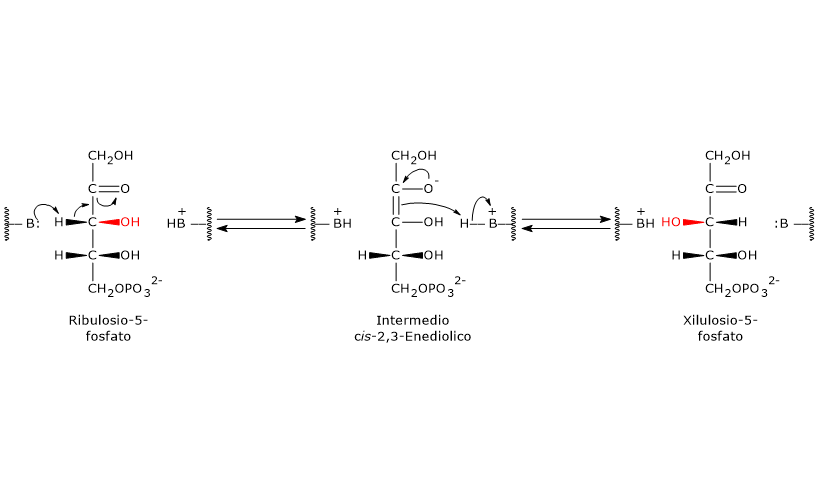
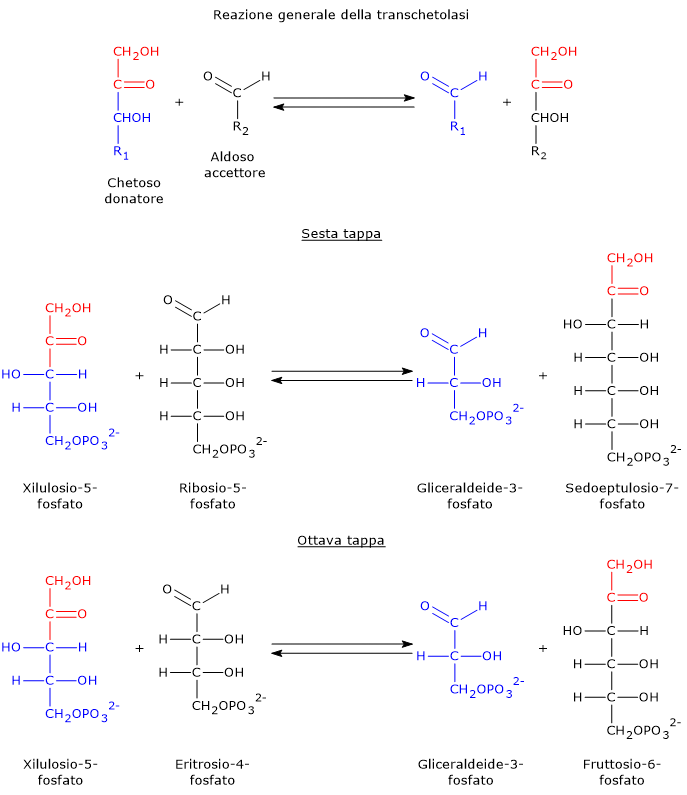
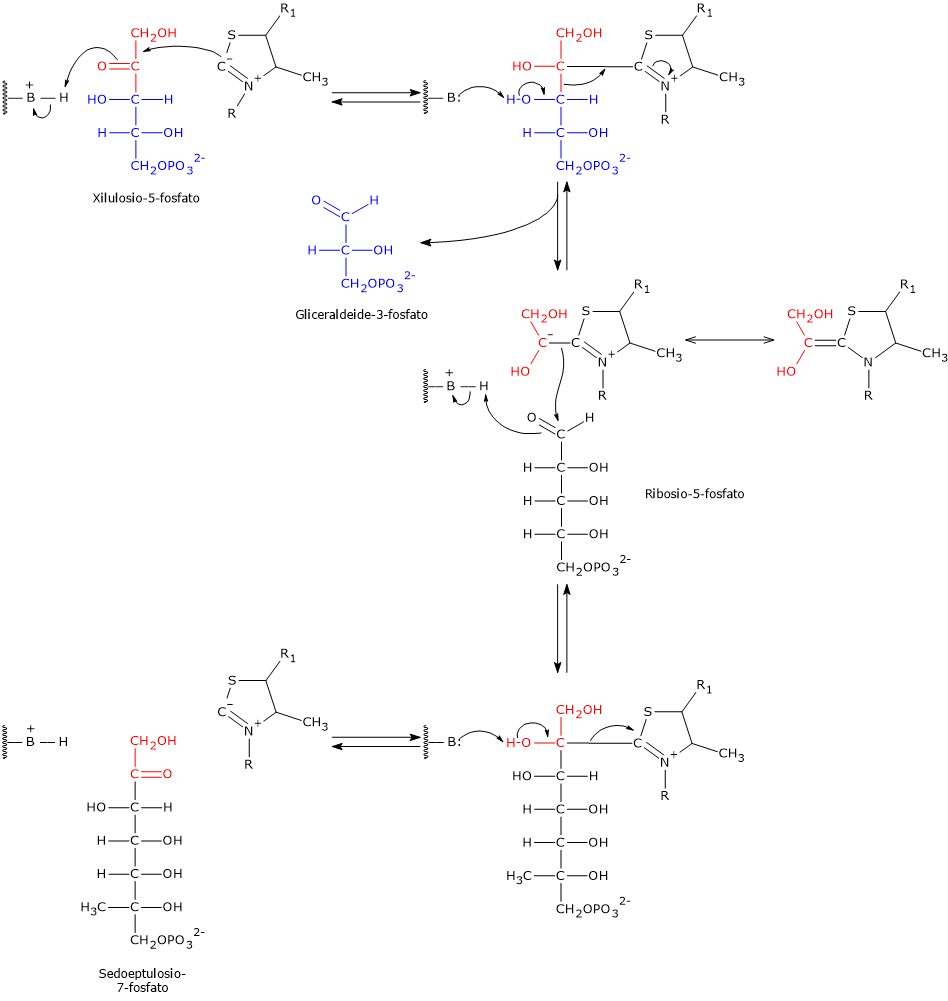
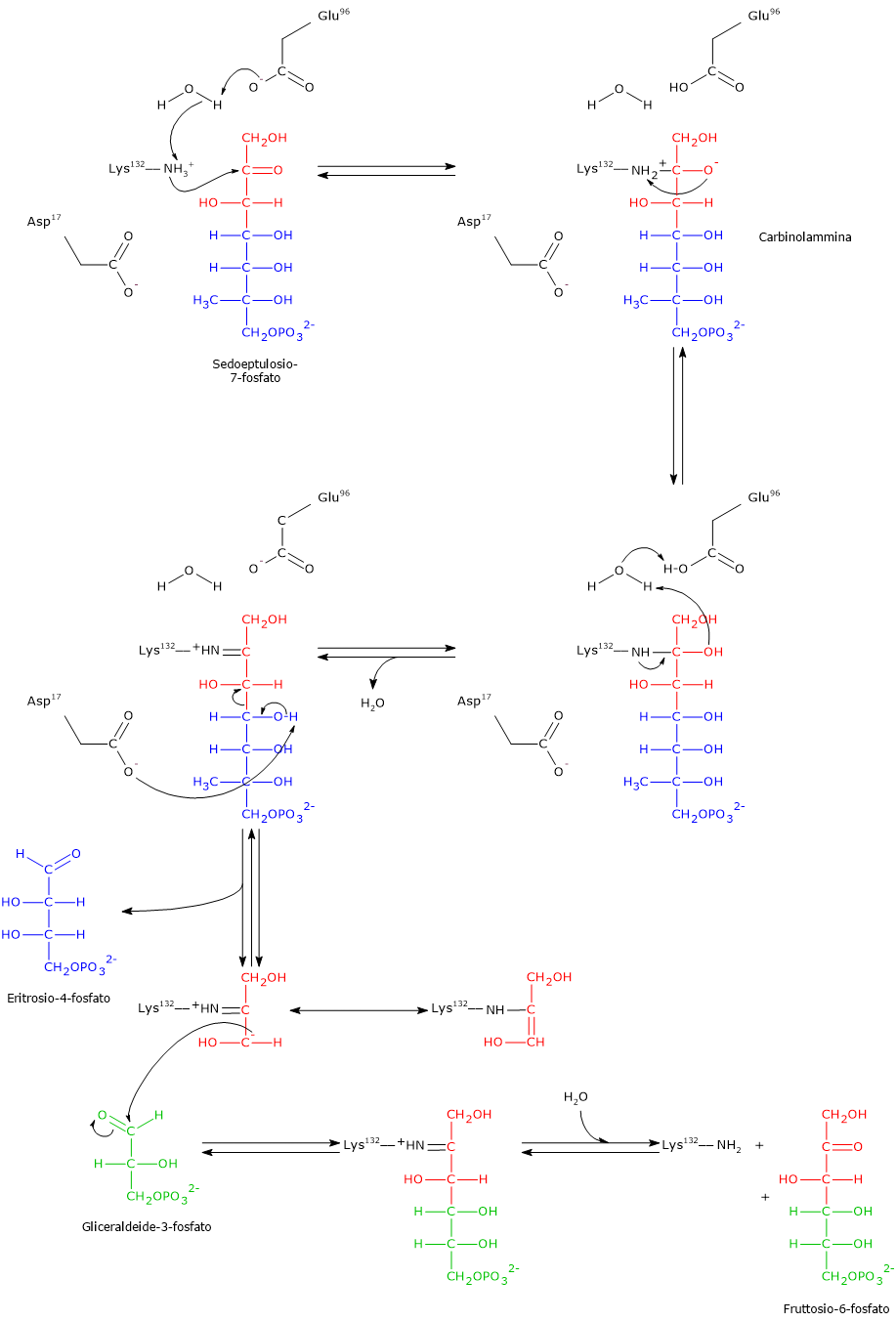
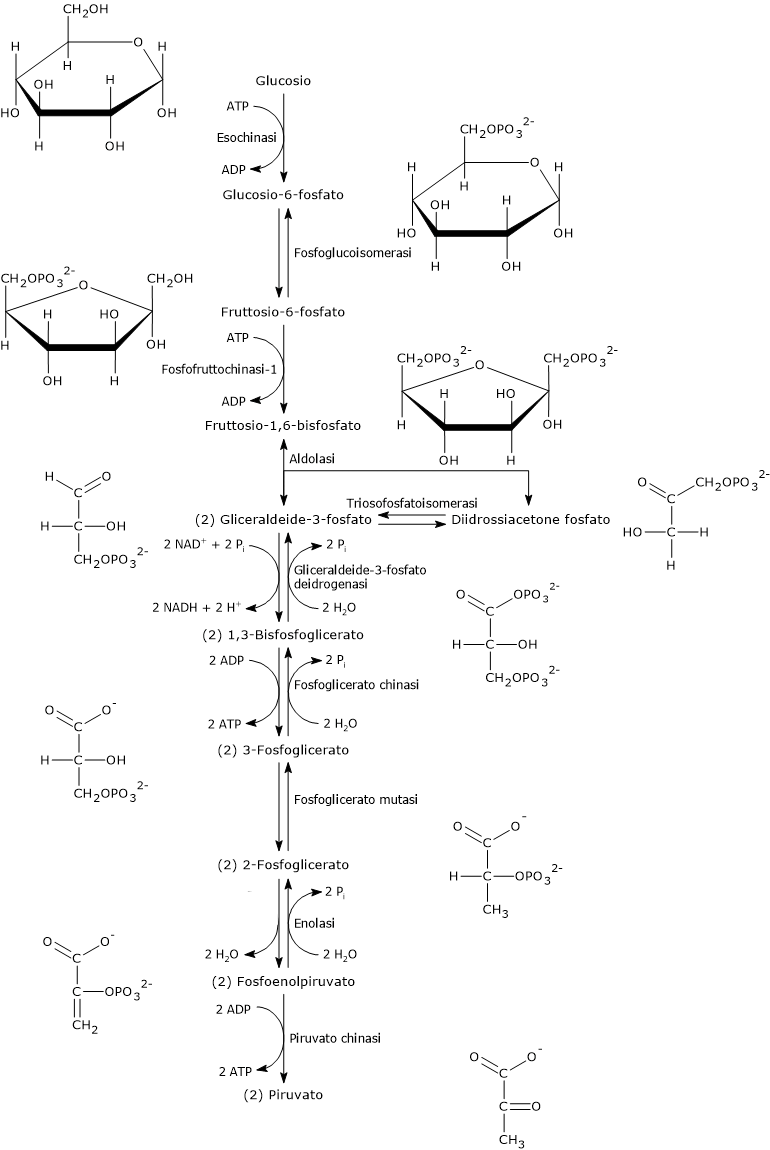
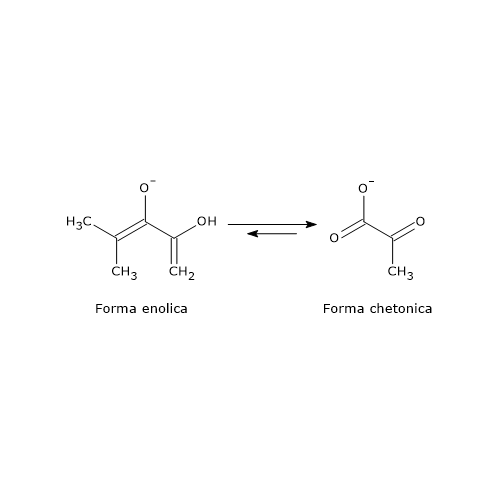
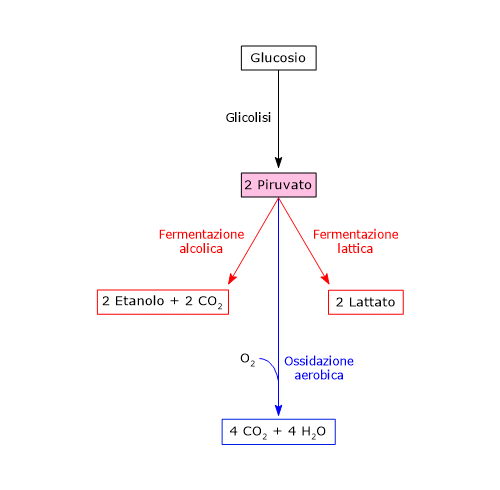
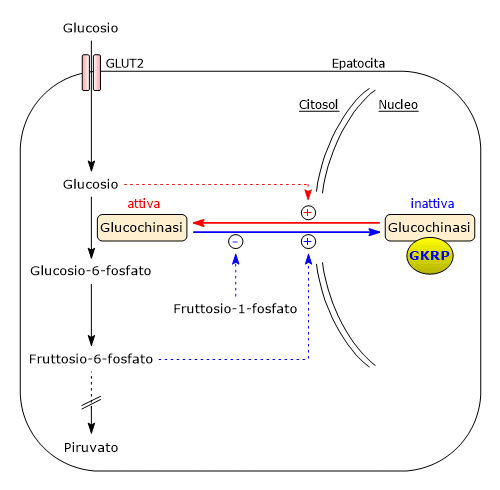
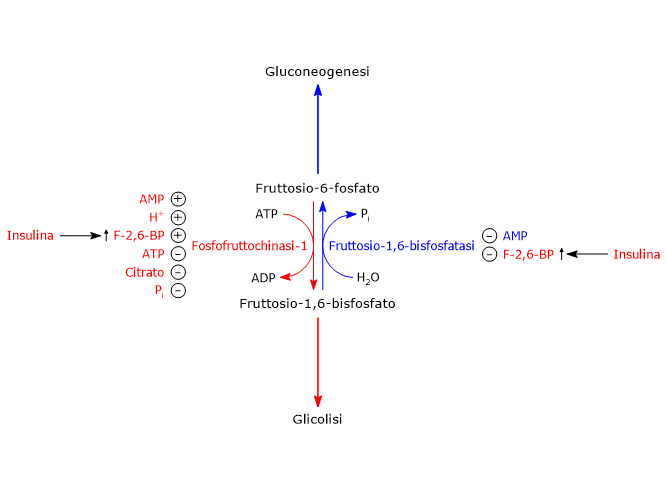
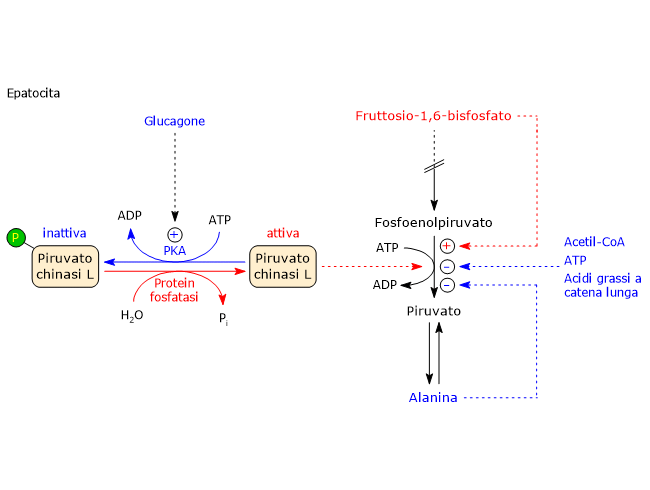
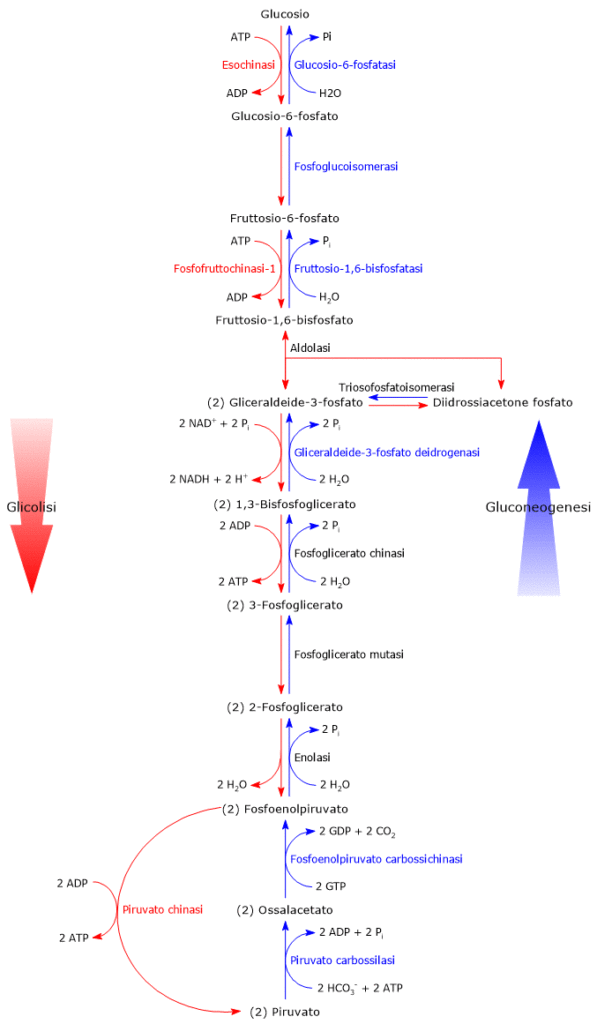
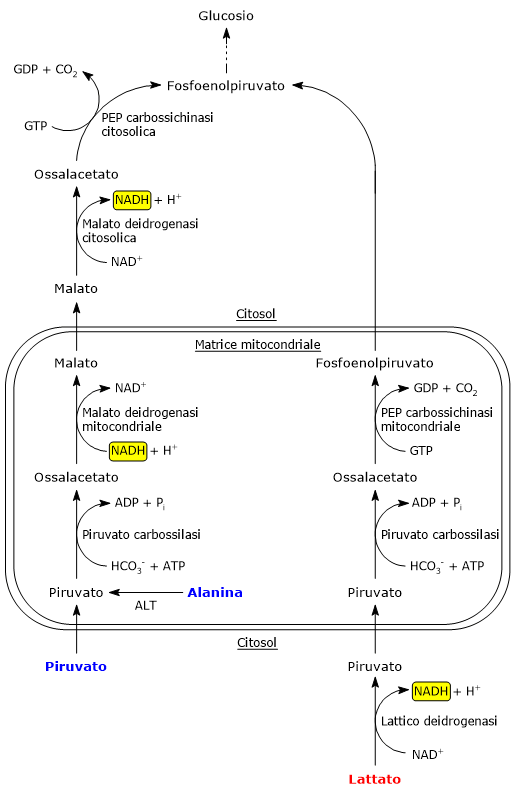
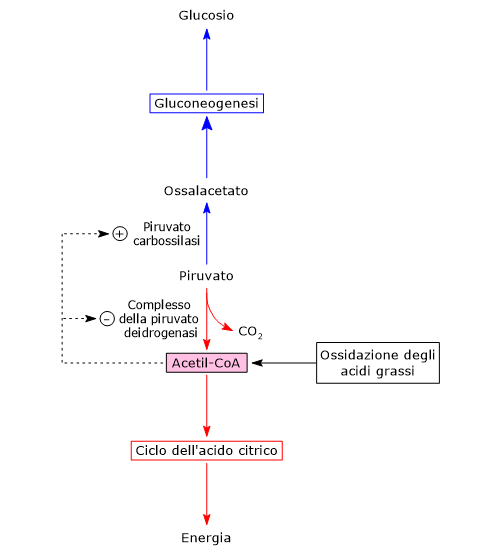
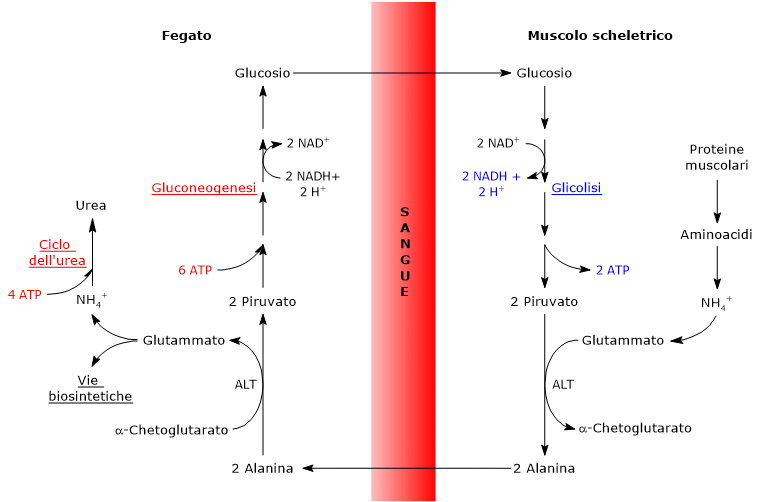
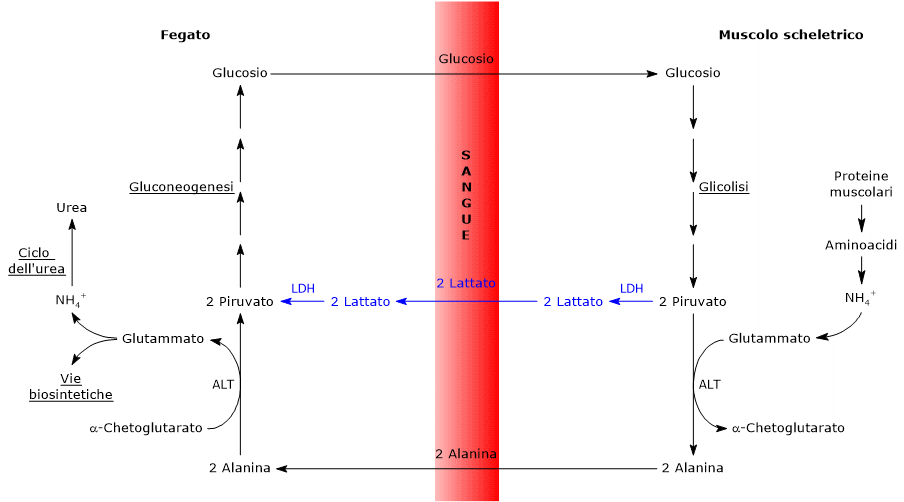
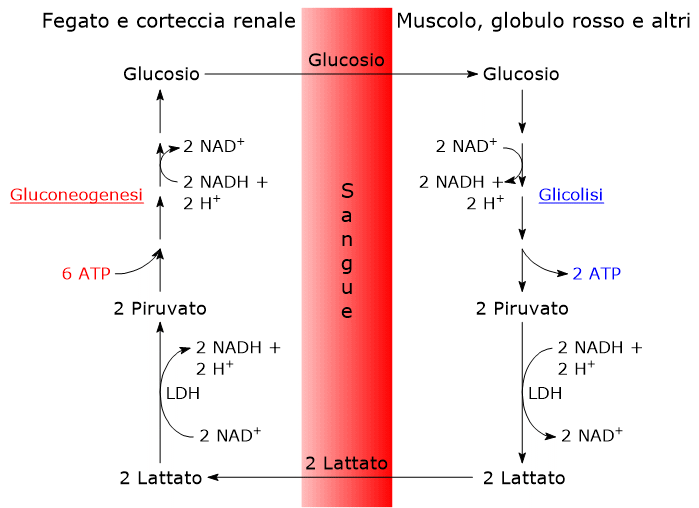
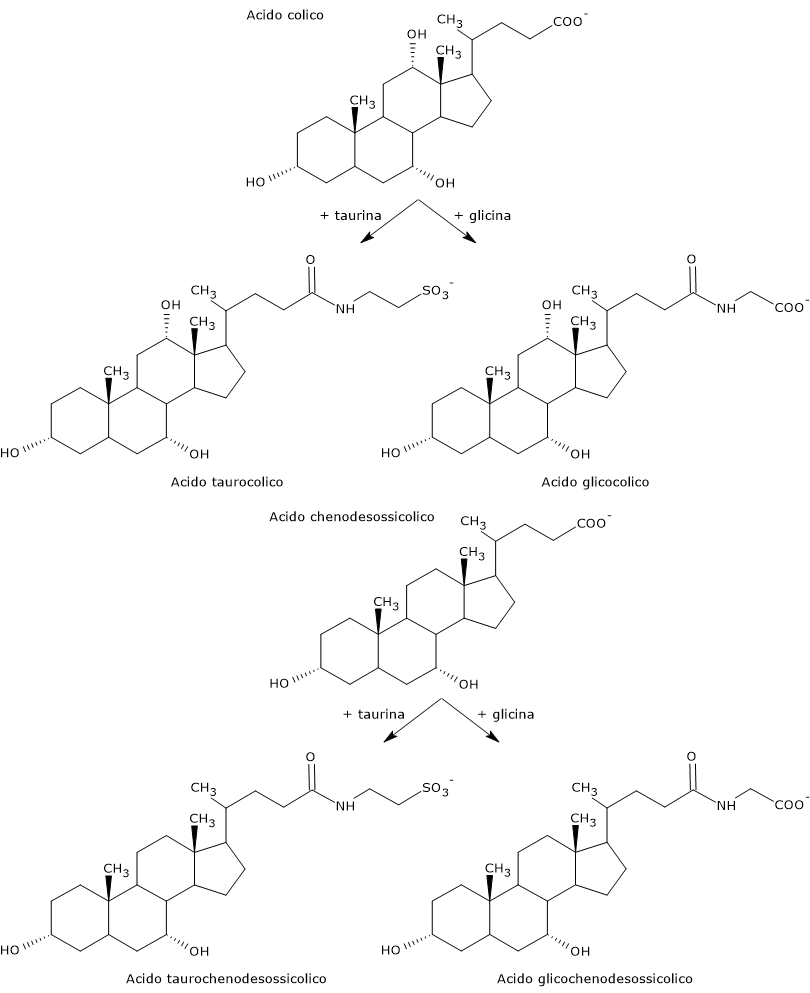
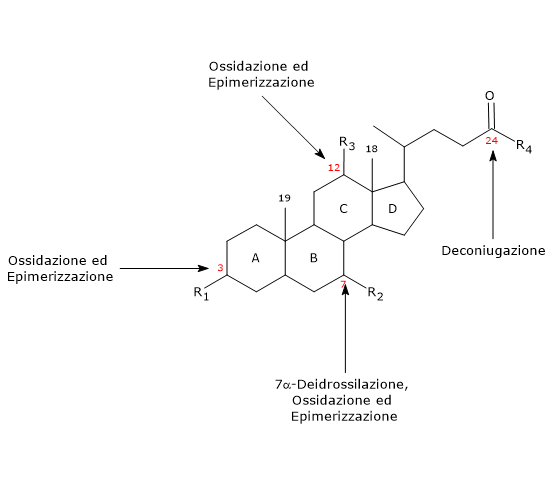
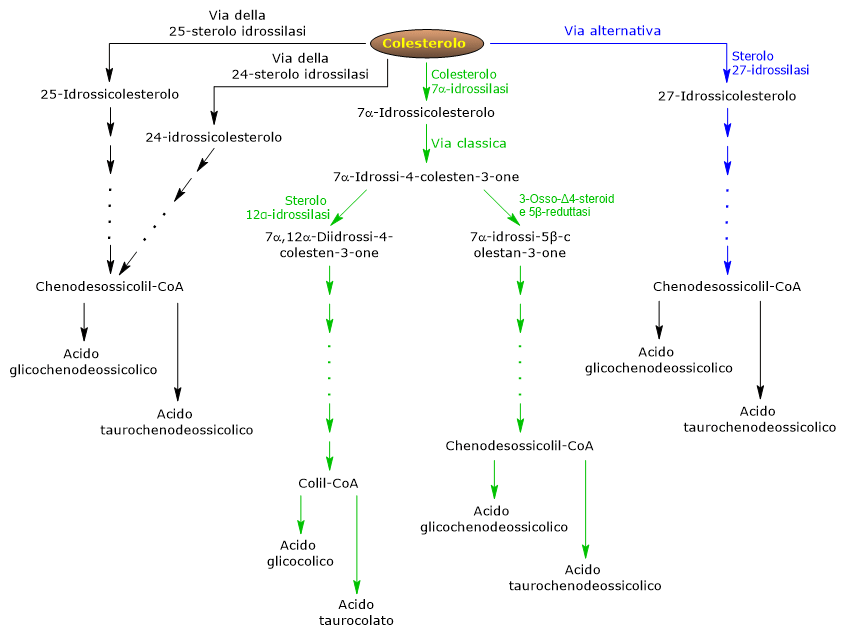
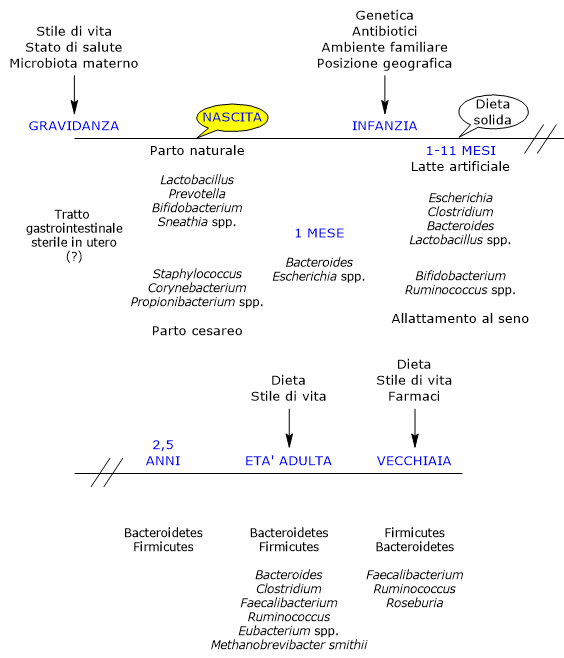
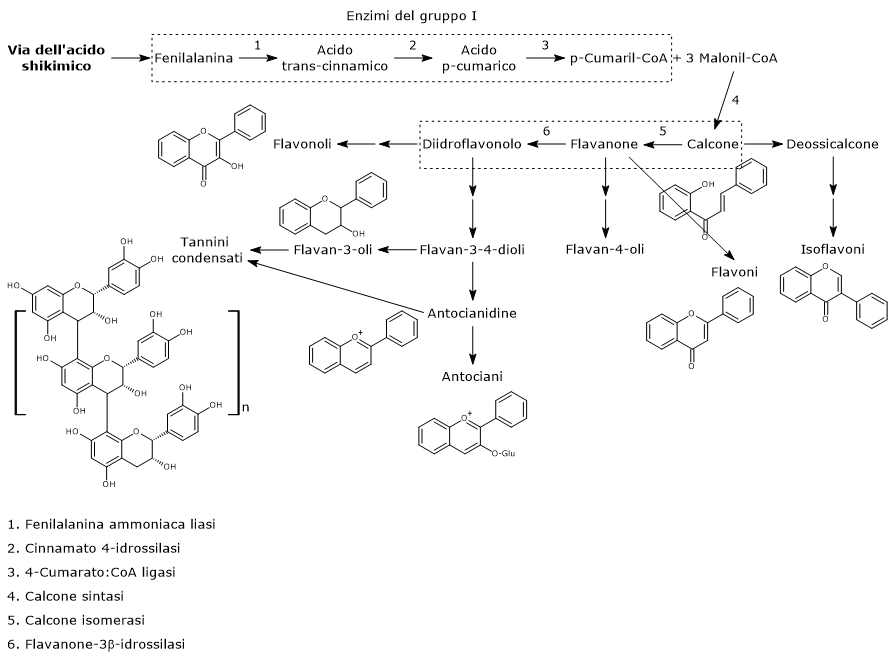
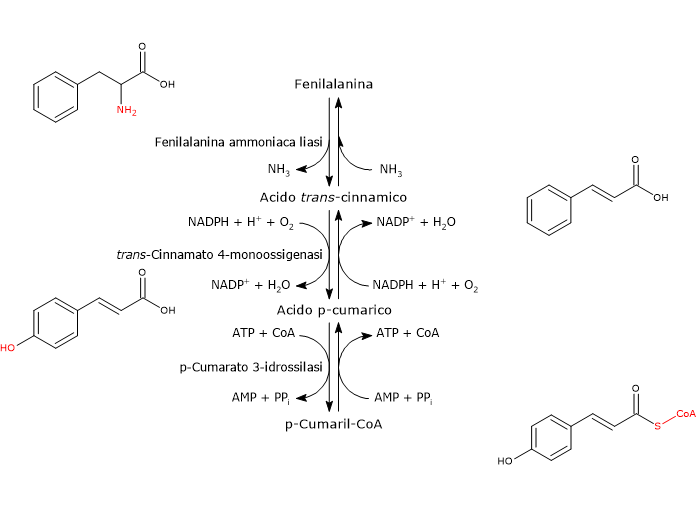
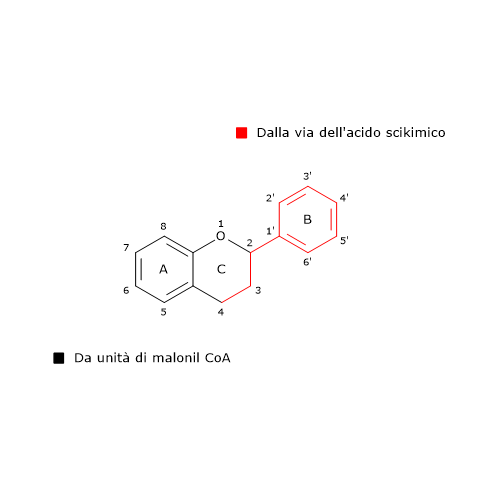
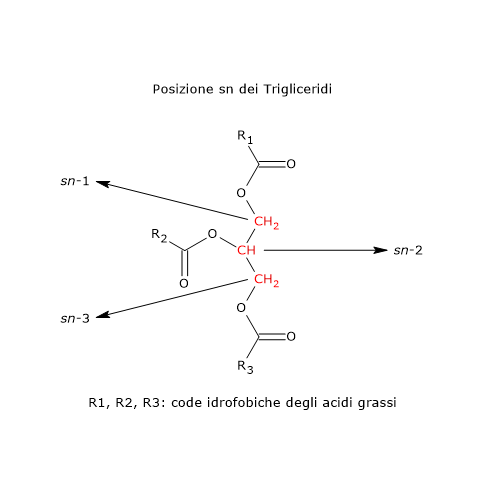
 Altre molecole presenti in quantità inferiori o tracce sono ad esempio il stigmasterolo, 2%, colesterolo, brassicasterolo, ergosterolo.
Altre molecole presenti in quantità inferiori o tracce sono ad esempio il stigmasterolo, 2%, colesterolo, brassicasterolo, ergosterolo.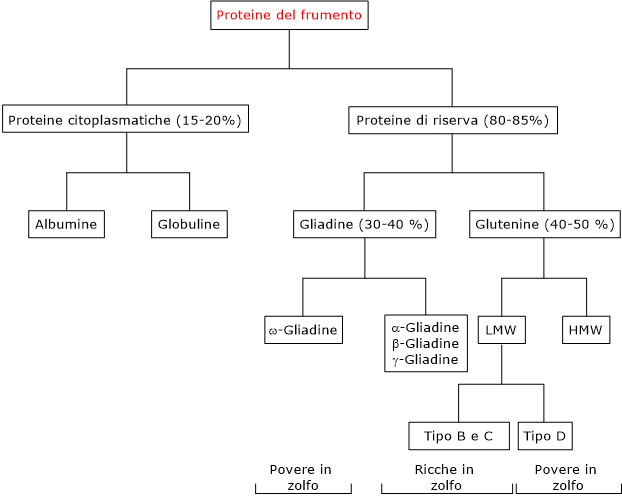
 Nella corsa il runner va incontro a un consumo calorico e a una perdita di acqua e sali minerali che dipendono da diversi fattori quali la tecnica, il grado di allenamento, le condizioni ambientali e le caratteristiche fisiologiche proprie di ciascun atleta. Ne consegue che la loro conoscenza permette di impostare un’alimentazione adeguata sia nella fase di allenamento che in quella di recupero tra un allenamento e il successivo, con l’obbiettivo di ottimizzare la performance.
Nella corsa il runner va incontro a un consumo calorico e a una perdita di acqua e sali minerali che dipendono da diversi fattori quali la tecnica, il grado di allenamento, le condizioni ambientali e le caratteristiche fisiologiche proprie di ciascun atleta. Ne consegue che la loro conoscenza permette di impostare un’alimentazione adeguata sia nella fase di allenamento che in quella di recupero tra un allenamento e il successivo, con l’obbiettivo di ottimizzare la performance.

 Il consumo limitato a questi quantitativi deve essere ottenuto con l’assunzione di bevande a basso contenuto alcolico, preferibilmente ai pasti (bere quantità anche piccole o moderate al di fuori dei pasti aumenta la probabilità di sviluppare ipertensione): ciò significa non più di 680 ml di una birra normale o 280 ml di vino (12% vol), soprattutto in caso di ipertensione; per le donne e i soggetti esili il consumo dovrebbe essere dimezzato1.
Il consumo limitato a questi quantitativi deve essere ottenuto con l’assunzione di bevande a basso contenuto alcolico, preferibilmente ai pasti (bere quantità anche piccole o moderate al di fuori dei pasti aumenta la probabilità di sviluppare ipertensione): ciò significa non più di 680 ml di una birra normale o 280 ml di vino (12% vol), soprattutto in caso di ipertensione; per le donne e i soggetti esili il consumo dovrebbe essere dimezzato1.
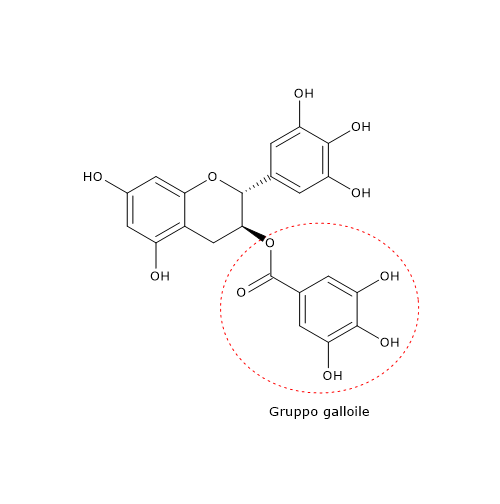

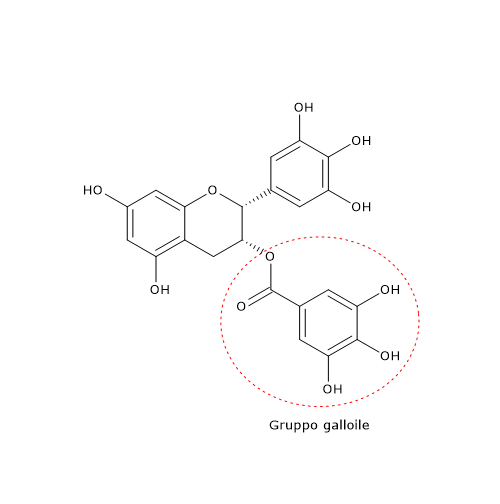
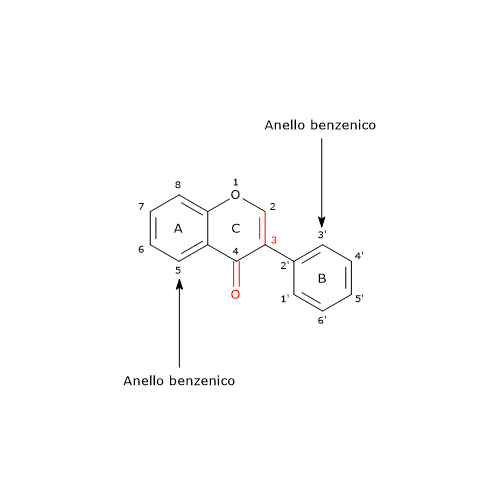
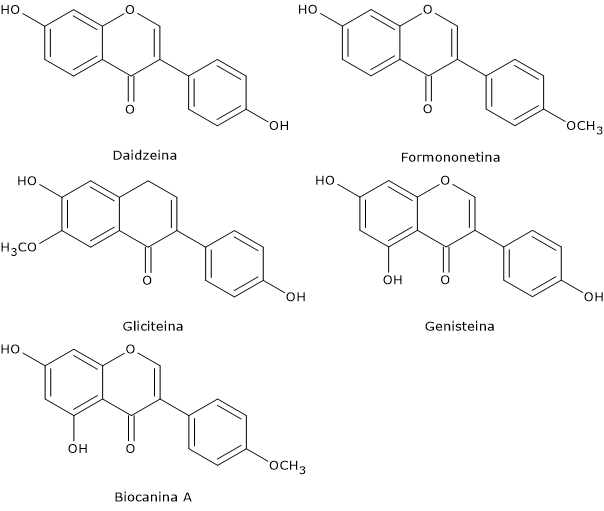



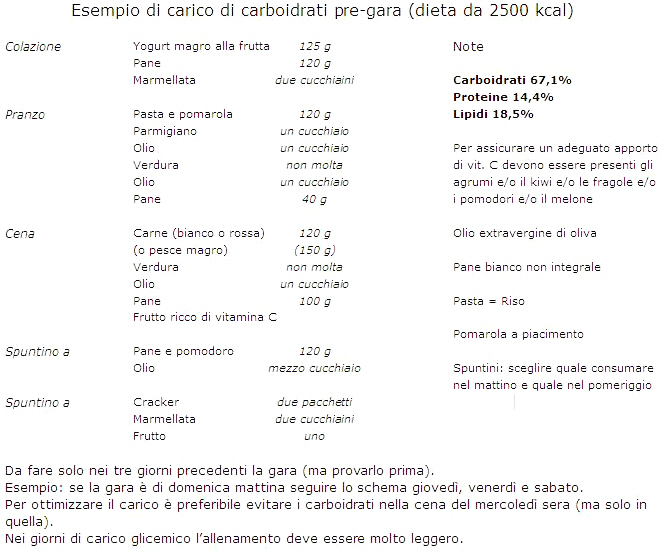
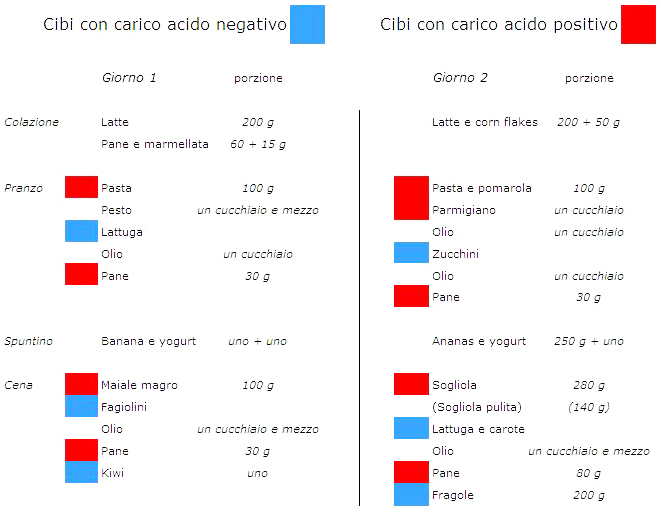
 Il glucosio potrà così lasciare l’epatocita, per mezzo di un trasportatore di membrana detto GLUT2, e riversarsi nel circolo sanguigno per essere distribuito alle cellule extraepatiche, in primis neuroni e globuli rossi per i quali rappresenta la principale, e per i globuli rossi l’unica fonte di energia. I neuroni, con l’esclusione di quelli presenti in alcune zone del cervello che non possono fare a meno del glucosio, sono in grado di utilizzare un’altra fonte di energia che diviene preponderante nei periodi di digiuno prolungato, i corpi chetonici.
Il glucosio potrà così lasciare l’epatocita, per mezzo di un trasportatore di membrana detto GLUT2, e riversarsi nel circolo sanguigno per essere distribuito alle cellule extraepatiche, in primis neuroni e globuli rossi per i quali rappresenta la principale, e per i globuli rossi l’unica fonte di energia. I neuroni, con l’esclusione di quelli presenti in alcune zone del cervello che non possono fare a meno del glucosio, sono in grado di utilizzare un’altra fonte di energia che diviene preponderante nei periodi di digiuno prolungato, i corpi chetonici.
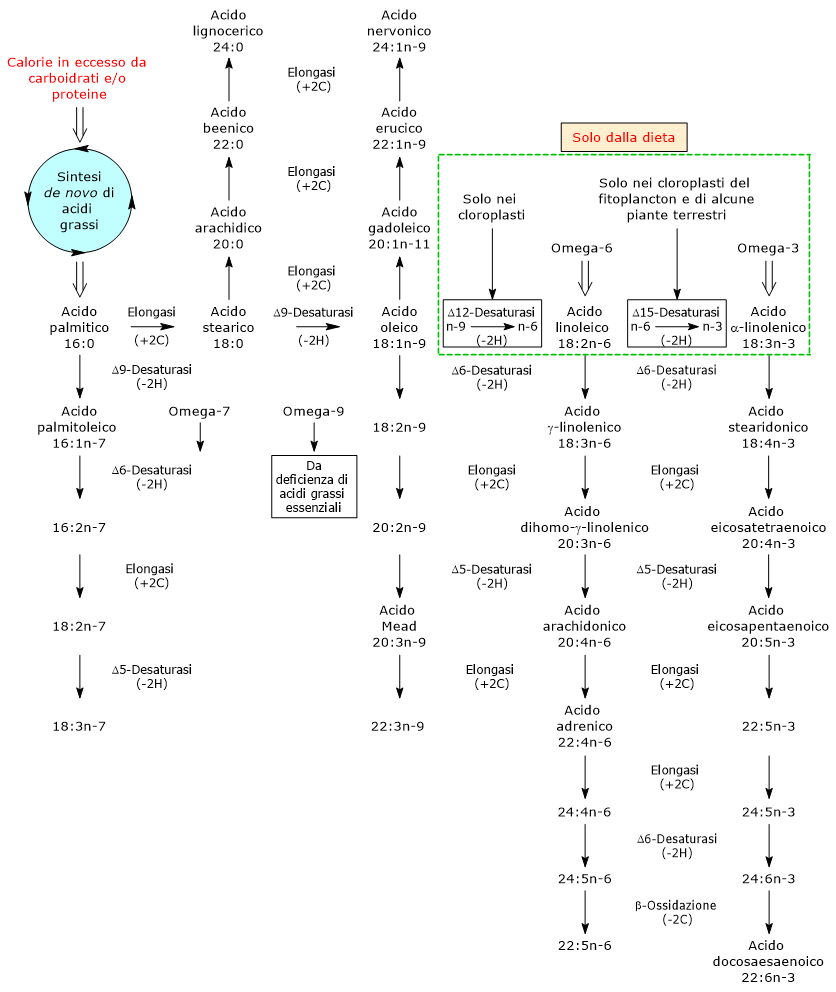
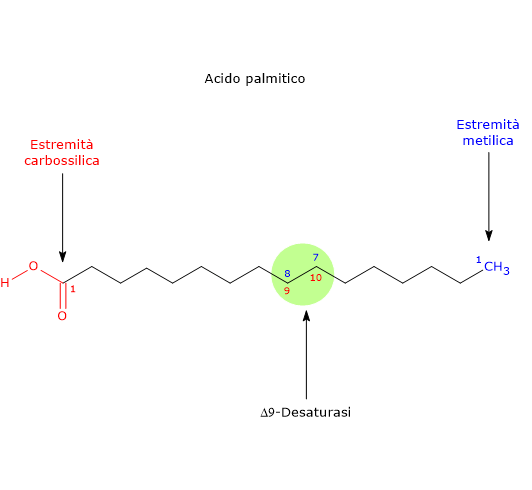 Se il substrato è l’acido stearico il doppio legame apparirà tra la posizione posizione n-9 e n-10 della catena, e verrà prodotto l’acido oleico.
Se il substrato è l’acido stearico il doppio legame apparirà tra la posizione posizione n-9 e n-10 della catena, e verrà prodotto l’acido oleico.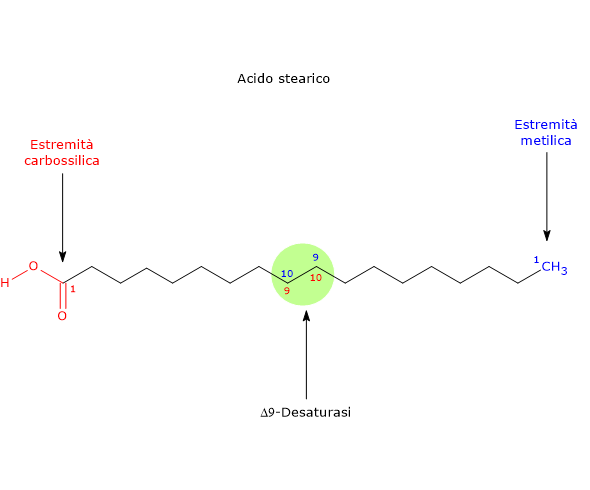
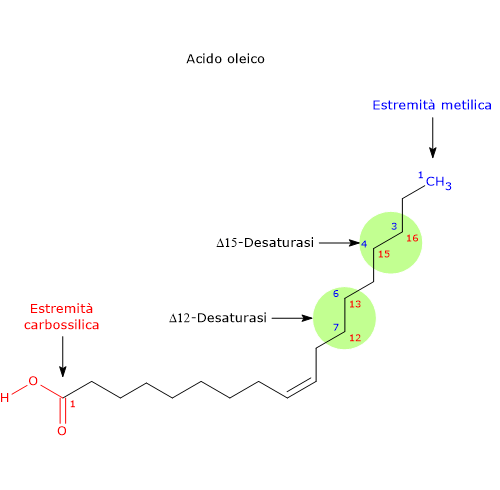
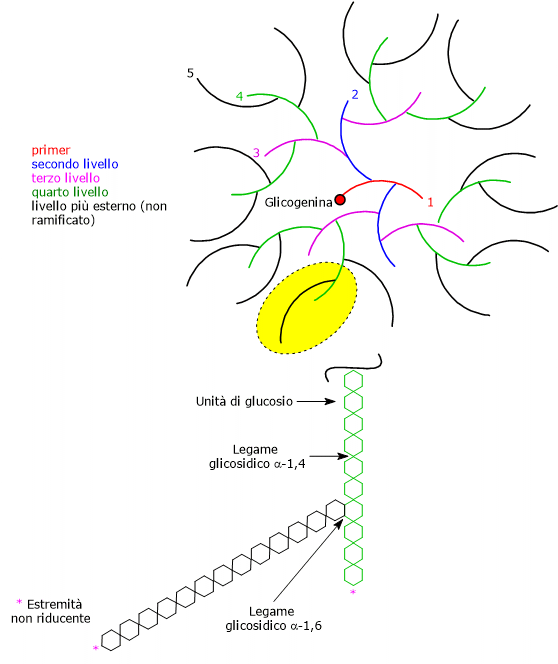 Per produrre glicogeno è indispensabile assumere carboidrati; ma quanti, quali, quando e con che frequenza?
Per produrre glicogeno è indispensabile assumere carboidrati; ma quanti, quali, quando e con che frequenza?
 Inoltre, a seconda del prodotto, può essere presente anche saccarosio non scisso.
Inoltre, a seconda del prodotto, può essere presente anche saccarosio non scisso.